
A Non-Coding RNA Promotes Bacterial Persistence and Decreases Virulence by Regulating a Regulator in
Staphylococcus aureus is a commensal and an opportunistic pathogen that causes a large range of community and hospital-acquired infections. The bacteria produce an array of virulence factors, the expression of which is regulated by a set of regulators including proteins and RNAs. In recent years, a large number of small non-coding RNAs encoded by the S. aureus genome have been identified but determination of their function is still lagging behind. This study shows that RsaA, a staphylococcal conserved non-coding RNA, operates at the post-transcriptional level by repressing the translation of the master regulatory protein MgrA. The repression is based on a direct interaction of RsaA with the ribosome binding site of mgrA mRNA. Through MgrA regulation, RsaA activates biofilm formation and inhibits capsule synthesis. Using appropriate animal models, we showed that RsaA acts as a suppressor of virulence because the deletion of its gene increases the invasiveness of S. aureus in the mice sepsis model. RsaA is thus part of complex regulatory network that modify the interactions of S. aureus with the eukaryotic immune system. These findings illustrate how small RNAs can have a major impact in bacterial biology.
Published in the journal:
. PLoS Pathog 10(3): e32767. doi:10.1371/journal.ppat.1003979
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003979
Summary
Staphylococcus aureus is a commensal and an opportunistic pathogen that causes a large range of community and hospital-acquired infections. The bacteria produce an array of virulence factors, the expression of which is regulated by a set of regulators including proteins and RNAs. In recent years, a large number of small non-coding RNAs encoded by the S. aureus genome have been identified but determination of their function is still lagging behind. This study shows that RsaA, a staphylococcal conserved non-coding RNA, operates at the post-transcriptional level by repressing the translation of the master regulatory protein MgrA. The repression is based on a direct interaction of RsaA with the ribosome binding site of mgrA mRNA. Through MgrA regulation, RsaA activates biofilm formation and inhibits capsule synthesis. Using appropriate animal models, we showed that RsaA acts as a suppressor of virulence because the deletion of its gene increases the invasiveness of S. aureus in the mice sepsis model. RsaA is thus part of complex regulatory network that modify the interactions of S. aureus with the eukaryotic immune system. These findings illustrate how small RNAs can have a major impact in bacterial biology.
Introduction
Staphylococcus aureus is an opportunistic pathogen that has evolved complex regulatory circuits allowing rapid adaption of cell growth in response to its diverse hosts and ecological niches. Present in a large proportion of the population as a commensal of skin and nose, the bacteria is also responsible for a large range of hospital-acquired and community infections [1]. A successful infection by S. aureus largely depends on the coordinated and sequential expression of a multitude of virulence factors and accessory genes. Over the last decade, it has been established that S. aureus genes are regulated at many different levels by a variety of trans-acting regulators, which act in a coordinated manner [2], [3]. Among them, RNAs are now recognized as important players in virulence and many physiological and adaptive responses [4], [5]. The first regulatory RNA that was discovered in 1993 is RNAIII, the main intracellular effector of the quorum sensing agr system [6]. This multi-functional regulatory RNA binds to several target mRNAs to regulate their translation and decay [7]–[11]. Later on, several teams have experimentally identified a large number of small RNAs (sRNA) that are issued from the core genome and from mobile and accessory elements (e.g., [12], [13]). These sRNAs include cis-acting regulatory regions of mRNAs (the so-called riboswitches), cis-encoding antisense RNAs (asRNA), and non-coding RNAs (ncRNAs). In addition, sRNAs carrying small open reading frames (sORF) have been recently identified [14]–[17] and one of them was shown to express a small cytolytic peptide [17]. The functional and mechanistic studies of trans-acting sRNAs are still lagging behind their discovery, but some of them are known to sense population density and various environmental changes, modify cell surface properties, adjust the bacterial metabolism during cell growth, regulate virulence gene expression, and respond to antibiotic treatment [4], [18].
We have previously identified a group of sRNAs that carry a conserved and unpaired UCCC sequence suggesting that they act through a common regulatory mechanism [12]–[14], [19]–[23]. This sequence motif is located in several hairpin loops of S. aureus RNAIII and was demonstrated to be the seed sequence, which recognizes the ribosome binding sites of mRNA targets to repress translation [5]. Here, using a combination of in vivo and in vitro approaches, we have elucidated the function and the mechanism of action of one of these sRNAs called RsaA [20]. We show that RsaA, a Sigma B (σB)-dependent sRNA, represses the translation of mgrA mRNA, which codes for a master regulator of transcription [24]–[28]. Through MgrA regulation, RsaA activates biofilm formation and inhibits capsule synthesis. Experimental animal models showed that RsaA attenuates the severity of acute systemic infections and enhances chronic catheter infection. These data indicate that RsaA is part of a regulatory circuit that contributes to the complex interactions of S. aureus with the eukaryotic immune system.
Results
RsaA affects the synthesis of MgrA and of proteins regulated by MgrA
To assess the function of RsaA, we first constructed an HG001-derived strain, in which the rsaA gene was deleted and replaced by aphA-3 (aminoglycoside 3′-phosphotransferase, kanamycin resistance). We have verified that RsaA expression in HG001, which is σB-dependent, was maximal at the late-exponential phase of growth and was abolished in the mutant ΔrsaA strain. In BHI medium, growth was not affected by rsaA deletion. We then analyzed the effect of the ΔrsaA mutation on protein synthesis using comparative proteomics performed on cytosolic protein extracts prepared from wild-type (HG001) and mutant ΔrsaA strains grown in BHI medium to late-exponential growth phase. Protein samples were pre-labeled with two different fluorescent dyes (Cy3 and Cy5) and were separated by a two-dimensional difference gel electrophoresis (DiGE). Even though only a small proportion of cytosolic proteins (around 100 proteins) were analyzed, we identified more than 10 proteins with significantly altered expression patterns (Figures 1A, S1). Among the proteins whose synthesis was significantly increased in the mutant strain, was SpoVG, a protein involved in capsule synthesis [29]. Other proteins involved in intermediary metabolism and in virulence were also affected by the ΔrsaA mutation. Several of these proteins belong to the regulon of the pleiotropic regulator MgrA [25], [26] but the effect of RsaA was opposite to that of MgrA.

RsaA contains an unpaired and conserved C-rich sequence, suitable for interaction with ribosome binding sites (RBS) of mRNAs, as previously shown for several other regulatory RNAs in S. aureus [12]–[14], [19]–[23]. Using IntaRNA [30] and targetRNA [31], we searched for potential intermolecular base pairings between the ribosome binding sites (RBS) of mRNAs (30 nucleotides (nts) downstream and 20 nts upstream of the AUG codon) and the conserved unpaired region of RsaA. Interestingly, the best mRNA candidate obtained by the two programs was mgrA mRNA (Table S1). In addition, two mRNAs encoding SpoVG and glycyl-tRNA synthetase, whose protein levels were decreased in an RsaA-dependent fashion, were predicted to form base pairings with RsaA (Figure 1B).
Because MgrA protein could not be detected by the differential proteomic analysis, two alternative methods were used to analyze whether the synthesis of MgrA was under the control of RsaA. Protein extracts, prepared from the WT strain (HG001), the ΔrsaA-HG001 mutant strain, and the same strain complemented with a plasmid expressing RsaA under its own promoter were analyzed by Western blot using monoclonal antibodies (generously provided by A. Cheung) and by LC-MS-MS coupled to spectral count analysis (Figure 1C, D). The data obtained by the two methods were well correlated and showed that the yield of MgrA protein was decreased by two-fold in the WT strain as compared to the ΔrsaA mutant strain while the repression was even more pronounced (about 10-fold) in the mutant strain expressing RsaA from a plasmid. This effect was specific since the yield of the ribosomal protein L21 remained identical in the various strains (Figure 1C).
We have previously shown that S. aureus RNAIII, which represses translation of target mRNAs, also affects their turnover. Therefore, we analyzed the steady-state level of mgrA mRNA using quantitative RT-PCR analysis on total RNA extracts prepared from the WT (HG001 and Becker) strains, the ΔrsaA mutant strains, and the same mutant strains transformed with a plasmid expressing RsaA (Figure 1E). The data showed that the level of mgrA mRNA was strongly enhanced in the ΔrsaA mutant strains while complementation caused an even greater decrease in mRNA yield than in the isogenic WT strains (Figure 1E). Because mgrA is transcribed in two transcripts [27], which initiate at −124 or −302 nts upstream of the initiation codon, northern experiments were also performed (Figure 1F). The level of the longest mRNA (from P2 promoter) decreased significantly at the late exponential phase of growth in both the WT and ΔrsaA derivative of HG001, indicating that the growth phase-dependent regulation of the P2 promoter is not under the control of RsaA (Figure 1F). Conversely, the level of the second and smaller transcript (from P1 promoter) was strongly enhanced in the mutant strain lacking the rsaA gene (ΔrsaA). These data suggested that the mgrA mRNA transcribed from the P1 promoter is under the control of RsaA while the mRNA formed from the P2 promoter is regulated independently of RsaA.
All in all, these data advocated that RsaA has a significant effect on the S. aureus proteome most probably through indirect interactions via the regulation of mgrA.
RsaA binds to mgrA mRNA at two distinct regions
The predicted base pairing between RsaA (nts 65 to 108) and mgrA mRNA (nts −28 to +16) suggested that the RsaA-dependent repression of mgrA mRNA was governed by direct RsaA-mRNA pairing. To map more precisely the regions of interaction, we performed footprinting experiments using enzymatic and chemical probing. The regions of interaction between mgrA mRNA and RsaA were probed using RNase T1 (specific for unpaired guanines), RNase T2 (specific for unpaired nucleotides with a preference for adenines), RNase V1 and the endoribonuclease III (specific for double stranded regions), and lead(II)-induced cleavages (specific for unpaired nucleotides). We also used dimethylsulfate, which methylates adenines at the N1 position and cytosines at the N3 position to probe the structure of mgrA mRNA and its interactions with RsaA. The cleavage sites were analyzed using 5′ end labeled RsaA while for mgrA mRNA, the cleavages and chemical modifications were analyzed by primer extension using reverse transcriptase. Experiments are shown in Figures 2A–B.

The probing data obtained with RsaA alone correlated well with the existence of three hairpin loops (Figure S2A) as previously published [12]–[14], [19]–[23]. Indeed, single-strand specific RNase cleavages were mainly located in the apical loops and unpaired regions while RNase V1 cuts were predominantly found in the helices of RsaA. Furthermore, RNase III specifically cleaved helix H1, in agreement with the observation that RsaA decay is mediated by RNase III in vivo [23]. Enzymatic hydrolysis was then performed on 5′ end labeled RsaA incubated with increasing concentrations of mgrA mRNA. Binding of mgrA mRNA affected the cleavages at two distant regions of RsaA. Strong protections were observed close to the C-rich motif of RsaA, i.e. at A83–85 against RNase T2, at U82-A94 against lead(II)-induced cleavages, and at G86 against RNase T1 (Figure 2C). Concomitantly, several enhanced RNase V1 cuts were found at U88-C89, and two strong RNase III cleavages were observed at U82 and U87 of RsaA (Figure 2A). Unexpectedly, binding of mgrA mRNA also induced strong protections against single-strand specific probes in the hairpin loop H1 at residues A27 to G33 of RsaA revealing the existence of a second RNA binding site (Figure 2A, C).
The same experiments were performed on an mgrA fragment encompassing 124 nts upstream of the initiation codon and 258 nts of the coding sequence (Figure 2B). The enzymatic cleavages obtained on mgrA mRNA alone supported the existence of the long hairpin IV within the coding sequence as well as the stem-loop III structure, which partially sequestered the SD sequence (Figure S1B). The guanines of the Shine and Dalgarno (SD) sequence were indeed weakly accessible towards RNases T1 and T2. Binding of RsaA caused changes in the RNase cleavage patterns at two distant regions of the mRNA, namely around the RBS and in the hairpin loop IV located in the coding sequence (Figure 2C). In particular, strong RNase V1 cleavages occurred at positions C13 to A17, C70–C72 and C75, and two strong RNase III cleavages were observed at A+1 and C-5 of mgrA mRNA (Figure S2C). Concomitantly, RsaA-induced protections against RNase T2 were observed in the hairpin loop IV (at A67, A68, A74) and at nucleotide A-9 close to the SD sequence, and strong reduction of reactivities of adenines at N1 against dimethylsulfate were located at A+1, A-1, A-6, A-7 and A-9 in the RBS (Figures 2B, 2C). Interestingly, the hairpin loop IV carries the unpaired nucleotides UCGCUACU, which are fully complementary to the hairpin loop I of RsaA (AGUAGCGA) (Figure 2C).
All together, these data revealed that the mgrA mRNA-RsaA complex is composed of a bipartite site, which implies the formation of an imperfect duplex sequestering the RBS of mgrA and a loop-loop interaction. Furthermore, the long imperfect duplex creates a specific site for the RNase III.
RsaA inhibits ribosome binding in vitro and regulates mgrA translation in vivo
Structure probing strongly suggested that RsaA would repress translation initiation of mgrA. Toeprinting assays were first used to monitor the effect of RsaA binding on the formation of the initiation ribosomal complex involving the 30S ribosomal subunits, the initiator tRNAMet, and mgrA mRNA. Formation of the ternary complex, which blocked the elongation of a cDNA primer by reverse transcriptase, produced a toeprint at A+16 (Figure 3A). Binding of RsaA completely abolished the toeprint at a ratio of 1∶1 (RsaA:mgrA mRNA). These data indicate that the RsaA-mgrA mRNA complex is rapidly formed and sufficiently stable to prevent the formation of the ribosomal initiation complex.

To further validate the in vivo relevance of RsaA-dependent repression of mgrA mRNA, we analyzed the expression of a reporter construct in S. aureus HG001-ΔrsaA strain expressing the wild-type (WT) RsaA. Mutations were introduced in both mgrA and RsaA at the two binding sites (S1 and S2), and were designed to disrupt or restore base pairings (Figure 2C). The entire leader regulatory region of the mgrA gene produced from P1 (124 nts upstream of the initiation codon) including 258 nts of the coding sequence was cloned in-frame with the lacZ gene into the pTCV-lac shuttle vector (Table S2). This construct is under the control of a constitutive promoter (PrpoB). ß-galactosidase activity assays were first performed in the S. aureus ΔrsaA mutant strain transformed with the pCN38 vector expressing WT RsaA from its own promoter. The ß-galactosidase activity was 4-fold lower in the ΔrsaA mutant strain expressing WT RsaA than in the same strain transformed with the control vector (Figure 3B). To probe the importance of the first binding site, we introduced mutations in the stem-loop structure H2 of RsaA where 86GUUCU90 was substituted by CAAGA (RsaA-S1) to weaken the interaction with the RBS of mgrA mRNA (Figure 2C). The ß-galactosidase activity was reproducibly enhanced 3-fold in the ΔrsaA mutant strain expressing RsaA-S1 as compared to the ΔrsaA strain expressing WT RsaA (Figure 3B). Thus, disruption of the predicted base pairings alleviated the repression of the reporter mgrA-WT-lacZ gene. We then introduced changes in the RBS of mgrA-lacZ reporter gene where -9AGAAC-5 was replaced by UCUUG to restore base pairing with RsaA-S1. This mutation weakened the strength of the SD-aSD helix since the first adenine of the SD sequence was substituted by a guanine. Indeed, the resulting mutated mgrA-S1-lacZ fusion produced less ß-galactosidase than the WT fusion (Figure 3B). The expression of WT RsaA reduced only slightly the ß-galactosidase synthesis while RsaA-S1 caused a significant reduction of the ß-galactosidase synthesis from the fusion mgrA-S1-lacZ by a reproducible 4-fold factor (Figure 3B). The contribution of the second binding site in regulation was also analyzed. Mutations were introduced in the hairpin loop H1 of RsaA (RsaA-S2, 28GUAGCG33 changed to CAUCGC) to destabilize the loop-loop interaction. The data showed that the ß-galactosidase synthesis obtained from mgrA-WT-lacZ was strongly enhanced in the ΔrsaA strain expressing RsaA-S2 as compared to the same strain expressing the WT RsaA. Hence, disruption of the loop-loop interaction significantly alleviated repression of mgrA (Figure 3B).
Taken together, these data showed that RsaA represses the synthesis of MgrA at the post-transcriptional level through the formation of an RsaA-mRNA duplex, which sequesters the RBS of mgrA mRNA. Furthermore, the two distant regions of interaction contribute to the repression of MgrA synthesis.
The two binding sites are essential for RsaA-dependent translation inhibition of mgrA
The effects of mutations that disrupt or restore base pairings were further analyzed for their ability to form RNA-RNA interactions and their ability to inhibit mgrA translation. The formation of the RsaA-mRNA complex was followed using gel retardation assays (Figure 4B). In vitro 5′ end-labeled RsaA was incubated with increasing concentrations of mgrA mRNA. This experiment showed that mgrA mRNA binds to RsaA with a Kd value of around 20 nM. The initial rate of RsaA binding was estimated from a time-course analysis and resulted in an association rate constant of 5×105 M−1s−1 indicating that the complex is rapidly formed. Mutations of the RBS of mgrA (mgrA-S1) had a strong effect on binding to WT RsaA while compensatory mutations performed in RsaA-S1 restored the binding albeit with less efficiency (Kd value around 50 nM). Disruption of the loop-loop interaction caused a strong effect on the binding of mgrA-S2 to WT RsaA (Figure 4B). Therefore, both regions of interaction contribute to the stability of the RsaA-mRNA complex indicating that they act in a cooperative manner.

We then performed in vitro translation assays (PUREsystem) using the whole mgrA mRNA (expressed either from the P1 or the P2 promoter) carrying the sequence encoding a FLAG tag to visualize the protein by Western blot using FLAG-specific monoclonal antibodies. Addition of WT RsaA specifically repressed the translation of both mgrA mRNAs (Figure 4C). These data were well correlated with the in vivo reporter assays (Figure 3B). The same experiment was performed with mutated versions of mgrA mRNA (mgrA-S1, mgrA-S2) expressed from the P1 promoter. Consistent with the binding experiments, the addition of WT RsaA had no effect on the translation of mgrA-S1 while RsaA-S1 efficiently inhibited the translation of mgrA-S1 (Figure 4C). Furthermore, destabilization of the loop-loop interaction had a significant effect on translation repression because higher concentrations of WT RsaA were required to achieve inhibition with mgrA-S2 compared to WT mgrA (Figure 4C).
All in all, these data show that RsaA inhibits initiation of translation of mgrA and that the two regions of interaction contribute to binding and regulation.
Deletion of RsaA reduces biofilm formation
We then analyzed whether RsaA is able to counteract the action of the master regulator of transcription MgrA. The effect of RsaA was monitored on biofilm and capsule formation, which are two processes known to be regulated by MgrA [24], [26], [32], [33].
The biofilm Ring Test was used to follow the kinetics of early biofilm formation. This approach focuses on adhesion and aggregation of bacteria to a substrate (Figure 5A). We analyzed various strain backgrounds, namely RN6390 (σB−), HG001 (σB+), Newman and Becker (clinical isolates, σB+) as well as their rsaA mutant and complemented counterparts. For all genetic backgrounds, the rsaA inactivation induced a delay in the kinetic of biofilm formation but this delay was variable according to the genetic background (Figure 5A). The delay in the kinetics of early biofilm formation of mutant vs WT strains was equal to 10 min for RN6390, 30 min for HG001, 1 h 30 for Becker and 2 h 10 for Newman. The rsaA complementation restored fully (RN6390 and HG001) or partially (Becker and Newman) the parental phenotype. The Becker ΔmgrA strain, generously provided by C. Lee, was used as a control. As expected [32], the inactivation of mgrA reduced the delay of early biofilm formation by about 1 h 30 in comparison to the WT strain. The double mutant ΔmgrA-ΔrsaA Becker strain did not further reduce the delay of biofilm formation, supporting that RsaA regulated the biofilm formation via mgrA inhibition. Of note, RsaA plasmid complementation of the ΔmgrA-ΔrsaA Becker strain did not change the biofilm phenotype obtained with the double mutant.

Quantification of mature biofilm after 24 h was also performed for all strains (Figure 5B). The level of mature biofilm was extremely abundant for HG001, intermediate for Becker and RN6390, and moderate for Newman. The inactivation of rsaA was associated with a significant decrease in biofilm thickness in the three strain backgrounds. Conversely, the plasmid complementation restored the parental WT phenotype and biofilm formation was even slightly higher due to the overexpression of RsaA. As a negative control, transformation with the empty plasmid showed no effect (Figure 5B). Furthermore, mgrA inactivation in the Becker strain induced a high increase in mature biofilm formation, as previously published [32]. As expected, the double mutant ΔrsaA-ΔmgrA Becker strain, and the same strain complemented with a plasmid expressing RsaA, produced a phenotype similar to that of the single ΔmgrA Becker strain.
The icaADBC operon encoding the extracellular polysaccharide adhesin PIA/PNAG (polysaccharide intracellular adhesion/poly-N-acetylglucosamine) is responsible for some of the biofilm-positive phenotypes in S. aureus [34]. However, S. aureus can produce an alternative ica-independent biofilm via the expression of mgrA [32]. To rule out an involvement of icaADBC in the RsaA-dependent biofilm formation, we quantified PIA/PNAG production using an anti-PIA/PNAG specific antibody in the three strain backgrounds. We saw that the inactivation of rsaA had no effect on PIA/PNAG production for the equivalent quantity of bacterial lysates (Figure S3).
In summary, these data suggested that RsaA is acting upstream of MgrA and activates biofilm formation through the inhibition of MgrA synthesis.
RsaA inhibits capsule production and prevents opsonization of S. aureus
To study whether RsaA was involved in capsule regulation, we quantified capsule production after an overnight culture using anti-CP5 or anti-CP8 specific antibodies. Using equivalent amounts of bacterial lysates, we found that the Becker strain (capsular polysaccharide type 8, CP8) produced more capsular polysaccharides than Newman and HG001 (capsular polysaccharide type 5, CP5). The inactivation of rsaA induced a significant increase in CP8 biosynthesis in the Becker strain and in CP5 in the Newman strain while only a slight increase in CP5 was detectable for HG001 (Figure 6A). The expression of RsaA from a plasmid restored the parental WT phenotype for all strains. The same results were obtained after 6, 9 and 12 h of culture (data not shown). To verify that the regulation of capsule production by RsaA resulted from mgrA repression, we again analyzed the phenotype of the double mutant ΔrsaA-ΔmgrA in the Becker strain background. Compared to the WT, the level of CP8 production was considerably reduced in the single ΔmgrA mutant strain as well as in the double mutant ΔrsaA-ΔmgrA strain. Noticeably, we reproducibly found that the complementation of the double mutant with a plasmid expressing RsaA further reduced the level of CP8 production, suggesting the existence of an mgrA-independent repression effect of capsule production. This additional effect might be attributed to the RsaA-dependent inhibition of SpoVG (Figures 1, S1), a protein involved in the capsule formation [29].

To illustrate the accumulation of capsular polysaccharides in ΔrsaA mutant strains, indirect fluorescence was performed to investigate the capsule formation of the Becker strain and its derivatives after 7 h of growth in BHI. Strikingly, the capsule was highly labeled only in the ΔrsaA mutant Becker strain contrasting with the WT parental and complemented strains (Figure 6B). To verify the specificity of the secondary antibody, the same experiment was performed without the primary CP8 antibody, and as expected, all strains were only labeled in green (data not shown).
Because capsular polysaccharides protect S. aureus against opsonophagocytic killing by polymorphonuclear leukocytes (PMNs) [35], [36], we measured the percentage of bacteria killed after contact with serum and PMNs in strains expressing or deleted for RsaA. The data were not statistically significant because a large variability of bacterial survival was observed due to inter-PMNs donor differences. However, in the mutant strains, fewer bacteria were killed than in the parental strains expressing RsaA (particularly in the Becker strain background) in each replicate (Figure S4).
These data showed that RsaA significantly represses capsule synthesis in all strain backgrounds and renders S. aureus more sensitive to opsonophagocytic killing by PMNs.
RsaA attenuates virulence in mice infectious models
Because RsaA impacts both biofilm and capsular formation, we selected two animal models in mice that were considered relevant with respect to these processes, namely, the catheter infection model (for biofilm) and the intraperitoneal infection model (for the capsule).
For the in vivo catheter infection model, sections of polyurethane catheter (1 cm) were implanted sub-cutaneously into C57BL/6 mice and filled with about 4×104 bacteria/25 µl (Becker WT and ΔrsaA mutant strains) in PBS (6 mice/group). The catheters were implanted for 3, 6 or 10 days. No difference in weight was observed between the mice infected by the WT and mutant strains (data not shown). Both strains led to a local infection with swelling but the size of the swelling was not significantly different between the studied strains (data not shown). At day 3, the concentration of colony-forming units observed on the catheters was significantly higher in mice infected with the WT strain than those infected by the ΔrsaA mutant strain (p = 0.0173) (Figure 7A). Between days 6 and 10, this difference was no longer observed in mice infected with WT and mutant strains, likely due to the saturation of the growth within the catheters (Figure 7A). However, between days 3, 6 and 10, more colony-forming units were observed in the surrounding tissues of the mice infected with the WT strain while the number of colony-forming units of the mutant ΔrsaA strain remained unchanged (Figure 7B).

For the bacteremia model, we included the two major capsular types of S. aureus (type 5 and 8), namely Newman (type 5) and Becker (type 8), their ΔrsaA mutant derivatives, as well as their complemented counterparts (studied at the early time point only). Mice were challenged with sub-lethal doses of each strain by intra-peritoneal injection (1×106 bacteria/500 µl). Animals challenged with the strongly encapsulated strains (Becker-ΔrsaA) showed a significantly (p<0.005) higher bacteremia level at 1 h and 3 h after inoculation compared with animals infected with the parental and complemented strains (both less encapsulated) (Figures 7C and S5A). Furthermore, after 1 h and 3 h, the blood concentrations of ΔrsaA bacteria were significantly higher (p<0.05) than those of parental and complemented strains (Figures 7D and S5B). The same results were obtained with Newman background (Figures S5C–F). Evolution of the infection was followed by collecting the mice spleen for bacterial numeration. After 1 and 2 days, the bacterial counts in the spleen were generally higher in the ΔrsaA mutant strain - vs WT strain-infected animals for both Becker (Figure S5H) and Newman (Figure S5G) backgrounds. However, this difference only reached significance in the spleen after 1 day with Newman strain and after 2 days with Becker strain.
Our data obtained with both animal models suggested that RsaA diminishes invasiveness but favors chronic infections and local colonization.
RsaA is expressed by clinical isolates of S. aureus
The observation that RsaA impacted oppositely biofilm formation and invasiveness raised the question as to whether clinical isolates would express differential levels of RsaA depending on the clinical situation in which they had been sampled. To this end, a series of 18 isolates reflecting the commensal status of S. aureus (nasal colonization) as well as S. aureus diseases in various forms (chronic lung infection in cystic fibrosis patients, non-invasive acute skin and soft tissue infections, severe invasive disease illustrated by community-acquired pneumonia and infective endocarditis), were tested for RsaA expression at mid and late-exponential phases by RT-qPCR. The results revealed no specific correlation between RsaA expression levels and invasiveness, both at the mid and late-exponential state of growth (Figure S6A). Of note, all reference strains tested (HG001, Becker, Newman, USA300 LAC and SF8300, and the European CA-MRSA ST80) showed equivalent levels of RsaA production (Figure S6B).
Discussion
RsaA regulates the synthesis of the master global regulator MgrA
The virulence of S. aureus is in part conferred by numerous effector proteins that interact with the host. The levels of these virulence factors are controlled by a set of more than 20 transcriptional regulatory proteins [2]. However, S. aureus strains are genetically highly diverse and it is well recognized that their regulatory networks are not uniform. In order to determine the virulence traits of a defined strain, we must understand how the regulators interact with each other and how novel regulators such as non-coding RNAs influence these networks.
In this work, we show that the small non-coding RNA RsaA functionally links two global regulators, the σB and MgrA proteins. The transcription of RsaA was shown to be under the control of σB protein [20], while its fate was determined by two endoribonucleases, the double-strand specific RNase III [23] and RNase Y [37]. Our data indicate that the mgrA mRNA is repressed at the stationary phase of growth in an RsaA-dependent and RsaA-independent manner. The mgrA mRNA is transcribed from two promoters, and the mRNA with a longer 5′UTR was no longer produced at the late-exponential phase when RsaA was produced (Figure 1F). Whether the regulation is mediated by a protein - or another sRNA remains to be studied. Using a combination of approaches in vivo and in vitro, we showed that the primary effect of RsaA is to repress the translation of mgrA mRNA by preventing the formation of the ribosomal initiation complex. The steady state level of the mRNA was also substantially enhanced in the ΔrsaA strain suggesting subsequent degradation of the repressed mRNA. A reduction of mgrA mRNA from the P1 promoter was also observed at the post-exponential phase of growth in various σB+ strains, and the levels of mgrA mRNA were found to be higher in RN6390 (σB−) than in SH1000 (σB+) [38]. This was correlated in vivo with the level of MgrA protein, which was found to be higher in the ΔrsaA mutant strain than in the isogenic WT strain (Figures 1C, D). In vitro binding assays showed that RsaA binds to mgrA mRNA with a fast association rate constant, which is essential for efficient repression because the sRNA has to bind to its target mRNA within a short time frame before the stable ribosomal initiation complex is formed [39]. Two stretches of highly conserved and unpaired nucleotides of RsaA bind to two distant regions of mgrA, a C-rich motif of RsaA masks the SD sequence of mgrA while a loop-loop interaction occurs between two hairpin loop structures found in the 5′ end of RsaA and in the coding sequence of mgrA (Figure 8A). Mutational analysis revealed that the two RsaA binding sites are required for optimal binding and mgrA translation repression in vivo (Figures 3 and 4).
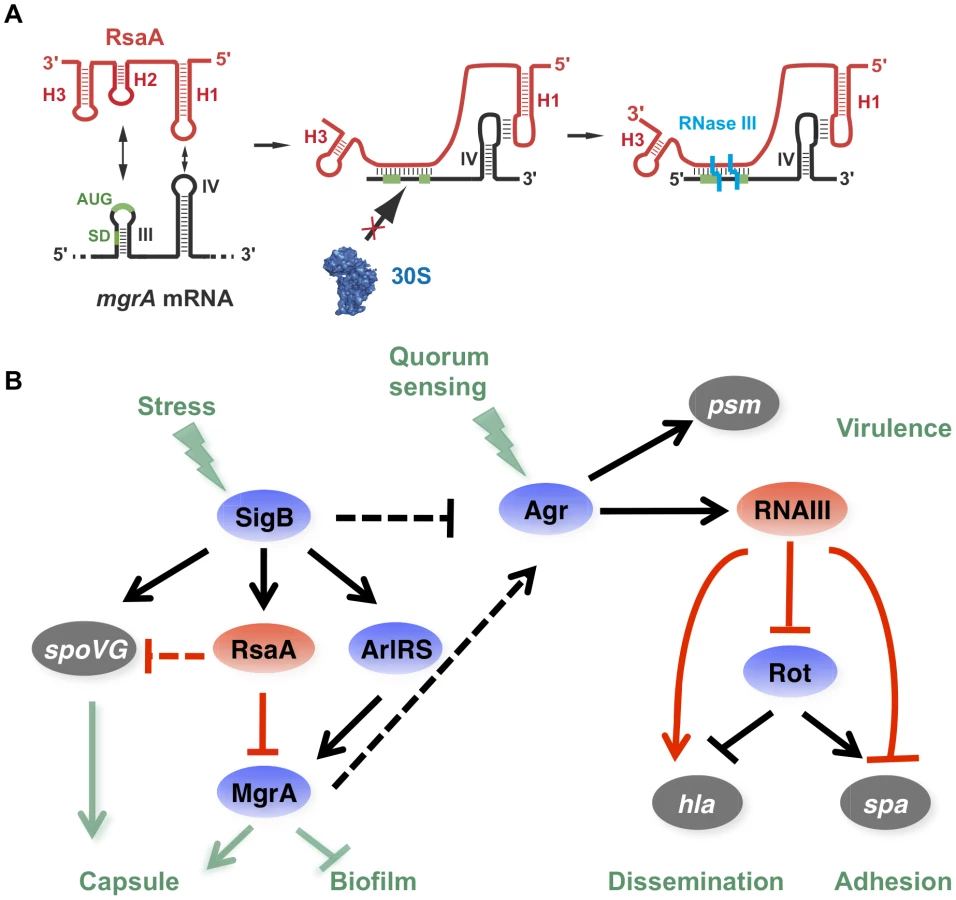
It is worth noting that RsaA and RNAIII share very similar regulatory mechanisms [11], [40] suggesting that the importance of bi-partite RNA-RNA binding sites in translation repression appears to be more general than previously expected in S. aureus (Figure 8A). In both cases, their mRNA targets are characterized by two regulatory modules: the SD sequence interacts with a C-rich motif within the regulatory RNAs to prevent ribosome binding and to create a specific RNase III binding site, while a hairpin motif constitutes either a second RNA binding site to stabilize the RNA-RNA interaction or to promote a specific binding site for RNase III [5], [10]. For the RsaA-mgrA complex, the second interaction site involves a loop-loop interaction, which greatly enhances the stability of the complex and the efficiency of translation repression (Figure 4). RNase III-dependent cleavages were observed at the site of interaction formed between the RBS of mgrA and the C-rich motif of RsaA. Note that mgrA mRNA and RsaA were co-immunoprecipitated with RNase III and the half-life of RsaA was significantly enhanced in the Δrnc mutant strain [23]. As it was shown for RNAIII, it is tempting to propose that RNase III also contributes to the degradation of the translationally repressed mgrA mRNA. In addition, since MgrA positively autoregulates its own transcription [27], RsaA-dependent repression of mgrA translation might indirectly inhibit mgrA transcription.
RsaA-dependent regulatory circuit might be linked to the quorum sensing system
By regulating the translation of MgrA, RsaA participates in a complex regulatory network involving two global regulators, which modulate the levels of RNAIII, thus affecting biofilm formation and capsule synthesis (Figure 8). RsaA expression is positively regulated by the alternative σB factor [20], an observation that is well correlated with our data. Indeed, rsaA inactivation affected less strongly the kinetics of biofilm development in RN6390 (σB−, 10 min) than in HG001 (σB+, 30 min) due to the fact that the sRNA was not strongly expressed in RN6390 [20]. RsaA synthesis follows the same pattern as σB, which is activated at a basal level at the exponential phase and strongly enhanced at the stationary phase [41]. Furthermore, σB affected the transcription of the virulence genes in an opposite manner to RNAIII, and this effect would be in part the consequence of lower RNAIII levels in strains harbouring a fully active σB [41]. σB was also shown to enhance resistance to oxidative and UV stresses, biofilm, and capsule formation. The σB-dependent activation of biofilm formation occurs via the icaADBC operon [42], [43]. We show that RsaA does not perturb the production of the PIA/PNAG polysaccharides produced by the icaADBC operon (Figure S2), but affects pathways that are predominantly dependent on mgrA. σB is also known to activate the capsule synthesis via the two-component system ArlRS and yabJ-spoVG [33]. It is worth noting that yabJ-spoVG is a potential mRNA target of RsaA whose expression was altered in the proteome analysis, and for which base pairings with RsaA have been predicted (Figure 1). The main target of RsaA, which is the transcription factor MgrA, regulates more than 350 genes [26]. MgrA is activated by the arlRS operon, whose expression is controlled by σB [26]. Taken together, the expression of MgrA is under the control of an incoherent feed-forward loop, involving a positive loop mediated through ArlRS and a negative loop mediated through RsaA (Figure 8). In addition, a recent study revealed that the DNA binding activity of MgrA is modulated through specific phosphorylation by the eukaryotic like kinase phosphatase pair Stk1-Stp1 [44]. Hence, the expression of this master regulator is controlled through multiple pathways. MgrA is central to the regulation of biofilm and represses the production of two out of the three components of the biofilm, i.e. surface proteins and exogenous DNA release [32]. Indeed, MgrA negatively regulates the DNA release by suppressing cidA expression and activating lrgAB encoding a peptidoglycan hydrolase and an inhibitor of murein hydrolase activity, respectively [32]. Through its synergistic action on these two loci, MgrA inhibits cell autolysis and the release of DNA within the biofilm [32]. The second control exerted by MgrA is to activate the agr system, in a quorum sensing-independent pathway, to inhibit the synthesis of surface proteins essential for adhesion [25], [26]. Although mgrA mutations significantly reduced RNAIII levels [25], [27], the protein also bypasses the quorum sensing system by directly activating the hla gene and repressing spa gene expression via sarS. As with the agr system, MgrA also has a positive effect on capsule synthesis [26], and in a rat infective endocarditis model, MgrA was shown to be the major regulator of capsule formation in vivo [28]. In correlation with these studies, we showed that inactivation of rsaA alleviates the repression of mgrA to increase capsule synthesis (Figure 6). Therefore, we propose that RsaA functionally links the global regulators σB and mgrA, with potential consequences on the temporal expression of virulence determinants ([25], [26]; Figure 8).
We demonstrate that RsaA activates biofilm formation (Figure 5) and represses capsule synthesis (Figure 6) most probably via the repression of mgrA translation. The regulation of these two phenotypes has been extensively studied in pathogenic bacteria demonstrating a complex interdependency. Studies in Staphylococcus haemolyticus and Streptococcus pneumoniae highlighted that encapsulated strains produced little or no biofilm, whereas non-capsulated strains produced a strong biofilm [45]. Moreover, inactivation of the cap operon of S. haemolyticus JCS1435 (sharing 57% of sequence similarity with S. aureus cap) induced biofilm formation [46] suggesting that the capsule would partially mask or inhibit the surface proteins essential for biofilm formation. In our study, we demonstrated this imbalance in favour of biofilm production for the HG001 strain that is minimally encapsulated but produces lots of biofilm; or in favour of the capsule synthesis for Becker and Newman strains that produce small amounts of biofilm but abundant capsule (Figures 5 and 6). The mechanistic details of this imbalance are not totally understood and may certainly involve other factors besides MgrA and RsaA.
RsaA favors chronic infection and attenuates invasiveness
The pathogenic bacterial chromosome is composed of the core genome plus accessory elements. The accessory genome contains genes that have been horizontally acquired through mobile genetic elements (plasmids, phages, pathogenicity islands). The acquisition of these elements, which often encode virulence determinants and antibiotic resistance, has been accompanied by subsequent adaptation of the bacteria to successfully incorporate these novel elements [47]. Some pathogenic bacteria harbour genes that can hinder the expression of acquired virulence factors, and certain genes must be inactivated for the expression of full pathogenicity [48]. Other bacterial genes lead to hypervirulence when they are inactivated [49]. This family of genes was identified in several pathogenic bacteria such as Listeria monocytogenes [50] or Salmonella enterica [51]. For instance, in S. enterica the leucin-responsive regulatory protein (Lrp) represses key virulence factors involved in the invasion of host cells [51]. These genes encode proteins, but recently an antisense RNA in S. typhimurium was shown to kinetically control the virulence determinant MgtC, which is required for defense against the host immune system [52]. Deletion of this antisense RNA causes an hypervirulent phenotype. In the present study, we demonstrate that RsaA, a non-essential ncRNA present in all Staphylococcus species [20] reduces the virulence of S. aureus. Indeed, the deletion of rsaA increases the invasiveness of the bacteria in the mice sepsis model and resistance to opsonophagocytic killing. Conversely, RsaA promotes biofilm formation and catheter colonization in mice, potentially promoting harmful colonization and chronic infections in human. Noticeably, quantification of RsaA expression from human clinical isolates revealed that Rsa is functional in all clinical isolates of S. aureus studied so far [53] but is not differentially expressed depending on the level of invasiveness ranging from commensalism to deep-seated infections (Figure S6). Of note, we also showed previously by direct quantification from clinical samples that RsaA is expressed in all clinical samples including nasal secretion [53]. Since Staphylococci are mainly present as commensal bacteria of warm-blooded organisms (e.g. mammals and birds), the functionality of RsaA as a suppressor of virulence may have been positively selected through evolution because it is favorable for commensalism and saprophytic interactions with the host. We thus postulate that mutations leading to RsaA deficiency would be counter selective, perhaps because hypervirulence would be deleterious to the human host that is the natural reservoir of this bacterium.
Materials and Methods
Ethics statement
All mouse protocols were carried out in strict accordance with the Directive 2010/63/EU revising Directive 86/609/EEC on the protection of animals used for scientific purposes. This directive was translated in the French regulation as the Décrêt n°2013-118 of Feb 2013 under the jurisdiction of the Ministry of Education, Research and Technology. The protocols were approved by the CECCAPP (Comité d'Evaluation Commun au Centre Léon Bérard, à l'Animalerie de transit de l'ENS, au PBES et au laboratoire P4) under identification numbers ENS_2011_033 and ENS_2012_051 for biofilm and bacteremia models, respectively.
Strains, plasmids and growth conditions
The Staphylococcus aureus strains, clinical isolates, and plasmids used in this study are listed in Table S2. Escherichia coli strain DH5α (Promega, Madison, USA) was used as a host strain for plasmid constructions. S. aureus strain RN4220 [54] was used as the recipient in electroporation of the constructed plasmids. Staphylococci were grown either on GP agar plates (1% peptone, 0,5% yeast extract, 0,5% NaCl, 0,1% glucose, 1,7% agar, pH 7,2) or in brain-heart infusion (BHI) supplemented with erythromycin (5 µg/ml) or chloramphenicol (10 µg/ml) when appropriate. E. coli strains were cultivated in Luria-Bertani medium (1% peptone, 0,5% yeast extract, 1% NaCl). Total DNA and plasmid DNA were prepared using DNAeasy tissue Kit Qiagen and Qiaprep Miniprep respectively (Qiagen, Valencia, USA). Transformation of E. coli DH5α was performed by thermic shock and S. aureus strains were transformed by electroporation (Bio-Rad gene Pulser).
The deletions/replacements ΔrsaA/aphA-3 mutants of S. aureus RN6390 (LUG1450), HG001 (LUG1630), Newman (LUG1680), Becker (LUG2010) and CYL1040 (LUG2009) strains were obtained using pMAD [55]. The kanamycin resistance gene aphA-3 was cloned in pMAD between two DNA fragments corresponding to the chromosomal regions upstream and downstream of the rsaA coding sequence using primers rsaA-76/rsaA-1120 and rsaA-1246/rsaA-2194, respectively (Table S3). The resulting plasmid (pLUG754) was electroporated into RN4220 recipient strain and then transferred to RN6390, HG001, Newman, Becker and CYL1040. Growth at non-permissive temperature (44°C) was followed by several subcultures at 30°C and 37°C to favor double crossing over as previously described [10]. To complement these mutant strains, the rsaA gene was amplified using rsaA-76/rsaA-2194 oligonucleotides and inserted into pLUG274 forming pLUG959. Complementation of mutant strains was performed using pE194, pCN51 or pCN38 carrying rsaA gene (Table S2). The CYL1040 was generously provided by CY Lee [24].
Preparation of RNAs
PCR fragments containing RsaA and its derivatives (RsaA-S1, RsaA-S2) were cloned into pUC18 vector, and then were transcribed in vitro using T7 RNA polymerase after plasmid linearization. The WT mgrA mRNA (−124 to +258) and its derivative (mgrA-S1, mgrA-S2) fragments were transcribed from PCR fragments (Table S3). The transcribed RNAs were purified by 8% polyacrylamide-8 M urea gel electrophoresis. After elution in 0.5 M ammonium acetate/1 mM EDTA buffer, the RNAs were precipitated twice with ethanol. The 5′ end-labeling of dephosphorylated RNA or DNA oligonucleotides was performed with T4 polynucleotide kinase and [γ-32P]ATP. Before use, RNAs were renatured by incubation at 90°C for 2 min in the absence of magnesium and salt, 1 min on ice, followed by an incubation step at 37°C for 15 min in TMN buffer (20 mM Tris-acetate pH 7.5, 10 mM magnesium-acetate, 150 mM Na-acetate).
Relative quantification of sRNA from S. aureus by RT-PCR
RNA extraction was performed from mid (OD600 = 0.5) or post-exponential phase (OD600 = 6) grown bacteria using RNeasy Plus mini kit (QIAGEN) as described [53]. One mg of total RNA was transcribed into cDNA using Reverse transcription system (Promega). Subsequently, the cDNA was used as a template for the real-time PCR amplification using a Realpex2 instrument (Eppendorf) and the specific primers shown in Table S1. The amplification products were detected using SYBR Green. The relative amounts of amplicons for each gene were determined using quantitative PCR relative to an internal standard (gyrB encoding Gyrase B subunit) as described [53]. The expression levels of the sRNA were expressed as n-fold differences relative to the calibrator.
In vivo ß-galactosidase assays
Translation fusions were constructed with plasmid pLUG220, a derivative of pTCV-lac, a low-copy-number promoter-less lacZ vector (Table S2). The whole leader region of mgrA (nts −124 to +258) mRNA (from P1 promoter) including 258 nts of the coding sequence, was cloned downstream the rpoB promoter in frame with lacZ. ß-galactosidase activity was measured four times on duplicate cultures with the Enzyme Assay System (Promega).
Northern blots
Electrophoresis of total RNA (10 µg) was done on a 1% agarose gel containing 25 mM guanidine thiocyanate and vacuum transfer to nylon membrane. Hybridizations with specific digoxigenin-labeled RNA probes complementary to mgrA mRNA or RsaA and luminescent detection were carried out as described previously [10]. For all experiments, we verified the quantity of 5S rRNA using a digoxigenin-labeled RNA probe.
Gel retardation assays
Binding rate constant of RNAIII-mgrA mRNA complex was measured as described previously [10]. Binding of 5′ end-labeled mgrA mRNA to a ten-fold excess of unlabeled RsaA or its derivatives was performed at 37°C in TMN buffer. Samples were withdrawn at various time points (0–10 min), added to gel application buffer and loaded onto a native 5% polyacrylamide gel under non denaturing conditions. The gel was run at 4°C and constant voltage (300 V) for 3 h and subsequently dried. Bands corresponding to the RsaA-mgrA mRNA complex and free RsaA, respectively, were quantified using the SAFA algorithm. For determination of the dissociation rate constant of RsaA-mgrA mRNA complex, end-labeled mgrA mRNA was incubated with an increased concentrations of WT RsaA or its variants for 15 min at 37°C in TMN buffer. All experiments were done four times.
Proteomics
Growth conditions, preparation of protein extracts, and separation of proteins using two-dimensional (2-D) gel electrophoresis concerted with protein identification by mass spectrometry approach have been described in [20]. Experimental details are given in the supplementary materials (Supplementary Protocol S1).
Relative MgrA protein expression level was defined by a spectral counting strategy (see Supplementary Protocol S1). Triplicate protein extracts from HG001 (WT), ΔrsaA mutant strain (LUG1630), and the same strain complemented with a plasmid expressing RsaA, were analyzed in separate LC/MS experiments and the MS/MS spectra number were compared for each protein. In parallel to MgrA, we analyzed several ribosomal proteins (L21, L22, L24) as internal controls of constitutively expressed proteins.
Western blot analysis
Cell extracts from the post-exponential (OD600 = 4) phase of growth were prepared from various staphylococcal strains as described in Supplementary Protocol S1. The concentration of total proteins from clear lysates was determined using the Bio-Rad protein estimation kit and BSA as the standard. Equal amounts of total cellular proteins were separated on 15% polyacrylamide-SDS gels and transferred onto PVDF membranes. Blots were incubated at 20°C with an appropriate dilution (1∶1000) of anti-MgrA-specific monoclonal for 1 h (generous gift from A. Cheung, USA), followed by another 1 h incubation with a 1∶2500 dilution of goat anti-mouse peroxidase (HRP) conjugate. Prestained protein standards were used for molecular mass estimations, and the gel was stained by Coomassie blue to verify that the quantity of proteins was homogenous in each sample. Each experiment was repeated at least three times with different samples.
RNA structure probing using enzymes
RNAIII-mgrA mRNA formation was carried out at 37°C for 15 min in TMN buffer. Enzymatic hydrolysis was performed either on cold mgrA (1 pmole) or 5′ end-labeled RsaA (50000 cpm) in 10 µl of TMN, in the presence of 1 µg carrier tRNA at 37°C for 5 min: RNase T1 (0.0025 units), RNase V1 (0.5 units), RNase T2 (0.05 U), RNase III (500 nM). Lead(II) induced cleavages were performed on 5′ end-labeled RsaA at 20°C in 20 µl of TMN buffer containing 2 µg of carrier tRNA with 2.5 µl of different concentrations of lead(II)-acetate (12, 40, and 80 mM) for 5 min at 37°C. The reactions were stopped with 5 µl of EDTA 0.1 M for lead(II)-induced cleavages and all other reactions were stopped by phenol extraction followed by RNA precipitation. The end-labeled RsaA fragments were sized on 12% polyacrylamide/8 M urea slab gels. For mgrA mRNA, the enzymatic cleavages were detected by primer extension with reverse transcriptase [56].
Toeprinting assays
The preparation of the S. aureus 30S subunits, the formation of a simplified translational initiation complex with mRNA, and the extension inhibition conditions were described according to [57]. Experimental details are given in the supplementary materials (Supplementary Protocol S1).
In vitro transcription-translation assays
The in vitro translation assays were carried out using the full-length WT mgrA or mgrA-S1 or mgrA-S2 mRNAs and the PURESYSTEMII classic kit (Cosmo Bio Co, Japon). All constructs carry an additional sequence corresponding to the Flag peptide, which was inserted at the 5′ end of mgrA. The reaction was done at 37°C for 1 h in the presence of 25 µl of the commercial solution A and 10 µl of the commercial solution B in the presence of 10 pmoles of mRNA. Experiments were also carried out in the presence of increasing concentrations of WT or mutant RsaA (5 to 30 pmoles). The tubes were placed on ice to stop the reactions, and the proteins were detected by western blot using antibodies against the FLAG tag.
Biofilm Crystal Violet Staining and Biofilm Ring Test
The biofilm formation was detected by tissue culture plate method that allowed semi-quantitative measurement of biofilm formation as described previously by Heilmann et al. [58]. All tests were carried out three times and average OD value was calculated for each strain and for negative controls. Experimental details are given in the supplementary materials (Supplementary Protocol S1).
The kinetics of early biofilm was evaluated by Biofilm Ring test (BRT) (BioFilm Control, Saint Beauzire, France) as described [59]. This assay is based on the immobilization of magnetic beads embedded by bacterial aggregates with enough strength to overcome the magnetic attraction forces applied on them. Thus, in the absence of sessile cells, all the beads are gathered by a magnet centered under the well, giving an easily detectable red spot [59]. Three experiments with two repeats (2 wells per slide) were performed per strain and per incubation time. Experimental details are given in the supplementary materials (Supplementary Protocol S1).
Quantification of PIA/PNAG production
The PIA/PNAG polysaccharide of biofilm was quantified by the immuno-slot-blotting method with rabbit anti-PIA/PNAG antiserum as described by Cuçarella et al. [60].
Quantification and visualization of the capsular polysaccharides
Capsular polysaccharides type 5 (CP5) or 8 (CP8) were quantified by the immuno-slot-blotting method with rabbit anti-CP5 or anti-CP8 antisera, essentially as described by Luong et al. [24]. CP8 production was determined by indirect immunofluorescence. After culture for 8 h in BHI medium at 37°C, bacteria were spotted in diagnostic slides. The slide was incubated with rabbit anti-CP8 antiserum for 1 h at 37°C in humid chamber. After 2 washes, the slide was incubated with secondary antibody, AlexaFluor 647 goat anti-rabbit immunoglobulin G (Invitrogen, Paisley, UK). Total bacteria were labeled with a 1∶1 mixture of vancomycin and vancomycin-Bodipy FL (VBFL) fluorochrome (Invitrogen) at a concentration of 0.8 µg/ml for 15 min. Images were analyzed with confocal microscopy Leica SP5 X (Leica, Solms, Germany) and treated with Leica software: LAS AF Lite (Leica Application Suite 2.6.0).
CP5 or CP8 were quantified by the immuno-slot-blotting method with rabbit anti-CP5 or anti-CP8 antisera, essentially as described by Luong et al. [24]. Experimental details are given in the supplementary material (Supplementary Protocol S1).
Model of catheter infection in mice
The catheter infection model was performed essentially as described by [61]. C57Bl/6 female mice (11–14 weeks old) were purchased from Charles River Laboratories (Charles River, Wilmington, USA) and maintained in pathogen-free conditions at the “Plateau de Biologie Expérimentale de la Souris” (PBES, Ecole Normale Supérieure de Lyon, Lyon, France). This study and procedure were approved by the “Comité Rhône-Alpes d'Ethique pour l'Expérimentation Animale” (CECCAPP, Lyon, France).
Mice were anesthetized with 2–3% of isofluran. After depilating and disinfecting the back of the mouse, a sterile catheter was implanted subcutaneously on the back of each mouse. Twenty-five microliters of PBS containing approximately 4×104 bacteria (Becker or Becker-ΔrsaA) were then injected into the catheter. The wound was sewed by a clip. Each day, mice were weighed and the size of the swelling was measured with an electronic caliper. On days 3, 6 and 10, mice were euthanized by cervical dislocation, the catheter and surrounding tissues were carefully removed and transferred separately in sterile tubes for bacterial quantification by plate counting as described. Experimental details are given in the supplementary materials (Supplementary Protocol S1).
Bacteremia model
CD-1 female mice (8 to 10 weeks old) were obtained from Charles River Laboratories and maintained in pathogen-free conditions at the PBES. This study and procedure were approved by the CECCAPP. Groups of six mice were inoculated intraperitoneally with 5×106 CFU/mouse in 0.5 ml of PBS. After 1 and 3 h, mice were sacrificed and 4 ml of PBS were intraperitoneally injected to recover the surviving bacteria. Blood samples were also collected in heparinized tubes from intracardiac puncture. Spleen and kidneys were collected after 24 and 48 h. After addition of 1 ml of NaCl 0,9%, organs were crushed with a homogenizer T18 basic. Bacterial quantification of all samples was performed by plate counting.
Statistical analysis
All the data were expressed as mean ± SD. The statistical analyses were performed using GraphPad Prism 6 software. A one-way analysis of variance was realized to test the results obtained in mature biofilm quantification. Other data were analyzed using Mann-Whitney test. A value of p<0.05, <0.005 or <0.0005 was considered to be statistically significant (*), highly significant (**), or extremely significant (***), respectively.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. ChambersHF, DeleoFR (2009) Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol 7 : 629–641.
2. PriestNK, RudkinJK, FeilEJ, van den ElsenJM, CheungA, et al. (2012) From genotype to phenotype: can systems biology be used to predict Staphylococcus aureus virulence? Nat Rev Microbiol 10 : 791–797.
3. NovickRP, GeisingerE (2008) Quorum sensing in staphylococci. Annu Rev Genet 42 : 541–564.
4. FeldenB, VandeneschF, BoulocP, RombyP (2011) The Staphylococcus aureus RNome and its commitment to virulence. PLoS Pathog 7: e1002006.
5. RomillyC, CaldelariI, ParmentierD, LioliouE, RombyP, et al. (2012) Current knowledge on regulatory RNAs and their machineries in Staphylococcus aureus. RNA Biol 9 : 402–413.
6. NovickRP, RossHF, ProjanSJ, KornblumJ, KreiswirthB, et al. (1993) Synthesis of staphylococcal virulence factors is controlled by a regulatory RNA molecule. EMBO J 12 : 3967–3975.
7. MorfeldtE, TaylorD, von GabainA, ArvidsonS (1995) Activation of alpha-toxin translation in Staphylococcus aureus by the trans-encoded antisense RNA, RNAIII. EMBO J 14 : 4569–4577.
8. GeisingerE, AdhikariRP, JinR, RossHF, NovickRP (2006) Inhibition of rot translation by RNAIII, a key feature of agr function. Mol Microbiol 61 : 1038–1048.
9. HuntzingerE, BoissetS, SaveanuC, BenitoY, GeissmannT, et al. (2005) Staphylococcus aureus RNAIII and the endoribonuclease III coordinately regulate spa gene expression. EMBO J 24 : 824–835.
10. BoissetS, GeissmannT, HuntzingerE, FechterP, BendridiN, et al. (2007) Staphylococcus aureus RNAIII coordinately represses the synthesis of virulence factors and the transcription regulator Rot by an antisense mechanism. Genes Dev 21 : 1353–1366.
11. ChevalierC, BoissetS, RomillyC, MasquidaB, FechterP, et al. (2010) Staphylococcus aureus RNAIII binds to two distant regions of coa mRNA to arrest translation and promote mRNA degradation. PLoS Pathog 6: e1000809.
12. PichonC, FeldenB (2005) Small RNA genes expressed from Staphylococcus aureus genomic and pathogenicity islands with specific expression among pathogenic strains. Proc Natl Acad Sci USA 102 : 14249–14254.
13. RobertsC, AndersonKL, MurphyE, ProjanSJ, MountsW, et al. (2006) Characterizing the effect of the Staphylococcus aureus virulence factor regulator, SarA, on log-phase mRNA half-lives. J Bacteriol 188 : 2593–2603.
14. BeaumeM, HernandezD, FarinelliL, DeluenC, LinderP, et al. (2010) Cartography of methicillin-resistant S. aureus transcripts: detection, orientation and temporal expression during growth phase and stress conditions. PLoS One 5: e10725.
15. NielsenJS, LarsenMH, LillebaekEM, BergholzTM, ChristiansenMH, et al. (2011) A small RNA controls expression of the chitinase ChiA in Listeria monocytogenes. PLoS One 6: e19019.
16. SayedN, JousselinA, FeldenB (2012) A cis-antisense RNA acts in trans in Staphylococcus aureus to control translation of a human cytolytic peptide. Nat Struct Mol Biol 19 : 105–112.
17. SayedN, Nonin-LecomteS, RetyS, FeldenB (2012) Functional and structural insights of a Staphylococcus aureus apoptotic-like membrane peptide from a toxin-antitoxin module. J Biol Chem 287 : 43454–43463.
18. HowdenBP, BeaumeM, HarrisonPF, HernandezD, SchrenzelJ, et al. (2013) Analysis of the small RNA transcriptional response in multidrug-resistant Staphylococcus aureus after antimicrobial exposure. Antimicrob Agents Chemother 57 : 3864–3874.
19. Abu-QatousehLF, ChinniSV, SeggewissJ, ProctorRA, BrosiusJ, et al. (2010) Identification of differentially expressed small non-protein-coding RNAs in Staphylococcus aureus displaying both the normal and the small-colony variant phenotype. J Mol Med (Berl) 88 : 565–575.
20. GeissmannT, MarziS, RombyP (2009) The role of mRNA structure in translational control in bacteria. RNA Biol 6 : 153–160.
21. BohnC, RigoulayC, ChabelskayaS, SharmaCM, MarchaisA, et al. (2010) Experimental discovery of small RNAs in Staphylococcus aureus reveals a riboregulator of central metabolism. Nucleic Acids Res 38 : 6620–6636.
22. LasaI, Toledo-AranaA, DobinA, VillanuevaM, de los MozosIR, et al. (2011) Genome-wide antisense transcription drives mRNA processing in bacteria. Proc Natl Acad Sci USA 108 : 20172–20177.
23. LioliouE, SharmaCM, CaldelariI, HelferAC, FechterP, et al. (2012) Global regulatory functions of the Staphylococcus aureus endoribonuclease III in gene expression. PLoS Genet 8: e1002782.
24. LuongTT, NewellSW, LeeCY (2003) Mgr, a novel global regulator in Staphylococcus aureus. J Bacteriol 185 : 3703–3710.
25. IngavaleS, van WamelW, LuongTT, LeeCY, CheungAL (2005) Rat/MgrA, a regulator of autolysis, is a regulator of virulence genes in Staphylococcus aureus. Infect Immun 73 : 1423–1431.
26. LuongTT, DunmanPM, MurphyE, ProjanSJ, LeeCY (2006) Transcription Profiling of the mgrA Regulon in Staphylococcus aureus. J Bacteriol 188 : 1899–1910.
27. IngavaleSS, Van WamelW, CheungAL (2003) Characterization of RAT, an autolysis regulator in Staphylococcus aureus. Mol Microbiol 48 : 1451–1466.
28. GuptaRK, AlbaJ, XiongYQ, BayerAS, LeeCY (2013) MgrA activates expression of capsule genes, but not the alpha-toxin gene in experimental Staphylococcus aureus endocarditis. J Infect Dis
29. MeierS, GoerkeC, WolzC, SeidlK, HomerovaD, et al. (2007) sigmaB and the sigmaB-dependent arlRS and yabJ-spoVG loci affect capsule formation in Staphylococcus aureus. Infect Immun 75 : 4562–4571.
30. BuschA, RichterAS, BackofenR (2008) IntaRNA: efficient prediction of bacterial sRNA targets incorporating target site accessibility and seed regions. Bioinformatics 24 : 2849–2856.
31. TjadenB (2008) TargetRNA: a tool for predicting targets of small RNA action in bacteria. Nucleic Acids Res 36: W109–13.
32. TrotondaMP, TamberS, MemmiG, CheungAL (2008) MgrA represses biofilm formation in Staphylococcus aureus. Infect Immun 76 : 5645–5654.
33. LuongTT, LeeCY (2006) The arl locus positively regulates Staphylococcus aureus type 5 capsule via an mgrA-dependent pathway. Microbiology 152 : 3123–3131.
34. VuongC, VoyichJM, FischerER, BraughtonKR, WhitneyAR, et al. (2004) Polysaccharide intercellular adhesin (PIA) protects Staphylococcus epidermidis against major components of the human innate immune system. Cell Microbiol 6 : 269–275.
35. ThakkerM, ParkJS, CareyV, LeeJC (1998) Staphylococcus aureus serotype 5 capsular polysaccharide is antiphagocytic and enhances bacterial virulence in a murine bacteremia model. Infect Immun 66 : 5183–5189.
36. XuS, ArbeitRD, LeeJC (1992) Phagocytic killing of encapsulated and microencapsulated Staphylococcus aureus by human polymorphonuclear leukocytes. Infect Immun 60 : 1358–1362.
37. MarincolaG, SchaferT, BehlerJ, BernhardtJ, OhlsenK, et al. (2012) RNase Y of Staphylococcus aureus and its role in the activation of virulence genes. Mol Microbiol 85 : 817–832.
38. BallalA, MannaAC (2009) Expression of the sarA family of genes in different strains of Staphylococcus aureus. Microbiology 155 : 2342–2352.
39. WagnerEG, AltuviaS, RombyP (2002) Antisense RNAs in bacteria and their genetic elements. Adv Genet 46 : 361–398.
40. LioliouE, SharmaCM, AltuviaY, CaldelariI, RomillyC, et al. (2013) In vivo mapping of RNA-RNA interactions in Staphylococcus aureus using the endoribonuclease III. Methods 63 : 135–143.
41. BischoffM, DunmanP, KormanecJ, MacapagalD, MurphyE, et al. (2004) Microarray-based analysis of the Staphylococcus aureus sigmaB regulon. J Bacteriol 186 : 4085–4099.
42. RashidMH, RumbaughK, PassadorL, DaviesDG, HamoodAN, et al. (2000) Polyphosphate kinase is essential for biofilm development, quorum sensing, and virulence of Pseudomonas aeruginosa. Proc Natl Acad Sci USA 97 : 9636–9641.
43. ValleJ, Toledo-AranaA, BerasainC, GhigoJM, AmorenaB, et al. (2003) SarA and not sigmaB is essential for biofilm development by Staphylococcus aureus. Mol Microbiol 48 : 1075–1087.
44. SunF, DingY, JiQ, LiangZ, DengX, et al. (2012) Protein cysteine phosphorylation of SarA/MgrA family transcriptional regulators mediates bacterial virulence and antibiotic resistance. Proc Natl Acad Sci USA 109 : 15461–15466.
45. MoscosoM, GarciaE, LopezR (2006) Biofilm formation by Streptococcus pneumoniae: role of choline, extracellular DNA, and capsular polysaccharide in microbial accretion. J Bacteriol 188 : 7785–7795.
46. FlahautS, VinogradovE, KelleyKA, BrennanS, HiramatsuK, et al. (2008) Structural and biological characterization of a capsular polysaccharide produced by Staphylococcus haemolyticus. J Bacteriol 190 : 1649–1657.
47. NovickRP, ChristieGE, PenadesJR (2010) The phage-related chromosomal islands of Gram-positive bacteria. Nat Rev Microbiol 8 : 541–551.
48. MaurelliAT (2007) Black holes, antivirulence genes, and gene inactivation in the evolution of bacterial pathogens. FEMS Microbiol Lett 267 : 1–8.
49. BlivenKA, MaurelliAT (2012) Antivirulence genes: insights into pathogen evolution through gene loss. Infect Immun 80 : 4061–4070.
50. Toledo-AranaA, DussurgetO, NikitasG, SestoN, Guet-RevilletH, et al. (2009) The Listeria transcriptional landscape from saprophytism to virulence. Nature 459 : 950–956.
51. BaekCH, WangS, RolandKL, CurtissRr (2009) Leucine-responsive regulatory protein (Lrp) acts as a virulence repressor in Salmonella enterica serovar Typhimurium. J Bacteriol 191 : 1278–1292.
52. LeeEJ, GroismanEA (2010) An antisense RNA that governs the expression kinetics of a multifunctional virulence gene. Mol Microbiol 76 : 1020–1033.
53. SongJ, LaysC, VandeneschF, BenitoY, BesM, et al. (2012) The expression of small regulatory RNAs in clinical samples reflects the different life styles of Staphylococcus aureus in colonization vs. infection. PLoS One 7: e37294.
54. KreiswirthBN, LofdahlS, BetleyMJ, O'ReillyM, SchlievertPM, et al. (1983) The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305 : 709–712.
55. ArnaudM, ChastanetA, DebarbouilleM (2004) New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl Environ Microbiol 70 : 6887–6891.
56. ChevalierC, GeissmannT, HelferAC, RombyP (2009) Probing mRNA structure and sRNA-mRNA interactions in bacteria using enzymes and lead(II). Methods Mol Biol 540 : 215–232.
57. FechterP, ChevalierC, YusupovaG, YusupovM, RombyP, et al. (2009) Ribosomal initiation complexes probed by toeprinting and effect of trans-acting translational regulators in bacteria. Methods Mol Biol 540 : 247–263.
58. HeilmannC, SchweitzerO, GerkeC, VanittanakomN, MackD, et al. (1996) Molecular basis of intercellular adhesion in the biofilm-forming Staphylococcus epidermidis. Mol Microbiol 20 : 1083–1091.
59. ChavantP, Gaillard-MartinieB, TalonR, HebraudM, BernardiT (2007) A new device for rapid evaluation of biofilm formation potential by bacteria. J Microbiol Methods 68 : 605–612.
60. CucarellaC, SolanoC, ValleJ, AmorenaB, LasaI, et al. (2001) Bap, a Staphylococcus aureus surface protein involved in biofilm formation. J Bacteriol 183 : 2888–2896.
61. KristianSA, BirkenstockTA, SauderU, MackD, GotzF, et al. (2008) Biofilm formation induces C3a release and protects Staphylococcus aureus from IgG and complement deposition and from neutrophil-dependent killing. J Infect Dis 197 : 1028–1035.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 3
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Cytomegalovirus m154 Hinders CD48 Cell-Surface Expression and Promotes Viral Escape from Host Natural Killer Cell Control
- Human African Trypanosomiasis and Immunological Memory: Effect on Phenotypic Lymphocyte Profiles and Humoral Immunity
- DHX36 Enhances RIG-I Signaling by Facilitating PKR-Mediated Antiviral Stress Granule Formation
- Conflicting Interests in the Pathogen–Host Tug of War: Fungal Micronutrient Scavenging Versus Mammalian Nutritional Immunity
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy