
Dengue Virus Non-structural Protein 1 Modulates Infectious Particle Production via Interaction with the Structural Proteins
Dengue virus (DENV) is a major arthropod-borne human pathogen, infecting more than 400 million individuals annually worldwide; however, neither a therapeutic drug nor a prophylactic vaccine is currently available. Amongst the DENV proteins, non-structural protein 1 (NS1) is one of the most enigmatic, being required for RNA replication, but also secreted from infected cells to counteract antiviral immune response, thus contributing to pathogenesis. Despite its essential role at early stages of the viral replication cycle, the molecular determinants governing NS1 functions are unknown. Here, we used a combination of genetic, high-resolution imaging and biochemical approaches and found that NS1 additionally plays an important role for the production of infectious virus particles. By using a novel trans-complementation system with fully functional epitope-tagged NS1, we show that NS1 interacts with the structural proteins residing in the envelope of the virus particle. An NS1 variant retained in the endoplasmic reticulum still supported efficient DENV particle production, demonstrating that secretion of NS1 is dispensable for virion production. This study expands the list of functions exerted by NS1 for the DENV replication cycle. Given this multi-functional nature, NS1 appears to be an attractive target for antiviral therapy.
Published in the journal:
. PLoS Pathog 11(11): e32767. doi:10.1371/journal.ppat.1005277
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1005277
Summary
Dengue virus (DENV) is a major arthropod-borne human pathogen, infecting more than 400 million individuals annually worldwide; however, neither a therapeutic drug nor a prophylactic vaccine is currently available. Amongst the DENV proteins, non-structural protein 1 (NS1) is one of the most enigmatic, being required for RNA replication, but also secreted from infected cells to counteract antiviral immune response, thus contributing to pathogenesis. Despite its essential role at early stages of the viral replication cycle, the molecular determinants governing NS1 functions are unknown. Here, we used a combination of genetic, high-resolution imaging and biochemical approaches and found that NS1 additionally plays an important role for the production of infectious virus particles. By using a novel trans-complementation system with fully functional epitope-tagged NS1, we show that NS1 interacts with the structural proteins residing in the envelope of the virus particle. An NS1 variant retained in the endoplasmic reticulum still supported efficient DENV particle production, demonstrating that secretion of NS1 is dispensable for virion production. This study expands the list of functions exerted by NS1 for the DENV replication cycle. Given this multi-functional nature, NS1 appears to be an attractive target for antiviral therapy.
Introduction
Dengue is the most prevalent arthropod-borne viral disease affecting around 400 million people worldwide and causing around 25,000 deaths per year [1]. Dengue virus (DENV) infections can lead to a wide range of clinical manifestations, ranging from asymptomatic to life-threatening dengue hemorrhagic fever and shock syndrome. However, in spite of its high medical relevance, no prophylactic vaccines or antiviral therapies are currently available and therefore a better understanding of the flavivirus life cycle is essential to promote the development of effective therapeutic regimens.
DENV has a single stranded RNA genome of positive polarity, encoding for a polyprotein that is co - and post-translationally processed into three structural proteins (capsid, prM, and envelope) and seven nonstructural proteins (NS1-NS2A-NS2B-NS3-NS4A-NS4B-NS5) [2]. After viral entry and release of the genomic RNA into the cytoplasm of infected cells, newly synthesized viral proteins induce massive remodeling of intracellular membranes, creating distinct intracellular structures where viral RNA replication and virion assembly take place [3,4]. Nucleocapsid formation, thought to occur in close proximity to replication sites, is likely accompanied by acquisition of a lipid envelope via budding into endoplasmic reticulum (ER) membranes enriched in the envelope protein E and prM [5,6], through as yet undefined mechanisms. Assembled virions, stored within ER stacks in highly ordered arrays, are then released from the cell via the conventional secretory pathway, where cleavage of the prM protein by furin, a protease residing in the trans-Golgi network (TGN), renders the viral particles infectious.
Flavivirus NS1 is a multifunctional 48-kDa glycoprotein that is translocated into the ER lumen co-translationally. Within the ER, NS1 promptly dimerizes upon addition of high-mannose carbohydrates [7], and is targeted to three destinations: the viral replication sites, the plasma membrane and the extracellular compartment. The majority of secreted NS1 is a soluble, proteolipid particle forming an open-barrel hexameric shell with a central channel occupied by lipids [8]. The three-dimensional high-resolution structure of the DENV NS1 dimer was recently solved by X-ray crystallography [9], providing valuable insights into the complex NS1 fold (Fig 1). The dimer contains three domains: first, a small β-roll domain formed by two intertwined β-hairpins; second, a Wing domain, composed of an α/β subdomain and a discontinuous connector that sits against the β-roll; third, a β-ladder domain, formed by 18 antiparallel β-strands (9 contributed by each monomer) assembled in a continuous β-sheet that runs along the whole length of the dimer (Fig 1A, left panel). The protrusion created by the β-roll and the connector subdomain renders one side of the dimer hydrophobic, and has been proposed to face the ER membrane and to interact with other transmembrane viral proteins [9,10]. Conversely, within the NS1 hexamer, the β-roll faces the interior of the lipoparticle, where it associates with the central lipid core (Fig 1A, right panel). On the opposite side of the β-roll, both in the dimeric and the hexameric form, the distal tips of the β-ladder and the Wing domain loops point outward, and are therefore exposed to the solvent.

Secreted NS1 as well as NS1 residing on the plasma membrane and within cells, plays important roles in immune evasion via binding to complement proteins and modifying or antagonizing their functions [11–14]. Besides its immune evasive functions, NS1 modulates early events in viral RNA replication, was shown to co-localize with double strand RNA (dsRNA) and to interact with NS4B [10,15–17]. Indeed, deletion of NS1 from the viral genome completely abrogates replication, but ectopic expression of NS1 in trans can efficiently rescue NS1-deleted (ΔNS1) viruses [18–21].
Because of its essential role early in RNA replication, genetic studies have thus far provided limited information on the molecular determinants of NS1 responsible for the viral replication cycle and did not investigate possible functions of the protein for assembly and release of infectious virus particles. By using a combination of genetic, high-resolution imaging and biochemical approaches we discovered a novel role of NS1 for the production of infectious DENV particles that is linked to NS1 interaction with the structural proteins, but independent from NS1 secretion.
Results
Identification of critical NS1 determinants required for DENV replication
Sequence analysis and visual inspection of the recently solved three-dimensional crystal structure [9] of NS1 were performed to assess the degree of conservation of amino acid residues and to identify the most relevant positions to be targeted by site-directed mutagenesis (Fig 1B). Based on their distribution within the NS1 dimer and their relative conservation across the Flavivirus genus, we selected 46 residues for alanine scanning mutagenesis, including five invariant cysteine residues (C4, C55, C179, C291, C312), recently shown to be engaged in disulfide bonds and playing an essential role in stabilizing the protein fold [9,22]. To dissect the impact of each individual mutation on the different steps of the viral replication cycle, we assessed viral RNA replication and virus spread by taking advantage of a DVR2A luciferase reporter virus genome (Fig 2A). VeroE6 cells were electroporated with in vitro transcripts of wild-type (WT) or a given NS1 mutant and viral replication was assessed by luciferase activity 24, 48, 72, 96 and 120 h later (Fig 2B). Additionally, a replication-deficient NS5 mutant (GND) with a lethal mutation affecting the RNA-dependent RNA polymerase activity was included as negative control. Based on the replication phenotypes, half of the NS1 mutants displayed only minor defects or replicated comparably to WT (Table 1). Conversely, 23 mutations, including those affecting cysteine residues engaged in disulfide bonds, severely or completely blocked viral RNA replication (Fig 2B and Table 1, underlined mutants). Most of these mutations clustered on the core of the Wing and β-ladder domain, affecting residues that point towards the β-roll (S1 Fig), which has been proposed to face the ER membrane [9]. Interestingly, in close proximity to the previously reported di-amino acid motif (N10-K11) suggested to mediate interaction with NS4B and association with ER membranes [10], a mutation targeting W8 within the NS1 β-roll domain completely abrogated viral RNA replication. Similarly, mutations within the greasy finger loop of the β-ladder, namely Y158A and G161A, resulted in a lethal phenotype as already shown for alanine substitutions at residues G159 and F160 [9]. Altogether, these results provide a comprehensive map of molecular determinants within NS1 essential for viral RNA replication, highlighting an important role for selected residues of the β-roll and β-ladder domains, clustering on hydrophobic protrusions within the NS1 dimer structure (S1 Fig).


Identification of NS1 as a critical factor for infectious particle production
To determine the impact of each mutation on the production of infectious virus particles, culture supernatants of transfected cells (Fig 2B) were harvested 72 h after transfection and used to infect naïve VeroE6 cells. Virus production was determined by luciferase assay 48 h later (Fig 3A). This experiment revealed a group of mutations (S114A, W115A, D180A, T301A) with minor effects on RNA replication, but massive impairment of virus production (up to ~2.5 Log10 reduction compared to WT) (Fig 3B). Noteworthy, a mutation targeting T117 within the unresolved stretch of the Wing domain and in close proximity to S114 and W115, slightly enhanced particle production. Altogether, these results suggest a previously undiscovered role of NS1 for the production of infectious DENV particles.

Next we wanted to corroborate this observation and rule out that impaired virus production was an indirect consequence of diminished replication fitness rather than a specific defect in assembly or release of viral progeny. To this end, we assessed the impact of these mutations on RNA replication in the context of a sub-genomic reporter replicon (sgDVR2A) that does not support virus production, thus measuring replication independent from a possible contribution of virus spread (Fig 4A). In this and all subsequent analyses we focused on mutants with selective alterations of virus production (S114A, W115A, D180A, T301A; Fig 3B) in order to avoid possible indirect effects resulting from impaired replication fitness (Fig 3B). Therefore, NS1 mutants with strong replication defects were excluded (Table 1). Moreover, since several NS1 mutants with a defect in virus production were slightly impaired in RNA replication we included as control the NS1 mutation R314A that caused minimal defect in replication, but did not affect virus production (Table 1). Furthermore, mutant T117A was included in the analysis because of its increased capacity to produce infectious DENV particles and its close proximity to some of the sites where mutations caused a selective reduction of virus production. VeroE6 cells were transfected with in vitro transcripts of the replicon constructs and replication was measured 24, 48 and 72 h later (Fig 4A). While at early times post transfection we observed a moderate reduction in luciferase activity for S114A, W115A, D180A and T301A compared to WT (2 to 5-fold; 24 h.p.t.), all mutants replicated comparably at 48 and 72 h.p.t., arguing for a minor contribution of RNA replication to the observed reduction in infectious particle production. As already observed in the context of the full-length reporter virus, the T117A mutant exhibited a replication profile comparable to WT also within the subgenomic sgDVR2A replicon whereas R314A exhibited an overall decrease in replication fitness, with a 50% to 75% reduction at any time point. Collectively, these results demonstrate a selective defect for a sub-group of NS1 mutants in assembly and/or release of virus particles.

To investigate further the phenotype of the NS1 mutants with respect to the production of infectious intra - and extracellular virus particles, and to corroborate these observations in human cells, we determined the infectivity profiles of each mutant in the context of a full-length DENV genome transfected into human hepatoma Huh7 cells. Three days post-transfection, titers of infectious virus released into the culture media or contained in cells were determined by limiting dilution assay with naïve cells. The results shown in Fig 4B demonstrate that the amounts of intra - and extracellular infectivity were altered in all mutants, albeit to very different degrees. In agreement with the results obtained with the reporter virus genome, we found that alanine substitutions at residues S114, W115, D180 and T301 reduced extracellular infectivity titers up to 100-fold, confirming an essential role of these amino acid residues in NS1 for particle production. Interestingly, the amounts of intracellular virus particles were also reduced 5 - to 10-fold. While this impairment argued for a defect of the NS1 mutants in virus assembly or maturation, the higher reduction of extracellular virus titers suggested an additional effect on particle release as inferred from the ratio of intra - to extracellular virus titers and comparison with the WT (Fig 4C). The R314A mutation reduced the virus titer only ~3.5-fold and did not affect the ratio of intra - to extracellular infectivity, consistent with a subtle effect on assembly or virus maturation (Fig 4C). Interestingly, the T117A mutant produced 12-fold more intracellular virus than WT, concomitant with a ~5-fold higher titer of extracellular virus particles, indicating accelerated assembly or infectivity maturation and reduced virus particle release. In conclusion, these results suggest that alanine substitutions at position S114, W115, T117, D180 or T301 of NS1 alter the production of infectious virus, supporting the notion that NS1 is a critical determinant for assembly or release of infectious virus particles.
NS1 secretion is dispensable for DENV virus entry, RNA replication and particle release
NS1 accumulates in extracellular fluids as a homo-hexamer with a lipidic core [8,23,24] and besides its immune evasive functions [11–13] was shown to enhance virus attachment upon entry [25]. In addition, some studies hypothesized a link between NS1 secretion and virus assembly or release [26]; however the lack of genetic tools in those days allowing the selective block of NS1 secretion in the context of a complete replication cycle precluded any functional investigation.
Prior studies have shown that YFV, KUNV and WNV mutants lacking NS1 do not replicate, but can be rescued when NS1 is complemented in trans by ectopic expression of the full-length protein [17,18,21,27]. To elucidate the possible role(s) of NS1 secretion in the DENV replication cycle, we utilized a similar approach and generated a DENV genome containing a 97 amino acids in-frame deletion within the NS1 gene (DVR2AΔNS1). This mutant retained the N-terminal 156 and the C-terminal 99 residues, respectively (Fig 5A, left panel). In parallel, we engineered a set of helper VeroE6 cell lines, constitutively expressing different NS1 variants after lentiviral transduction of expression vectors containing the complete NS1 coding region (NS1WT) or the empty pWPI vector (CTRL) that served as positive and negative controls, respectively. Furthermore, we engineered C-terminally tagged variants carrying a HA-affinity epitope (NS1HA) or the well-described KDEL motif (NS1KDEL), responsible for retrieval of ER luminal proteins from the Golgi apparatus by retrograde transport (Fig 5A, right panel). Correct protein expression and secretion of each NS1 variant was confirmed by western-blotting (Fig 5B). As expected, NS1HA had a slower electrophoretic mobility than the WT. Furthermore, both NS1WT and NS1HA were readily detected in the culture media, while NS1KDEL was effectively retained in the ER.

Next, we assessed rescue of viral RNA replication and particle production by the NS1 variants by using transfection of the DVR2AΔNS1 genome into each helper cell line. As shown in Fig 5C, the ΔNS1 genome was able to replicate in NS1WT, NS1HA and NS1KDEL helper cells, while no luciferase activity could be detected in CTRL cells lacking NS1. Of note, when each naïve helper cell line was infected with virus-containing culture fluids harvested 72 h.p.t, comparable levels of luciferase activity were detected in all conditions, indicating that C-terminally HA-tagged NS1 is fully functional (Fig 5D). Importantly, the rescue of particle production by NS1KDEL shows that secretion of NS1 is dispensable for infectious DENV particle production.
To address the relative efficiency of trans-complementation upon virus infection, NS1WT-, NS1HA - and NS1KDEL-expressing VeroE6 cells were infected with trans-complemented DVR2AΔNS1 particles (ΔNS1TCP), produced in VeroE6_NS1WT cells. Culture fluids were harvested 24, 48 and 72 h later and the amounts of produced particles were determined by focus-forming unit (FFU) assay on NS1WT cells (Fig 6A). Consistent with the luciferase assay data, ΔNS1TCP did not produce infectious virus on CTRL cells that do not express NS1, confirming that this mutant fails to replicate in the absence of NS1. Titers of infectious ΔNS1TCP particles released from VeroE6_NS1WT, NS1HA or NS1KDEL cells were higher than DVR2A wild-type infection on CTRL cells at early times post-infection (24 and 48 h p.i.), with no appreciable differences observed at later time points (72 h p.i.) (Fig 6B). Interestingly, while none of the trans-complemented TCPs gave rise to clearly visible plaques as previously reported [17], DENV-containing foci detected by immunostaining were larger than those produced by the full-length wild-type virus (Fig 6B, lower panel).
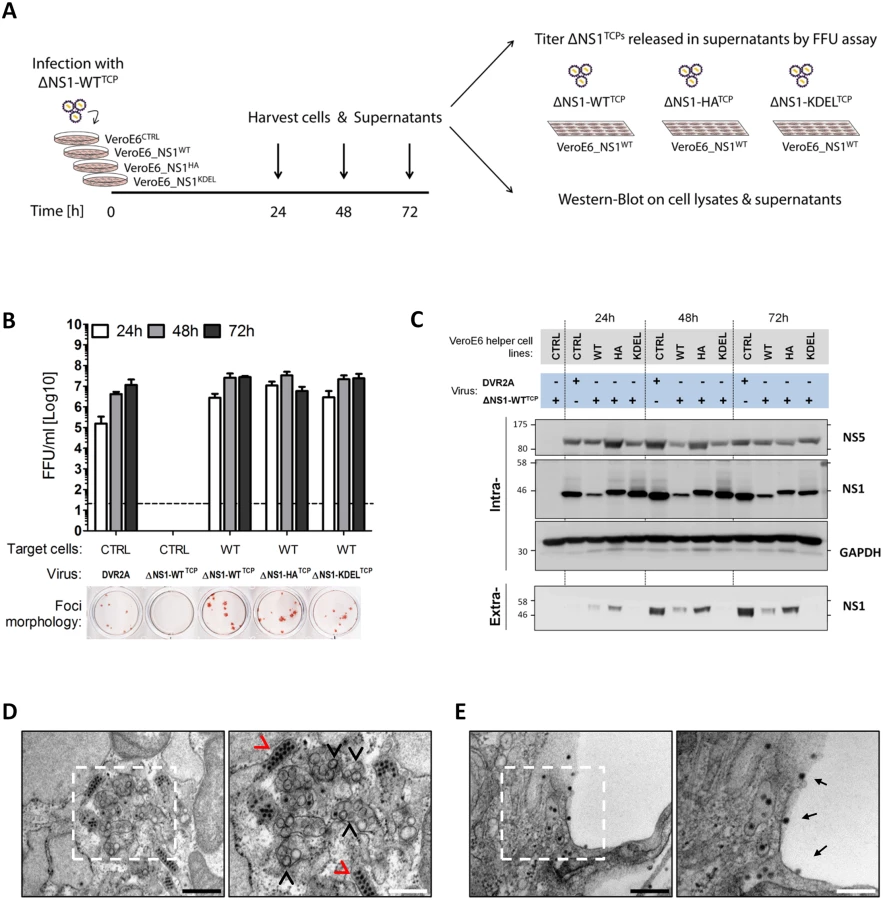
Additionally, lysates and culture supernatants of infected cells were harvested 24, 48 and 72 h p.i. to evaluate protein expression and secretion upon ΔNS1TCP infection. Under these conditions, virus replication in the various helper cell lines was comparable as judged by the intracellular protein levels of NS5 (Fig 6C; “Intra“). Furthermore, secretion profiles of HA - WT - and KDEL-tagged NS1 resembled those of uninfected cells, with the latter being efficiently retained intracellularly also upon DENV infection (Fig 6C; “Extra“).
To unequivocally confirm that the ΔNS1TCP system fully recapitulates wild-type DENV infections, we additionally investigated the ultrastructural morphology of NS1HA helper cells upon infection with DVR2AΔNS1 TCPs by using transmission electron microscopy. In agreement with the replication and infectious particle production data, NS1HA cells infected with DVR2AΔNS1 TCPs contained the characteristic membrane invaginations which have been proposed to represent viral replication factories (vRFs) [5,6] and electron-dense virus particles forming regular arrays within the ER (Fig 6D). Moreover, a large number of virus particles were observed on the plasma membrane or accumulating within the extracellular space between adjacent cells (Fig 6E). These structures were absent in both uninfected NS1HA cells and ΔNS1TCP-infected CTRL cells (S2 Fig).
In conclusion, these results demonstrate full functionality of HA-tagged and ER-retained NS1 supporting both DENV RNA replication and production of infectious virus particles.
NS1 associates with the viral envelope glycoproteins
Since NS1 secretion appeared functionally unlinked to infectious particle production, we next hypothesized that NS1 function(s) required for the late steps of the viral replication cycle might involve interactions between NS1 and the structural DENV proteins. To address this hypothesis we took advantage of our ΔNS1TCP system using HA-tagged NS1 for trans-complementation. DVR2AΔNS1 TCP stocks produced and titered in NS1WT helper cells were used to infect NS1HA target cells at an MOI of 1. Forty-eight hours later, cell lysates were subjected to HA-affinity capture using anti-HA agarose beads, and purified NS1 protein complexes or whole cell lysates were analyzed by western-blot using C-, prM-, E - and NS5-specific antibodies. Specificity of western-blot and immunoprecipitation analysis was monitored by including VeroE6_CTRL and VeroE6_NS1WT cells, respectively. As shown in Fig 7A, upon infection of NS1 helper cells with ΔNS1TCP, comparable amounts of structural proteins accumulated in both NS1WT and NS1HA cells, confirming that DVR2AΔNS1 replicates efficiently in both cell lines. Most interestingly, upon HA-immunoprecipitation, all three structural proteins (E, prM and C) specifically co-precipitated with NS1HA arguing for an interaction between NS1 and DENV virions and possibly also subviral particles. In spite of comparable protein amounts in the cell lysates, no specific signal was detected in case of the non-tagged NS1WT or for the NS5 protein, confirming specificity of the NS1HA-immunoprecipitation.

To corroborate the interaction between NS1 and the structural proteins, we performed analogous pull-down experiments using cells that had been infected with wild-type DENV. VeroE6 cells were mock-infected or infected with DENV-2 at an MOI of 1 and 48 hours later cell lysates were subjected to immunoprecipitation using a rabbit pre-immune serum (PIS) or an NS1-specific polyclonal antiserum (Fig 7B). Interestingly, also under these conditions E and prM were specifically co-immunoprecipitated with NS1 whereas C was not detected. The absence of C might be due to the overall lower efficiency of this immunocapture approach, to suboptimal conditions for antibody binding or to an altered ratio of subviral particles to infectious virions in the TCP system as compared to wild-type virus-infected cells (see discussion). Nevertheless, the interaction between E and NS1 was confirmed in a reciprocal approach by which the NS1 protein in DENV-2 infected cells could be specifically co-immunoprecipitated with envelope (S3 Fig). Altogether, these results support the notion that NS1 interacts with the viral envelope glycoproteins.
Mapping of the site in NS1 required for interaction with the DENV envelope glycoproteins
Based on the results described above, we hypothesized that the selective defect in infectious particle production observed for some of the NS1 point mutants was due to an altered association with the envelope glycoproteins. To investigate this hypothesis, we analyzed the association of selected NS1 mutants with the structural proteins by co-immunoprecipitation in the DVR2AΔNS1 TCP system, because it allowed highly efficient pull-down of NS1. We engineered stable VeroE6 cell lines expressing HA-tagged forms of the NS1 mutants S114A, W115A, T117A, D180A, T301A and R314A and infected these cell lines with ΔNS1TCP at an MOI of 1. Samples harvested 48 h.p.i., together with positive and negative controls, were subjected to HA-specific pull-down and cell lysates, immunocomplexes or culture supernatants were analyzed as described above. All cell lines expressed HA-tagged NS1 variants and despite small variations in the expression levels rescued DVR2AΔNS1 replication to similar extents as judged by the abundance of E, prM and C (Fig 8A) and the luciferase activity in the cell lysates (S5 Fig). Interestingly, analyses of NS1HA-immunocomplexes revealed marked differences in the interaction profiles of these mutants with the structural proteins. In case of NS1 mutants S114A and W115A, a significant reduction in E and prM co-precipitation, concomitant with a loss of the C-specific signal was observed, arguing for impaired association of these NS1 variants with DENV virions (Fig 8B; reduction of Envelope: ~7.5 - and 3.8-fold, respectively). In contrast, mutants D180A and T301A exhibited a selective loss of co-precipitated C while retaining high prM and E interaction, suggesting that NS1 contributes to virus production in an additional manner that is independent from interaction with the envelope glycoproteins. Consistent with their higher competence in supporting infectious particle production, T117A and R314A were still able to bind all three structural proteins, although the C-specific signal was lower as compared to wild-type.

These results were further corroborated by co-localization analyses of HA-tagged NS1 mutants with the envelope glycoproteins revealing reduced co-localization in case of the two mutants that were impaired in virus production (S114A and W115A; S6 Fig), but no significant change in case of all the other mutants. Importantly, no major differences could be observed with respect to NS1 secretion into the culture supernatants (Fig 8C), strengthening the notion that NS1 secretion is functionally unlinked to its role in the production of infectious extracellular virus particles.
NS1 co-localizes with assembled viral particles
To further investigate the interaction between NS1 and the structural proteins, we used immunofluorescence to visualize the sub-cellular colocalization of NS1 with the structural proteins. To allow detection of putative DENV assembly sites and/or assembled virus particles, we aimed to perform simultaneous immunostaining of capsid, envelope and NS1. Since we were limited by the availability of antibodies allowing for triple staining, we engineered a C-terminally mCherry-tagged NS1 variant (NS1mCherry) to be visualized without requirement for antibodies (Fig 9A). In the initial set of experiments, we confirmed correct NS1mCherry expression and sub-cellular distribution. Importantly, mCherry-tagged NS1 supported efficient DENV RNA replication and infectious particle production upon infection with ΔNS1TCP demonstrating full functionality of the fluorescently tagged NS1 (Fig 9B and 9C). Taking advantage of this approach, we analyzed infected cells by confocal microscopy and observed several discrete structures where Envelope, Capsid and NS1 colocalized (Fig 9D). Although the nature of these structures that might correspond to putative assembly sites or assembled virus particles is not clear, this colocalization provides additional evidence for a previously unreported association between NS1 and the structural DENV proteins.

To overcome the inherent limitations of fluorescence microscopy in allocating specific proteins to distinct subcellular structures and to elucidate the nature of these NS1-positive structures we performed correlative light-electron microscopy (CLEM) of VeroE6_NS1mCherry cells infected with ΔNS1TCP (Fig 10). MCherry-fluorescent structures were allocated by confocal microscopy of fixed cells grown on photo-etched gridded coverslips (Fig 10A and 10B), which were subsequently processed for EM (Fig 10C). After alignment of confocal and electron microscopy images we were able to identify NS1-enriched structures. As observed with NS1HA, NS1mCherry-infected cells also contained the characteristic DENV-induced membrane rearrangements. Indeed, we found that highly fluorescent mCherry-positive areas corresponded to ER, vesicle packets (VPs) and ER-associated bags containing arrays of DENV particles (Fig 9D and 9E). These results are consistent with our previous studies describing the association of NS1 with VPs as determined by immuno-EM [5]. Most importantly, the present data further support our assumption that NS1 associates with assembled virus particles (Fig 10Db and 10Ed), in agreement with our results from immunoprecipitation and confocal microscopy experiments.

Discussion
Flavivirus NS1 has emerged as one of the most enigmatic proteins forming distinct intra - and extracellular complexes and contributing to pathogenesis as well as viral replication. While soluble and cell-surface–associated NS1 was shown to modulate complement activation pathways through interactions with host proteins, such as the regulatory protein factor H, complement factor C4 or clusterin [11–13,28] and to induce cross-reacting antibodies to human proteins [29–32], the role of NS1 in the viral replication cycle has so far been elusive. NS1 was initially thought to be involved in virus assembly or maturation, given its subcellular localization within the ER lumen and its secretion profile, largely mirroring that of the structural proteins prM and E [33,34]. However, this hypothesis was challenged by the co-localization of NS1 with dsRNA, a marker for RNA replication sites and by biochemical and genetic evidences supporting an essential role of NS1 in viral RNA replication [10,35–37]. Additionally, NS1 has been reported to interact with NS4B and/or NS4A, which are assumed to relay NS1 signals to other components of the viral replicase through their ER luminal segments.
In the present study, we used a combination of genetic, biochemical, and imaging approaches to investigate the functional role of intra - and extracellular NS1 in the DENV replication cycle. In addition to the previously reported mutations within the N-linked glycosylation sites [35,38] and the cysteine residues engaged in disulfide bonds [39], we identified 18 additional residues that completely or severely reduced DENV replication. Interestingly, most of these mutations clustered on residues located towards the proposed ER binding site (S1 Fig). Among these, a conserved tryptophan residue at amino acid position 8 (W8A) was found to be essential for RNA replication. This residue is located in close proximity to the previously reported di-amino acid motif (N10K11) within the β-roll domain of WNV NS1 that is also critical for efficient RNA replication [10,40]. These residues are located in an exposed region of the NS1 dimer suggested to face the ER membrane and possibly mediating the interaction with NS4B. Moreover, within the NS1 hexamer, these residues point towards the inner cavity of the barrel and might contribute to the association of NS1 with its lipid cargo (S1B Fig) [9]. While these results are consistent with the proposed way how the NS1 dimer associates with intracellular membranes and provide a detailed genetic map of NS1 residues essential for viral RNA replication, further studies are required to decipher the impact of these mutations on protein-protein interactions and induction of ultra-structural membrane rearrangements.
Most interestingly, our mutagenesis approach identified a group of NS1 mutants with selective defects in infectious particle production. These mutants (S114A, W115A, D180A and T301A) had only minor or negligible defects in NS1 protein stability and viral RNA replication as judged by their replication kinetics in the context of a sub-genomic replicon (Fig 4A), but released up to 100-fold lower amounts of infectious DENV particles than the wild-type (Figs 3A and 4B). Of note, amounts of intracellular infectivity were reduced ~5 - to 10-fold, arguing that the NS1 mutations affected both assembly (as evidenced from intracellular virus titers) and release of infectious DENV particles (indicated by titer reduction in cell culture supernatants). Although we cannot precisely comment on the individual contribution of NS1 to virus assembly and particle release, the identification of an additional NS1 mutant (T117A) producing ~12-fold higher amounts of intracellular infectious virus strongly hints towards a primary role of NS1 in the assembly of infectious DENV particles.
Based on previous studies reporting the ability of ectopically-expressed NS1 to trans-complement YFV or WNV NS1 deletion mutants [18,21], we established a DENV-based ΔNS1TCP system to characterize NS1 functions. Taking advantage of this method, we probed the capacity of an intracellularly retained KDEL-tagged NS1 (NS1KDEL) to support production and release of viral particles and demonstrate that secretion of NS1 is dispensable for these functions. Of note, KDEL-tagged proteins are shuttling between the ER and the Golgi apparatus and thus, NS1KDEL could still assist early vesicular traffic of viral particles, i.e. from virion budding sites (ER) to early secretory compartments (ERGIC) (Fig 11). Recently it has been reported that depletion of the KDEL receptor or a subset of class II Arf proteins reduces secretion of non-infectious sub-viral particles and that prM—KDEL receptor interaction plays a role in virus secretion, arguing that flavivirus release from infected cells is assisted by specific sorting mechanisms [41,42]. However, additional viral or host factors appear to be required to coordinate these processes, since knock-down of KDEL receptor or Arf4+5 expression reduced YFV and DENV1-3 secretion less than 10-fold and had no effect on DENV4 and WNV particle production. Further studies will be needed to elucidate the possible contribution of NS1 trafficking from the ER to early or intermediate secretory compartments for DENV particle release.

By using HA-tagged NS1 variants (NS1HA) and ΔNS1 virus-infected cells, we identified a previously unreported interaction between NS1 and the envelope glycoproteins E and prM. Although more than two decades ago E—NS1 complexes were identified in insect cells infected with a selected group of flaviviruses, the interaction was suggested to result from unspecific protein aggregation, lacking functional significance and not reproducible in DENV-2 - and YFV-infected cells [26]. In contrast, several lines of evidence suggest that NS1 interaction with these structural proteins is specific and of importance for the production of infectious DENV particles: (i) the co-immunoprecipitation of NS1 with prM and E and, possibly indirectly, with C; (ii) the identification of triple-positive structures enriched in NS1, E and C by confocal microscopy; (iii) the detection of NS1 in compartments containing assembled virions by CLEM. Importantly, immunoprecipitation experiments using lysates of wild-type DENV-2-infected cells and an NS1-specific antibody confirmed the interaction of NS1 with both prM and E glycoproteins. Of note, while in this experimental set-up no specific signal could be detected for the capsid protein, it was well co-precipitated in the DVR2AΔNS1 TCP system. This discrepancy might be due to the overall lower NS1 capture efficiency in case of wild-type virus-infected cells. Alternatively, it is possible that in wild-type virus-infected cells subviral particles (SVPs) are produced in higher abundance relative to virus particles, whereas in the DVR2AΔNS1 TCP system production of virus particles might be favored relative to SVPs. Consistent with this assumption we observed a much faster and more efficient production of infectious virus particles in our TCP system than with wild-type virus-infected cells (Fig 6B). Assuming that NS1 can also interact with prM/E present on the surface of SVPs, in case of their excess production we would still observe coprecipitation of NS1 with the envelope glycoproteins, whereas C would no longer be co-precipitated. In contrast, when virus particles are produced in excess over SVPs, we would observe NS1 co-precipitation with prM/E and C, i.e. virions. This is consistent with the observed colocalization of NS1 with C and E (Fig 9) and the enrichment of NS1 in areas containing fully assembled DENV particles (Fig 10). Moreover, the conclusion is consistent with the membrane topology of E, prM and NS1 that are located in the lumen of the ER, while capsid resides on the cytoplasmic side of ER membranes (Fig 11). Since no tangible interactions between capsid and the glycoproteins were reported to date, the most likely explanation for the apparent NS1-C co-precipitation is an interaction between NS1 and assembled virions. While the precise mechanism remains to be determined, the specificity and functional relevance of these interactions was corroborated by the interaction profiles of NS1 mutants having defects in infectious particle production. Mutations affecting highly conserved residues of the flexible solvent-exposed loop in the NS1 Wing domain (S114A and W115A; Fig 8D) simultaneously abrogated C, prM and E association arguing that this NS1 loop is engaged in interaction with DENV virions. In contrast, mutations affecting residues in the β-ladder domain (D180A and T301A) preserved glycoprotein binding, but prevented association with capsid arguing for a second function of NS1 in the assembly of DENV particles that is independent from envelope protein interaction. While the exact mechanism remains to be established, it is tantalizing to speculate that NS1 might, directly or indirectly via these residues, interact with other viral or cellular factor(s) required for the formation of infectious DENV particles. Alternatively, NS1 might assist membrane budding or conformational changes in prM/E required for the envelopment of nucleocapsids. This hypothesis is supported by the localization of “capsid non-binder” mutants in the NS1 dimer structure. In fact, D180 resides at the intersection of the Wing and β-ladder domain and T301 points towards the ER membrane and is solvent exposed (Fig 8D). These mutations might affect the NS1 dimer fold, its affinity for membranes or its membrane-bending ability, while preserving prM/E-association through the distal tips of the Wing domain that points towards the ER lumen. Alternatively, these mutations might induce conformational constraints in prM/E complexes, reducing their plasticity and capability to envelope budding nucleocapsids, affect the recruitment of prM/E to assembly sites or only indirectly affect association with capsid, as a consequence of altered interactions with other viral [43,44] or host factors playing critical roles in virion morphogenesis. However, ultrastructural analysis of NS1 mutants failed to identify assembly intermediates, the only striking difference to the wild-type being a marked reduction in the overall amount of electron-dense particles found in proximity to or directly at the plasma membrane (S2 Fig). These results suggest that nucleocapsid formation and envelopment are coupled or that naked nucleocapsids have an extremely short half-life. Further experiments will be required to dissect the exact role of NS1 in the assembly process of infectious virus particles.
Assembly of flavivirus particles is a poorly understood process (reviewed in [45]). High-resolution imaging approaches have shed some light on the topological arrangement of viral RNA replication and virus particle assembly. The viral replicase machinery is assumed to reside in highly organized membranous structures, designated as vesicle packets (VPs). These structures are formed by ER invaginations containing pores that would allow the release of newly synthesized viral RNA to be used for packaging into virions [5,6]. While genetic evidence suggests the involvement of several non-structural proteins and host factors in flavivirus assembly [41,43,44,46–48], the underlying molecular mechanisms are not known. Besides the lack of tangible interactions between C and the prM/E complex, major limitations are posed by the difficulty to visualize assembly intermediates and the seemingly unspecific incorporation of genomic RNA into nucleocapsids [49,50]. Based on the results presented here, it is tempting to speculate that NS1 might assist virion morphogenesis via its lipid-remodeling activity [8,9], its affinity for membranes [9] and its ability to interact with both non-structural [10,15] and structural proteins. In this respect NS1 might provide essential lipids or recruit essential host factor required for the biogenesis of the replication complex, while coordinating the recruitment of E and prM to assembly sites juxtaposed to the viral replicase [5] (Fig 11).
In conclusion, the present study provides a comprehensive genetic map of NS1 determinants important for viral RNA replication and identifies a novel role of NS1 for the production of infectious DENV particles. We demonstrate that NS1 interacts with the envelope glycoproteins presumably on the surface of virions and these interactions are required for efficient production of infectious virus particles. Given its multiple roles in counteracting host defense, promoting RNA replication and enhancing production of virus particles, NS1 exemplifies the genetic economy of flaviviruses and emerges as attractive target for antiviral drugs.
Materials and Methods
Antibodies and sera
The mouse monoclonal antibody recognizing human GAPDH (sc-47724/0411) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). The mouse anti-Envelope monoclonal antibody (3H5-1) was purchased from ATCC. The mouse monoclonal antibody 6F3.1 reacting with the capsid protein was a kind gift of Dr. John G. Aaskov (Queensland University of Technology, Australia); rabbit polyclonal serum anti-capsid was a kind gift of Dr. Andrea Gamarnik (Fundación Instituto Leloir, Argentina). The rabbit polyclonal antibodies recognizing Envelope, NS1, NS5 and prM were previously described [5]. Rabbit anti-HA antibody (Ab9110) and mouse anti-NS1 antibody (ab41623) were purchased from Abcam. The mouse monoclonal anti-HA (H3663) and anti-Flag (F1804) antibodies, agarose anti-HA conjugated beads and secondary anti-mouse and anti-rabbit horse-radish peroxidase-conjugated antibodies were purchased from Sigma-Aldrich (Sigma-Aldrich, Saint Louis, MO).
Cell culture
Huh7 [51], HeLa [52], VeroE6 (ATCC #CRL-1586), and BHK-21 (ATCC #CCL-10) cells were maintained in Dulbecco's modified Eagle medium (DMEM; Invitrogen, Karlsruhe, Germany) supplemented with 2 mM l-glutamine, nonessential amino acids, 100 U/ml penicillin, 100 μg/ml streptomycin and 10% fetal calf serum. VeroE6 stably expressing NS1WT or tagged derivatives were cultured in the presence of 10 μg/ml of puromycin.
Immunoblot analysis
Samples were denatured in 2x protein sample buffer (200 mM Tris [pH 8.8], 5 mM EDTA, 0.1% Bromophenolblue, 10% sucrose, 3% SDS, 1 mM DTT) and incubated for 5 min at 95°C. Proteins were separated by SDS-polyacrylamide gel electrophoresis (PAGE) and transferred onto polyvinylidene difluorid membranes by using a MINI-SDS-PAGE wet-blotting apparatus (Bio-Rad, Munich, Germany). Membranes were blocked with 5% non-fat dry milk in PBS/0.5% Tween-20 (PBST) and incubated with primary antibodies (capsid 1 : 50; GAPDH 1 : 1,000; HA 1 : 1,000; E 1 : 1,000; prM 1 : 500; NS1 1 : 500) by over-night incubation at 4°C or for 1 h at room temperature. After 3 washes with PBST, membranes were incubated with secondary horse radish peroxidase-conjugated antibodies, developed with the Western Lightning Plus-ECL reagent (Perkin Elmer; Waltham, MA) and bands were imaged using an Intas ChemoCam Imager 3.2 (Intas, Göttingen).
Immunofluorescence analysis
VeroE6 helper cells expressing different forms of NS1 were infected with ΔNS1TCP at an MOI of 1. Two days later, cells were fixed with 4% PFA for 10 min at room temperature, permeabilized with 0.5% (vol/vol) Triton X-100 in PBS and aspecific biding sites blocked with PBS containing 5% FBS for 30 min at RT.
For staining of NS1mCherry cells, rabbit anti-NS1 (S3 Fig), or rabbit anti-C and mouse anti-Envelope antibodies (Fig 8) were used in combination with goat anti-mouse Alexa 647-conjugated and donkey anti-rabbit Alexa488-conjugated secondary antibodies. For staining of NS1HA cells (wild-type and mutants) (S4 Fig), rabbit anti-HA and mouse anti-Envelope antibodies were used in combination with goat anti-mouse Alexa 568-conjugated and donkey anti-rabbit Alexa488-conjugated secondary antibodies. Nuclear DNA was stained with 4′,6-diamidino-2-phenylindole (DAPI) (Molecular Probes, Karlsruhe, Germany). Coverslips were mounted in Fluoromount-G mounting medium (Southern Biotechnology Associates, Birmingham, AL). For 3D visualization of NS1mCherry, E and C, samples were imaged with an Ultraview ERS spinning disk (PerkinElmer Life Sciences) on a Nikon TE2000-E inverted confocal microscope using a Plan-Apochromat VC 100× objective (numeric aperture [NA], 1.4). Optical sections of 0.13 μm were acquired separately for each channel. Z-stacks were deconvolved with a theoretical point-spread function, and chromatic shifts between green and far-red dyes were corrected using Autoquant X3 software. 3D reconstructed images were created using the Imaris 8 software package. For colocalization analyses of HA-tagged NS1 mutants and envelope fluorescence signals, Pearson's correlation coefficient was calculated on single plane images, by using the integrated function in Fiji (ImageJ).
Infectivity assays
For determination of virus titers by limiting dilution assay, Huh7 target cells were seeded into 96-well plates (104 cells/well) the day before infection. Cells were inoculated with serial dilutions of virus-containing supernatants that had been filtered through a 0.45-μm-pore-size filter. Infected cells were detected by immune staining of the E protein using the mouse anti-E antibody (3H5-1; diluted 1 : 500) and secondary horseradish peroxidase-conjugated antibody (1 : 200). Virus titers (expressed as 50% tissue culture infective dose [TCID50]/ml) were calculated as previously reported [53]. For determination of virus titers by Focus forming unit (FFU) assay, VeroE6 or VeroE6_NS1WT cells (2x105 cells/well) seeded into 24-well plates, were infected with serial dilutions of 0.45-μm-filtered supernatants and incubated in the presence of 0.8% methylcellulose for 5 days. Monolayers were rinsed twice in PBS, fixed with 5% PFA and permeabilized with 0.2% (v/v) TritonX-100 in PBS for 15 min. Infected foci were detected by immune staining of the E protein using the mouse anti-E antibody (3H5-1; diluted 1 : 1,000 in PBS) and secondary horseradish peroxidase-conjugated antibody (1 : 200). Alternatively, virus titers were determined by standard plaque assay (PFU) on target VeroE6 cells as previously described [54]. To determine intracellular infectivity titers, transfected cells were disrupted by several freeze–thaw cycles as described earlier [55]. In brief, transfected Huh7 cells were extensively washed with PBS, scraped off the plate into PBS and centrifuged for 5 min at 700 × g. Cell pellets were resuspended in complete DMEM (containing 15 mM HEPES, pH 7.2–7.5) and subjected to three cycles of freezing and thawing by using liquid nitrogen and a thermo block set to 37°C. Cell debris was removed by centrifugation at 20,000 × g for 10 min at 4°C. Virus-containing culture supernatants from transfected cells were treated in the same way and infectivity was determined in parallel by limiting dilution assay as described above.
Co-immunoprecipitation assay
VeroE6 cells were seeded into 15-cm2 dishes (7.5x106 cells/dish). Twenty-four hours later, cell monolayers were either mock-infected or infected with DENV-2 (MOI = 1). Forty-eight hours later, cell monolayers were scraped into 1 ml lysis buffer (50 mM Tris-HCl [pH 8.0], 0.5% NP-40, 150 mM NaCl and protease inhibitor cocktail (cOmplete, Roche)). After 30 min incubation on ice, cell debris was removed by 15 min centrifugation at 13,800xg and 400 μl of clarified cell lysate was incubated with rabbit pre-immune serum (PIS) or rabbit anti-NS1 antiserum for 6 hours at 4°C in a head-to-head shaker. Samples were incubated with 40 μl of protein A beads slurry (Sigma Aldrich, St. Louis, USA) for 1 hour at 4°C, washed three times with 1 ml of lysis buffer and protein A-bound complexes were transferred into a fresh tube. After a final wash with 1 ml of lysis buffer for 15 min at 4°C, protein complexes were eluted at room-temperature by two consecutive steps with 75 μl of 0.1 M glycine [pH 2.5] for 5 min. Collected supernatants were immediately neutralized by adding 7.5 μl 1 M Tris-HCl [pH 8] and denatured for 5 min at 95°C in the presence of 33 μl of 6X SDS sample buffer. Alternatively, mouse anti-HA or mouse anti-E antibodies were used in combination with protein G beads slurry (Sigma Aldrich, St. Louis, USA) and the immunoprecipitation was carried exactly as described above. For ΔNS1TCP experiments, VeroE6 cells stably expressing an empty pWPI vector (CTRL), or wild-type NS1 (NS1WT), or HA-tagged NS1 (NS1HA) or derivatives thereof were seeded into 10-cm2 dishes (3x106 cells/dish). Twenty-four hours later, cell monolayers were infected with DVR2AΔNS1 (MOI = 1) for 4 h at 37°C. Forty-eight hours post-infection, cell monolayers were scraped into 1 ml lysis buffer (50 mM Tris-HCl [pH 8.0], 0.5% NP-40, 150 mM NaCl and protease inhibitor cocktail (cOmplete, Roche) as recommended by the manufacturer). After 30 min incubation on ice, cell debris was removed by 15 min centrifugation at 13,800xg. For HA-specific affinity capture, samples were incubated with HA-specific agarose beads (Sigma-Aldrich, St.Louis, USA) for 5 h by continuously inverting the tubes at 4°C. Beads were washed three times for 20 min with large volumes of lysis buffer at 4°C and samples were eluted at room-temperature in two consecutive steps with 3% SDS in PBS for 5 min and PBS for 5 min, respectively. The two eluates were pooled and precipitated over-night at -20°C with 4 volumes of ice-cold acetone. Samples were centrifuged for 30 min at 20,000xg, air-dried, resuspended in 2x SDS sample buffer and boiled for 5 min at 95°C. Alternatively, 4 h before ΔNS1TCP infection, VeroE6 cells stably expressing wild-type NS1 (NS1WT) or HA-tagged NS1 (NS1HA) were transfected with 10 μg of pcDNA 3.1(+) empty vector or pCMV_NS4B-FLAG (encoding a C-terminally Flag-tagged NS4B protein of Hepatitis C virus) by using the TransIT-LT1 transfection reagent (MirusBio LLC, Madison, WI, USA) as recommended by the manufacturer. Infection and HA-specific immunoprecipitation were carried out exactly as described above. Eluted proteins were further analyzed by western blot as specified in the results section. For analysis of secreted NS1, supernatants were clarified through 0.45 μm filters and incubated with 40 μl mouse anti-HA slurry beads over-night at 4°C, in a head-to-head shaker. Immunoprecipitates were washed three times with lysis buffer and eluted as described above. Alternatively cell culture supernatants containing NS1 were used undiluted for SDS-PAGE.
Electroporation of DV, DVR2A and sgDVR2A in vitro transcripts into mammalian cells
In vitro transcripts were generated as previously described [56]. For RNA transfection, single-cell suspensions were prepared by trypsinization, washed with PBS, and resuspended at a concentration of 1x107 cells (Huh7) or 1.5x107 cells (VeroE6) per ml in Cytomix, supplemented with 2 mM ATP and 5 mM glutathione. Five to 10 μg of subgenomic or genomic in vitro transcript was mixed with 400 μl of the cell suspension and transfected by electroporation using a Gene Pulser system (Bio-Rad) and a cuvette with a gap width of 0.4 cm (Bio-Rad) at 975 μF and 270 V. Cells were immediately diluted into 20 ml of DMEM cplt and seeded in the appropriate format (1ml/well in 24-well plates; 2 ml/well in 12-well plates; 15 ml/dish in 15 cm-diameter dishes).
Production of lentiviruses
Human immunodeficiency virus (HIV)-based particles that were pseudotyped with the vesicular stomatitis virus glycoprotein (VSV-G) were generated by transfection of 293T cells as described previously [53]. For production of transducing lentiviral particles, 293T cells were co-transfected with a transfer vector encoding the gene of interest and a puromycin resistance gene (pWPI_Puro), the HIV-1 packaging plasmid (pCMV) and a VSV-G expression vector (pMD.G) (ratio 3 : 3:1). Cells were transfected using the CalPhos mammalian transfection kit as recommended by the manufacturer (Becton Dickinson). After 48 and 72 h, supernatants were harvested, clarified through 0.45 μm pore size filters, pooled and stored in aliquots at -20°C until use. Titers of lentiviral particles were estimated by colony-forming unit (CFU) assay using HeLa cells and serial dilutions of each lentiviral stock. Inoculated cells were subjected to selection using the appropriate antibiotic for 5–7 days and surviving cell colonies were stained for 15min with a 1% crystal violet solution. Colonies were counted under a bright-field inverted microscope and lentivirus titers were calculated as CFU/ml.
Transient replication assay
Huh7 or VeroE6 cells transfected with full-length or subgenomic DVsR2A in vitro transcripts were seeded as specified in the results section (typically 12 - or 24-wells plates). Replication was determined by measuring luciferase activity in cell lysates 4, 24, 48 and 72 h after transfection. For determination of luciferase activity, cells were washed once with PBS and lysed by adding 200 μl of luciferase lysis buffer as previously described [57]. Cells were frozen immediately at −70°C and after thawing, lysates were resuspended by gentle pipetting. For each well 20 μl lysate, mixed with 400 μl assay buffer (25 mM glycylglycine, 15 mM MgSO4, 4 mM EGTA, 1 mM DTT, 2 mM ATP, 15 mM K2PO4 [pH 7.8], 1.42 μM coelenterazine H), were measured for 10 sec in a tube luminometer (Lumat LB9507, Berthold, Freiburg, Germany). In some cases (24-well plates, 100 μl lysis buffer per well) a plate luminometer was used (Mithras LB940, Berthold, Freiburg, Germany). Each well was measured in duplicate. To determine the amount of infectious virus particles released into culture supernatants 72 h after electroporation, naïve VeroE6 cells were inoculated with culture supernatants of transfected cells and 48 h later luciferase activity was determined. Kinetics of virus replication were calculated by normalizing the relative light units (RLU) measured at a given time point to the respective 4 h value.
Plasmids
The plasmid containing a synthetic version of the full-length DENV-2 strain 16681 (pFK-DVs) and subgenomic constructs derived therefrom without or with luciferase reporter were previously described [56]. For NS1-targeted site-directed mutagenesis external primers NS1_BamHI_f (5’-CTG GGA TTT TGG ATC CTT GGG AGG AG-3’) and NS1_KasI_r (5’-TCC GTC ATA GTG GCG CCT ACC ATA AC-3’) were used in combination with mutagenic forward and reverse primers (the full list of primers is available upon request). Amplicons containing the desired point mutation in the NS1 coding region were inserted into the full-length DVsR2A constructs via the BamHI-KasI restriction sites. Full-length non-reporter constructs containing selected NS1 mutations were generated by insertion of a DNA fragment that was excised via BamHI and KasI from pFK-DVsR2A into pFK-DVs; subgenomic luciferase reporter constructs were generated by DNA fragment exchange using MluI and KasI and insertion into pFK-sgDVsR2A. The pCMV_NS4B-FLAG expressing C-terminally Flag-tagged NS4B protein of Hepatitis C virus was previously described [58]. The pWPIpuro-based NS1 constructs used for production of lentiviral vectors were generated by PCR-based amplification of the NS1 encoding sequence plus the last 24 codons of the envelope coding region, by using full-length genomic constructs as template and the following primers: pWPIpuro_BamHI_NS1_HA_frw (5’-GCT GGG ATC C ACC ATG AGC ACC TCA CTG TCT GTG ACA CTA GTA TTG GTG-3’) and pWPIpuro_NS1_Stop_NheI_rev (5’-AGA TAG CTA GCC TAA GCT GTG ACC AAG GAG TTG ACC AAA TTC-3’). Amplicons were inserted into pWPI-Puro via BamHI and SpeI restriction sites. For variants encoding the C-terminal HA epitope or the KDEL ER retrieval sequence, the primer pWPIpuro_BamHI_NS1_HA_frw was used in combination with NheI_NS1-HA_stop_rev (5’-ATA GCT AGC CTA AGC GTA ATC TGG AAC ATC GTA TGG GTA TGA TCC AGC TGT GAC CAA GGA GTT GAC CAA ATT CTC TTC TTT CT-3’) or pWPIpuro_NS1_Stop_NheI_KDEL_rev (5’-AGA TAG CTA GCC TAT AGC TCG TCC TTA GCT GTG ACC AAG GAG TTG ACC-3’), respectively.
The pWPIpuro-based NS1mCherry expression construct was generated by amplifying the mCherry sequence contained in pFKI389neoNS3-3′δg_JFH-1_NS5A-aa2359_mCherry [59] with primers pWPIpuro_SpeI_mCherry_frw (5´-TCA ACT CCT TGG TCA CAG CTA CCG GTG GAT CGA TGG TGA GCA AGG GCG AGG A-3´) and pWPIpuro_SpeI_mCherry_rev (5´-AAA ACT AGT CTA CTT GTA CAG CTC GTC CAT GC-3´) and the NS1 coding sequence contained in pWPIpuro_NS1 with primers pWPIpuro_SpeI_NS1_frw (5´-GAC ACT AGT ATT GGT GGG AAT TGT GAC AC-3´) and pWPIpuro_SpeI_NS1_rev (5´-TCC TCG CCC TTG CTC ACC ATC GAT CCA CCG GTA GCT GTG ACC AAG GAG TTG A-3´). The two PCR fragments were used to generate an intermediate amplicon using primers pWPIpuro_SpeI_NS1_frw and pWPIpuro_SpeI_mCherry_rev, which was inserted into pWPIpuro via SpeI. Finally, the envelope leader peptide sequence was excised from pWPIpuro_NS1 wild-type and inserted upstream of the NS1mCherry sequence via BamHI-MluI.
The ΔNS1 full-length DVsR2A genome used for trans-complementation studies in NS1 helper cell lines, was created by insertion of a 97 codon in-frame deletion into the NS1 open reading frame using NS1_BamHI_f and NS1_KasI_rev as external primers and NS1_156_frw (5’-GAA TTC GTT GGA AGT TGA ACA CAA CTA TAG ACC AGG CTA-3’) and NS1_156_rev (5’-TAG CCT GGT CTA TAG TTG TGT TCA ACT TCC AAC GAA TTC-3’) as internal primers. Thus, in the final construct, the first 156 codons and the last 99 codons of NS1 were retained, respectively.
Sequence alignments and molecular graphics
Sequence alignment of NS1 open-reading frames were performed using the ClustalW algorithm available in the JalView Desktop software and a ClustalW scoring algorithm, with the following isolates (UniprotKB/Swiss-Prot accession numbers are given): DV-1 (Brazil/97-11/1997) P27909; DV-2 (Thailand/16681-PDK53) P29991; DV-3 (Martinique/1243/1999) Q6YMS3; DV-4 (Thailand/0348/1991) Q2YHF0; West Nile virus P06935; Yellow Fever (Ivory Coast/1999) Q6J3P1; Japanese Encephalitis virus (SA-14) P27395; Kunjin virus (MRM61C) P14335; St. Louis Encephalitis virus (MS1-7) P09732. Molecular graphics were performed with the UCSF Chimera package developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco [60] on the DENV-2 NS1 crystal structure (Protein Data Bank [PDB] accession no. 4O6B).
Analysis of infected cells by transmission electron microscopy
VeroE6-based helper cell lines expressing different NS1HA mutants were seeded onto glass coverslips (5x104 cells/well) and 16 h later, infected with 1 MOI of DVR2AΔNS1. After a 48 h incubation period, cells were fixed and prepared for transmission electron microscopy as described previously [61]. For correlative light-electron microscopy, VeroE6-based helper cell lines expressing NS1mCherry were seeded into glass-bottom culture dishes containing photo-etched gridded coverslips (MatTek Corporation, Ashland, MA) and infected with 1 MOI of DVR2AΔNS1. After 48 hours, cells were fixed with 4% PFA and 0.2% glutaraldehyde in PBS for 30 min at room temperature, washed three times with PBS, stained with DAPI and analyzed by fluorescence microscopy to acquire optical sections of 0.13 μm as described above. NS1mCherry-positive cells were imaged and their position on the gridded coverslip was recorded. Cells were then processed for analysis as described previously [61]. The DAPI signal was used for correlation purposes and images were adapted by using the Image J (version 1.46r) and Adobe Photoshop (version 12.1.1) software packages.
Statistical analyses
Statistical analyses were performed by applying the two-tailed, unpaired Student’s t-test available within the GraphPad Prism (ver. 5.0) software.
Supporting Information
Zdroje
1. Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, Scott TW, Farrar JJ, Hay SI (2013) The global distribution and burden of dengue. Nature 496 : 504–507. nature12060 [pii]; doi: 10.1038/nature12060 23563266
2. Nowak T, Farber PM, Wengler G, Wengler G (1989) Analyses of the terminal sequences of West Nile virus structural proteins and of the in vitro translation of these proteins allow the proposal of a complete scheme of the proteolytic cleavages involved in their synthesis. Virology 169 : 365–376. 2705302
3. Paul D, Bartenschlager R (2013) Architecture and biogenesis of plus-strand RNA virus replication factories. World J Virol 2 : 32–48. doi: 10.5501/wjv.v2.i2.32 24175228
4. Belov GA, van Kuppeveld FJ (2012) (+)RNA viruses rewire cellular pathways to build replication organelles. Curr Opin Virol 2 : 740–747. S1879-6257(12)00141-1 [pii]; doi: 10.1016/j.coviro.2012.09.006 23036609
5. Welsch S, Miller S, Romero-Brey I, Merz A, Bleck CK, Walther P, Fuller SD, Antony C, Krijnse-Locker J, Bartenschlager R (2009) Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe 5 : 365–375. S1931-3128(09)00098-5 [pii]; doi: 10.1016/j.chom.2009.03.007 19380115
6. Junjhon J, Pennington JG, Edwards TJ, Perera R, Lanman J, Kuhn RJ (2014) Ultrastructural characterization and three-dimensional architecture of replication sites in dengue virus-infected mosquito cells. J Virol 88 : 4687–4697. JVI.00118-14 [pii]; doi: 10.1128/JVI.00118-14 24522909
7. Winkler G, Randolph VB, Cleaves GR, Ryan TE, Stollar V (1988) Evidence that the mature form of the flavivirus nonstructural protein NS1 is a dimer. Virology 162 : 187–196. 2827377
8. Gutsche I, Coulibaly F, Voss JE, Salmon J, d'Alayer J, Ermonval M, Larquet E, Charneau P, Krey T, Megret F, Guittet E, Rey FA, Flamand M (2011) Secreted dengue virus nonstructural protein NS1 is an atypical barrel-shaped high-density lipoprotein. Proc Natl Acad Sci U S A 108 : 8003–8008. 1017338108 [pii]; doi: 10.1073/pnas.1017338108 21518917
9. Akey DL, Brown WC, Dutta S, Konwerski J, Jose J, Jurkiw TJ, DelProposto J, Ogata CM, Skiniotis G, Kuhn RJ, Smith JL (2014) Flavivirus NS1 structures reveal surfaces for associations with membranes and the immune system. Science 343 : 881–885. science.1247749 [pii]; doi: 10.1126/science.1247749 24505133
10. Youn S, Li T, McCune BT, Edeling MA, Fremont DH, Cristea IM, Diamond MS (2012) Evidence for a genetic and physical interaction between nonstructural proteins NS1 and NS4B that modulates replication of West Nile virus. J Virol 86 : 7360–7371. JVI.00157-12 [pii]; doi: 10.1128/JVI.00157-12 22553322
11. Avirutnan P, Hauhart RE, Somnuke P, Blom AM, Diamond MS, Atkinson JP (2011) Binding of flavivirus nonstructural protein NS1 to C4b binding protein modulates complement activation. J Immunol 187 : 424–433. jimmunol.1100750 [pii]; doi: 10.4049/jimmunol.1100750 21642539
12. Avirutnan P, Fuchs A, Hauhart RE, Somnuke P, Youn S, Diamond MS, Atkinson JP (2010) Antagonism of the complement component C4 by flavivirus nonstructural protein NS1. J Exp Med 207 : 793–806. jem.20092545 [pii]; doi: 10.1084/jem.20092545 20308361
13. Chung KM, Liszewski MK, Nybakken G, Davis AE, Townsend RR, Fremont DH, Atkinson JP, Diamond MS (2006) West Nile virus nonstructural protein NS1 inhibits complement activation by binding the regulatory protein factor H. Proc Natl Acad Sci U S A 103 : 19111–19116. 0605668103 [pii]; doi: 10.1073/pnas.0605668103 17132743
14. Wilson JR, de Sessions PF, Leon MA, Scholle F (2008) West Nile virus nonstructural protein 1 inhibits TLR3 signal transduction. J Virol 82 : 8262–8271. JVI.00226-08 [pii]; doi: 10.1128/JVI.00226-08 18562533
15. Chatel-Chaix L, Fischl W, Scaturro P, Cortese M, Kallis S, Bartenschlager M, Fischer B, Bartenschlager R (2015) A combined genetic-proteomic approach identifies residues within Dengue virus NS4B critical for interaction with NS3 and viral replication. J Virol. JVI.00867-15 [pii]; doi: 10.1128/JVI.00867-15
16. Mackenzie JM, Jones MK, Young PR (1996) Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology 220 : 232–240. S0042-6822(96)90307-4 [pii]; doi: 10.1006/viro.1996.0307 8659120
17. Youn S, Ambrose RL, Mackenzie JM, Diamond MS (2013) Non-structural protein-1 is required for West Nile virus replication complex formation and viral RNA synthesis. Virol J 10 : 339. 1743-422X-10-339 [pii]; doi: 10.1186/1743-422X-10-339 24245822
18. Lindenbach BD, Rice CM (1997) trans-Complementation of yellow fever virus NS1 reveals a role in early RNA replication. J Virol 71 : 9608–9617. 9371625
19. Khromykh AA, Kenney MT, Westaway EG (1998) trans-Complementation of flavivirus RNA polymerase gene NS5 by using Kunjin virus replicon-expressing BHK cells. J Virol 72 : 7270–7279. 9696822
20. Khromykh AA, Sedlak PL, Westaway EG (2000) cis - and trans-acting elements in flavivirus RNA replication. J Virol 74 : 3253–3263. 10708442
21. Khromykh AA, Sedlak PL, Guyatt KJ, Hall RA, Westaway EG (1999) Efficient trans-complementation of the flavivirus kunjin NS5 protein but not of the NS1 protein requires its coexpression with other components of the viral replicase. J Virol 73 : 10272–10280. 10559344
22. Edeling MA, Diamond MS, Fremont DH (2014) Structural basis of Flavivirus NS1 assembly and antibody recognition. Proc Natl Acad Sci U S A 111 : 4285–4290. 1322036111 [pii]; doi: 10.1073/pnas.1322036111 24594604
23. Flamand M, Megret F, Mathieu M, Lepault J, Rey FA, Deubel V (1999) Dengue virus type 1 nonstructural glycoprotein NS1 is secreted from mammalian cells as a soluble hexamer in a glycosylation-dependent fashion. J Virol 73 : 6104–6110. 10364366
24. Crooks AJ, Lee JM, Easterbrook LM, Timofeev AV, Stephenson JR (1994) The NS1 protein of tick-borne encephalitis virus forms multimeric species upon secretion from the host cell. J Gen Virol 75 (Pt 12): 3453–3460. 7527836
25. Alcon-LePoder S, Drouet MT, Roux P, Frenkiel MP, Arborio M, Durand-Schneider AM, Maurice M, Le B I, Gruenberg J, Flamand M (2005) The secreted form of dengue virus nonstructural protein NS1 is endocytosed by hepatocytes and accumulates in late endosomes: implications for viral infectivity. J Virol 79 : 11403–11411. 79/17/11403 [pii]; doi: 10.1128/JVI.79.17.11403-11411.2005 16103191
26. Blitvich BJ, Mackenzie JS, Coelen RJ, Howard MJ, Hall RA (1995) A novel complex formed between the flavivirus E and NS1 proteins: analysis of its structure and function. Arch Virol 140 : 145–156. 7646339
27. Young LB, Melian EB, Khromykh AA (2013) NS1' colocalizes with NS1 and can substitute for NS1 in West Nile virus replication. J Virol 87 : 9384–9390. JVI.01101-13 [pii]; doi: 10.1128/JVI.01101-13 23760245
28. Kurosu T, Chaichana P, Yamate M, Anantapreecha S, Ikuta K (2007) Secreted complement regulatory protein clusterin interacts with dengue virus nonstructural protein 1. Biochem Biophys Res Commun 362 : 1051–1056. S0006-291X(07)01834-7 [pii]; doi: 10.1016/j.bbrc.2007.08.137 17825259
29. Cheng HJ, Lin CF, Lei HY, Liu HS, Yeh TM, Luo YH, Lin YS (2009) Proteomic analysis of endothelial cell autoantigens recognized by anti-dengue virus nonstructural protein 1 antibodies. Exp Biol Med (Maywood) 234 : 63–73. 0805-RM-147 [pii]; doi: 10.3181/0805-RM-147
30. Falconar AK (2008) Monoclonal antibodies that bind to common epitopes on the dengue virus type 2 nonstructural-1 and envelope glycoproteins display weak neutralizing activity and differentiated responses to virulent strains: implications for pathogenesis and vaccines. Clin Vaccine Immunol 15 : 549–561. CVI.00351-07 [pii]; doi: 10.1128/CVI.00351-07 18160621
31. Falconar AK (1997) The dengue virus nonstructural-1 protein (NS1) generates antibodies to common epitopes on human blood clotting, integrin/adhesin proteins and binds to human endothelial cells: potential implications in haemorrhagic fever pathogenesis. Arch Virol 142 : 897–916. 9191856
32. Henchal EA, Henchal LS, Schlesinger JJ (1988) Synergistic interactions of anti-NS1 monoclonal antibodies protect passively immunized mice from lethal challenge with dengue 2 virus. J Gen Virol 69 (Pt 8): 2101–2107. 3404125
33. Lee JM, Crooks AJ, Stephenson JR (1989) The synthesis and maturation of a non-structural extracellular antigen from tick-borne encephalitis virus and its relationship to the intracellular NS1 protein. J Gen Virol 70 (Pt 2): 335–343. 2471787
34. Mason PW (1989) Maturation of Japanese encephalitis virus glycoproteins produced by infected mammalian and mosquito cells. Virology 169 : 354–364. 2523178
35. Muylaert IR, Chambers TJ, Galler R, Rice CM (1996) Mutagenesis of the N-linked glycosylation sites of the yellow fever virus NS1 protein: effects on virus replication and mouse neurovirulence. Virology 222 : 159–168. S0042-6822(96)90406-7 [pii]; doi: 10.1006/viro.1996.0406 8806496
36. Lindenbach BD, Rice CM (1999) Genetic interaction of flavivirus nonstructural proteins NS1 and NS4A as a determinant of replicase function. J Virol 73 : 4611–4621. 10233920
37. Suzuki R, de BL, Duarte dos Santos CN, Mason PW (2007) Construction of an infectious cDNA clone for a Brazilian prototype strain of dengue virus type 1: characterization of a temperature-sensitive mutation in NS1. Virology 362 : 374–383. S0042-6822(06)00875-0 [pii]; doi: 10.1016/j.virol.2006.11.026 17289102
38. Somnuke P, Hauhart RE, Atkinson JP, Diamond MS, Avirutnan P (2011) N-linked glycosylation of dengue virus NS1 protein modulates secretion, cell-surface expression, hexamer stability, and interactions with human complement. Virology 413 : 253–264. S0042-6822(11)00094-8 [pii]; doi: 10.1016/j.virol.2011.02.022 21429549
39. Wallis TP, Huang CY, Nimkar SB, Young PR, Gorman JJ (2004) Determination of the disulfide bond arrangement of dengue virus NS1 protein. J Biol Chem 279 : 20729–20741. doi: 10.1074/jbc.M312907200 ;M312907200 [pii]. 14981082
40. Youn S, Cho H, Fremont DH, Diamond MS (2010) A short N-terminal peptide motif on flavivirus nonstructural protein NS1 modulates cellular targeting and immune recognition. J Virol 84 : 9516–9532. JVI.00775-10 [pii]; doi: 10.1128/JVI.00775-10 20592095
41. Kudelko M, Brault JB, Kwok K, Li MY, Pardigon N, Peiris JS, Bruzzone R, Despres P, Nal B, Wang PG (2012) Class II ADP-ribosylation factors are required for efficient secretion of dengue viruses. J Biol Chem 287 : 767–777. M111.270579 [pii]; doi: 10.1074/jbc.M111.270579 22105072
42. Li MY, Grandadam M, Kwok K, Lagache T, Siu YL, Zhang JS, Sayteng K, Kudelko M, Qin CF, Olivo-Marin JC, Bruzzone R, Wang PG (2015) KDEL Receptors Assist Dengue Virus Exit from the Endoplasmic Reticulum. Cell Rep. S2211-1247(15)00167-9 [pii]; doi: 10.1016/j.celrep.2015.02.021
43. Kummerer BM, Rice CM (2002) Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J Virol 76 : 4773–4784. 11967294
44. Vossmann S, Wieseler J, Kerber R, Kummerer BM (2015) A Basic Cluster in the N Terminus of Yellow Fever Virus NS2A Contributes to Infectious Particle Production. J Virol 89 : 4951–4965. JVI.03351-14 [pii]; doi: 10.1128/JVI.03351-14 25694595
45. Apte-Sengupta S, Sirohi D, Kuhn RJ (2014) Coupling of replication and assembly in flaviviruses. Curr Opin Virol 9 : 134–142. S1879-6257(14)00202-8 [pii]; doi: 10.1016/j.coviro.2014.09.020 25462445
46. Mateo R, Nagamine CM, Spagnolo J, Mendez E, Rahe M, Gale M Jr., Yuan J, Kirkegaard K (2013) Inhibition of cellular autophagy deranges dengue virion maturation. J Virol 87 : 1312–1321. JVI.02177-12 [pii]; doi: 10.1128/JVI.02177-12 23175363
47. Heinz FX, Stiasny K, Puschner-Auer G, Holzmann H, Allison SL, Mandl CW, Kunz C (1994) Structural changes and functional control of the tick-borne encephalitis virus glycoprotein E by the heterodimeric association with protein prM. Virology 198 : 109–117. S0042-6822(84)71013-0 [pii]; doi: 10.1006/viro.1994.1013 8259646
48. Hsieh SC, Wu YC, Zou G, Nerurkar VR, Shi PY, Wang WK (2014) Highly conserved residues in the helical domain of dengue virus type 1 precursor membrane protein are involved in assembly, precursor membrane (prM) protein cleavage, and entry. J Biol Chem 289 : 33149–33160. M114.610428 [pii]; doi: 10.1074/jbc.M114.610428 25326389
49. Pong WL, Huang ZS, Teoh PG, Wang CC, Wu HN (2011) RNA binding property and RNA chaperone activity of dengue virus core protein and other viral RNA-interacting proteins. FEBS Lett 585 : 2575–2581. S0014-5793(11)00514-X [pii]; doi: 10.1016/j.febslet.2011.06.038 21771593
50. Teoh PG, Huang ZS, Pong WL, Chen PC, Wu HN (2014) Maintenance of dimer conformation by the dengue virus core protein alpha4-alpha4' helix pair is critical for nucleocapsid formation and virus production. J Virol 88 : 7998–8015. JVI.00940-14 [pii]; doi: 10.1128/JVI.00940-14 24807709
51. Nakabayashi H, Taketa K, Miyano K, Yamane T, Sato J (1982) Growth of human hepatoma cells lines with differentiated functions in chemically defined medium. Cancer Res 42 : 3858–3863. 6286115
52. Steigemann P, Wurzenberger C, Schmitz MH, Held M, Guizetti J, Maar S, Gerlich DW (2009) Aurora B-mediated abscission checkpoint protects against tetraploidization. Cell 136 : 473–484. S0092-8674(08)01601-2 [pii]; doi: 10.1016/j.cell.2008.12.020 19203582
53. Steinmann E, Brohm C, Kallis S, Bartenschlager R, Pietschmann T (2008) Efficient trans-encapsidation of hepatitis C virus RNAs into infectious virus-like particles. J Virol 82 : 7034–7046. JVI.00118-08 [pii]; doi: 10.1128/JVI.00118-08 18480457
54. Scaturro P, Trist IM, Paul D, Kumar A, Acosta EG, Byrd CM, Jordan R, Brancale A, Bartenschlager R (2014) Characterization of the mode of action of a potent dengue virus capsid inhibitor. J Virol 88 : 11540–11555. JVI.01745-14 [pii]; doi: 10.1128/JVI.01745-14 25056895
55. Gastaminza P, Kapadia SB, Chisari FV (2006) Differential biophysical properties of infectious intracellular and secreted hepatitis C virus particles. J Virol 80 : 11074–11081. JVI.01150-06 [pii]; doi: 10.1128/JVI.01150-06 16956946
56. Fischl W, Bartenschlager R (2013) High-throughput screening using dengue virus reporter genomes. Methods Mol Biol 1030 : 205–219. doi: 10.1007/978-1-62703-484-5_17 23821271
57. Kumar A, Buhler S, Selisko B, Davidson A, Mulder K, Canard B, Miller S, Bartenschlager R (2013) Nuclear localization of dengue virus nonstructural protein 5 does not strictly correlate with efficient viral RNA replication and inhibition of type I interferon signaling. J Virol 87 : 4545–4557. JVI.03083-12 [pii]; doi: 10.1128/JVI.03083-12 23408610
58. Gouttenoire J, Roingeard P, Penin F, Moradpour D (2010) Amphipathic alpha-helix AH2 is a major determinant for the oligomerization of hepatitis C virus nonstructural protein 4B. J Virol 84 : 12529–12537. JVI.01798-10 [pii]; doi: 10.1128/JVI.01798-10 20926561
59. Schaller T, Appel N, Koutsoudakis G, Kallis S, Lohmann V, Pietschmann T, Bartenschlager R (2007) Analysis of hepatitis C virus superinfection exclusion by using novel fluorochrome gene-tagged viral genomes. J Virol 81 : 4591–4603. JVI.02144-06 [pii]; doi: 10.1128/JVI.02144-06 17301154
60. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25 : 1605–1612. doi: 10.1002/jcc.20084 15264254
61. Paul D, Romero-Brey I, Gouttenoire J, Stoitsova S, Krijnse-Locker J, Moradpour D, Bartenschlager R (2011) NS4B self-interaction through conserved C-terminal elements is required for the establishment of functional hepatitis C virus replication complexes. J Virol 85 : 6963–6976. JVI.00502-11 [pii]; doi: 10.1128/JVI.00502-11 21543474
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2015 Číslo 11
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Dengue Virus Non-structural Protein 1 Modulates Infectious Particle Production via Interaction with the Structural Proteins
- On the Discovery of TOR As the Target of Rapamycin
- Parasite Glycobiology: A Bittersweet Symphony
- Broadening of Virus-Specific CD8 T-Cell Responses Is Indicative of Residual Viral Replication in Aviremic SIV Controllers
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy