
Lectin-Like Bacteriocins from spp. Utilise D-Rhamnose Containing Lipopolysaccharide as a Cellular Receptor
Lectin-like bacteriocins consist of tandem monocot mannose-binding domains and display a genus-specific killing activity. Here we show that pyocin L1, a novel member of this family from Pseudomonas aeruginosa, targets susceptible strains of this species through recognition of the common polysaccharide antigen (CPA) of P. aeruginosa lipopolysaccharide that is predominantly a homopolymer of d-rhamnose. Structural and biophysical analyses show that recognition of CPA occurs through the C-terminal carbohydrate-binding domain of pyocin L1 and that this interaction is a prerequisite for bactericidal activity. Further to this, we show that the previously described lectin-like bacteriocin putidacin L1 shows a similar carbohydrate-binding specificity, indicating that oligosaccharides containing d-rhamnose and not d-mannose, as was previously thought, are the physiologically relevant ligands for this group of bacteriocins. The widespread inclusion of d-rhamnose in the lipopolysaccharide of members of the genus Pseudomonas explains the unusual genus-specific activity of the lectin-like bacteriocins.
Published in the journal:
. PLoS Pathog 10(2): e32767. doi:10.1371/journal.ppat.1003898
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003898
Summary
Lectin-like bacteriocins consist of tandem monocot mannose-binding domains and display a genus-specific killing activity. Here we show that pyocin L1, a novel member of this family from Pseudomonas aeruginosa, targets susceptible strains of this species through recognition of the common polysaccharide antigen (CPA) of P. aeruginosa lipopolysaccharide that is predominantly a homopolymer of d-rhamnose. Structural and biophysical analyses show that recognition of CPA occurs through the C-terminal carbohydrate-binding domain of pyocin L1 and that this interaction is a prerequisite for bactericidal activity. Further to this, we show that the previously described lectin-like bacteriocin putidacin L1 shows a similar carbohydrate-binding specificity, indicating that oligosaccharides containing d-rhamnose and not d-mannose, as was previously thought, are the physiologically relevant ligands for this group of bacteriocins. The widespread inclusion of d-rhamnose in the lipopolysaccharide of members of the genus Pseudomonas explains the unusual genus-specific activity of the lectin-like bacteriocins.
Introduction
The ability to target a subgroup of pathogenic bacteria in a complex bacterial community has potential applications in medicine and agriculture where the maintenance of a ‘normal’ microbiome is beneficial. For example, the use of broad spectrum antibiotics to treat bacterial infections is known to cause a range of complications associated with collateral damage to the microbiome, including antibiotic associated diarrhea and Clostridium difficile infection [1], [2]. In addition, there is growing evidence to suggest that microbial dysbiosis may play a role in a range of chronic diseases such as inflammatory bowel disease, diabetes, obesity and rheumatoid arthritis [3], [4], [5], [6]. Indeed, for Crohn's disease, where the link with dysbiosis is well established, the administration of multiple courses of antibiotics is associated with an increased risk factor for the development of this chronic form of inflammatory bowel disease [7], [8], [9].
In contrast to the broad spectrum antibiotics that are widely used in medicine and agriculture, protein antibiotics known as bacteriocins often target a specific bacterial species or a group of closely related bacterial species [10], [11], [12], [13]. Well characterised bacteriocins include the S-type pyocins from P. aeruginosa and the closely related colicins of E. coli [12], [13]. The colicin-like bacteriocins form a diverse family of multidomain protein antibiotics which share similar mechanisms of uptake and kill cells through either a pore-forming activity, a specific nuclease activity against DNA, tRNA or rRNA or through inhibition of cell wall synthesis [14], [15], [16], [17]. In the case of S-type pyocins it is thought that their activity is limited to strains of P. aeruginosa, whereas colicins show activity against E. coli and some strains of closely related bacteria such as Salmonella spp. [18]. In the case of colicins and S-type pyocins, killing specificity is primarily determined by the presence of a specific outer membrane receptor on the cell surface. For example, the well characterised E group colicins utilise the TonB-dependent BtuB receptor, which has a normal physiological role in vitamin B12 uptake [19]. Colicin-like bacteriocins have also been shown to have a potent antibiofilm activity, indicating their potential as useful therapeutics for the treatment of chronic biofilm mediated infections [20], [21]. In the case of the opportunistic human pathogen P. aeruginosa there is an urgent requirement for the development of novel therapeutic options since its ability to form drug-resistant biofilms in combination with the presence of an outer membrane that is highly impermeable to many classes of antibiotics can make this pathogen essentially untreatable in some groups of patients. This is exemplified in cystic fibrosis patients where chronic lung infection with P. aeruginosa is the leading cause of mortality [22].
An interesting addition to this group of protein antibiotics is the recently discovered lectin-like bacteriocins that contain two carbohydrate-binding domains of the monocot mannose-binding lectin (MMBL) family [23], [24], [25], [26], [27]. Lectin-like bacteriocins from P. putida (putidacin L1 or LlpABW) P. syringae (LlpAPss642) and P. fluorescens (LlpA1Pf-5) have been characterised and have the unprecedented ability to kill strains of a broad range of bacterial species within the genus Pseudomonas, but are not active outside this genus [24], [26], [27]. Similarly the lectin-like bacteriocin LlpAXcm761 from Xanthomonas citri pv. malvacearum LMG 761 has the ability to kill various species within the genus Xanthomonas [24]. The molecular basis of this unusual genus specific activity has not been explained.
Lectins are a structurally and evolutionarily diverse class of proteins produced widely by prokaryotes and eukaryotes and are defined by their ability to recognise and bind carbohydrates. This binding is generally highly specific and mediates a range of diverse functions, including cell-cell interaction, immune recognition and cytotoxicity [28], [29] MMBLs represent a structurally conserved lectin subclass, of which the mannose-binding Galanthus nivalis agglutinin (GNA) was the first to be characterised [30]. The MMBL-fold consists of a three sided β-prism; each face of which contains a sugar binding motif with the conserved sequence QxDxNxVxY [31]. While originally identified in monocots like G. nivalis or Allium sativum, it is now recognised that proteins of this class are distributed widely throughout prokaryotes and eukaryotes, where they have evolved to mediate diverse functions [30], [32], [33], [34]. Structural and biochemical analysis of MMBLs has shown that they are generally translated as a single polypeptide chain containing tandem β-prism domains that are then proteolytically processed into monomers. These domains often form homo - or hetero-dimers by strand exchange and π-stacking [35].
The lectin-like bacteriocins are not proteolytically processed and thus consist of a single peptide chain, containing tandem β-prism domains. Sequence alignments of members of this class from Pseudomonas spp. show complete conservation of two sugar binding motifs on the C-terminal domain and partial conservation of two sites on the N-terminal domain [23]. Recent work by Ghequire et al [23] on the characterisation of putidacin L1 shows these motifs to be important for cytotoxicity. Mutagenesis of the first C-terminal motif has the most dramatic effect on activity, while mutagenesis of the second C-terminal and first N-terminal sugar binding motifs leads to a synergistic reduction in activity. This study also showed low-affinity binding between putidacin L1 and methyl-α-d-mannose or a range of mannose containing oligosaccharides. However, Kds for these protein-carbohydrate complexes were reported in the range from 46 mM for methyl-α-d-mannoside to 2 mM for a mannose containing pentasaccharide [23]. An extensive search for high affinity carbohydrate binding through the use of glycan arrays failed to detect high affinity carbohydrate binding for this lectin-like bacteriocin [23].
Despite progress in our understanding of the structure and host range of MMBL-like bacteriocins, the mechanism by which these bacteriocins target susceptible strains and exert their antimicrobial effects is unknown. Here we report on the discovery of a novel member of this family, pyocin L1 from P. aeruginosa, and show that it utilises lipopolysaccharide (LPS) as a surface receptor, specifically targeting the common polysaccharide antigen (CPA) that is a conserved homopolymer of d-rhamnose. Structural and biophysical analysis shows that the C-terminal carbohydrate binding motifs are responsible for d-rhamnose recognition and that these sites are specific for this sugar over d-mannose. Further to this, we show that the previously described putidacin L1 also selectively binds LPS from susceptible, but not from resistant, P. syringae isolates and shows selectivity for d-rhamnose over d-mannose. This work shows that the physiologically relevant ligand for the QxDxNxVxY carbohydrate binding site of the lectin-like bacteriocins is indeed d-rhamnose and not d-mannose as previously thought. As such, the genus-specific activity of lectin-like bacteriocins from Pseudomonas spp. can be attributed to the widespread inclusion of the rare d-rhamnose in the LPS of members of the genus Pseudomonas.
Results
Identification and characterisation of pyocin L1
As part of a wider project, aimed at identifying bacteriocins that could be used as novel therapeutics in the treatment of P. aeruginosa infections, we searched the genomes of 10 recently sequenced clinical and environmental isolates of P. aeruginosa for genes with homology to known bacteriocins. One putative bacteriocin gene identified in strain C1433, an isolate from a patient with cystic fibrosis, encodes a protein with 31% identity to the lectin-like bacteriocin LlpA1Pf-5, from P. fluorescens. This protein, designated pyocin L1, contained 256-amino acids with a predicted molecular mass of 28413 Da. Alignment of the pyocin L1 protein sequence with other lectin-like bacteriocins, LlpA1Pf-5, LlpAPss642, putidacin L1 (LlpABW), LlpAAu1504 from Burkholderia cenocepacia and LlpAXcm761 from Xanthomonas citri pv. malvacearum shows that pyocin L1 contains tandem MMBL domains with three conserved QxDxNxVxY MMBL sugar-binding motifs (Figure S1). Two of these motifs are located in the C-terminal domain of the protein and one in the N-terminal domain. Comparison with the sequences of other lectin-like bacteriocins shows that the C-terminal QxDxNxVxY motifs are highly conserved, with only LlpAXcm761 lacking one C-terminal motif. In contrast the N-terminal sugar-binding motifs are less well conserved with only LlpAAu1504 possessing two fully conserved QxDxNxVxY motifs (Figure S1).
In order to determine the killing spectrum of pyocin L1 we cloned the pyocin L1 open reading frame into the pET21a vector and expressed and purified the protein by nickel affinity, anion exchange and size exclusion chromatography. Purified pyocin L1 was tested for its ability to inhibit the growth of 32 environmental and clinical isolates of P. aeruginosa using an overlay spot plate method on LB agar [36]. Under these conditions, pyocin L1 showed killing activity against nine of the P. aeruginosa strains tested. Strain E2, an environmental isolate from a tomato plant for which the genome sequence is available, and strain P8, a clinical isolate from a cystic fibrosis patient, showed the greatest sensitivity to pyocin L1 with killing observed down to concentrations of 27 nM and 7 nM, respectively. Pyocin L1 also showed activity against 5 of the 11 P. syringae strains tested, although the effect was much weaker, with cell killing observed at high µM concentrations.
Pyocin L1 targets the common polysaccharide antigen (CPA) of P. aeruginosa LPS
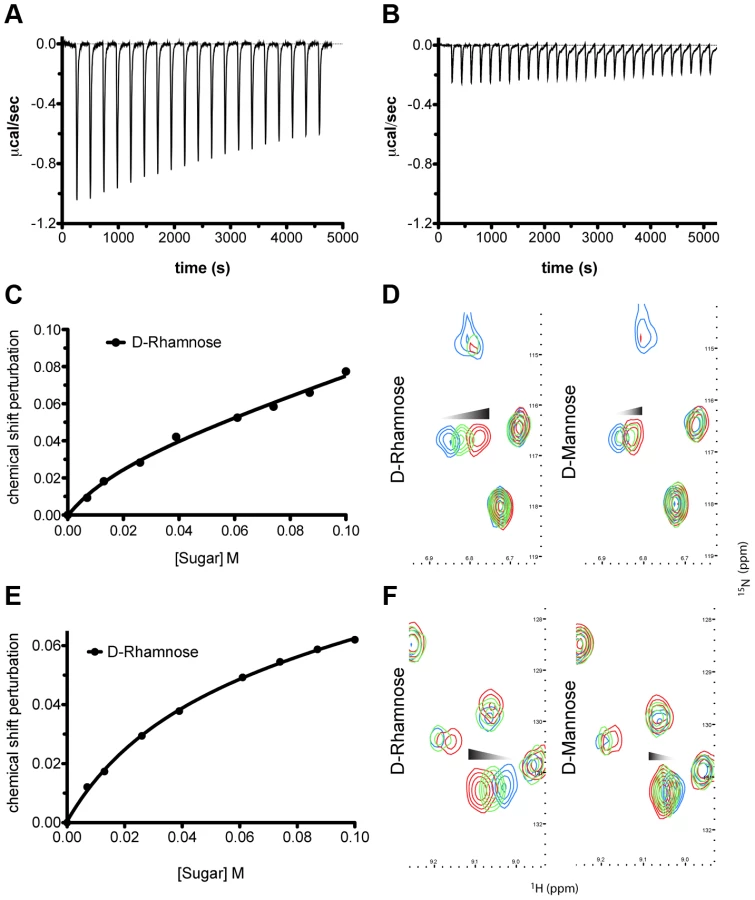
In order to gain insight into the bacteriocidal activity of pyocin L1, we subjected P. aeruginosa E2 to high concentrations of recombinant protein and recovered mutants with greatly increased tolerance to pyocin L1 (Figure 1A). The genomes of two of these mutants were sequenced and comparative analysis with the genome of wild-type E2 revealed a dinuclear deletion, C710 and T711, of the 1146-bp wbpZ gene. This deletion was common to both mutants. wbpZ encodes a glycosyltransferase of 381 amino acids that plays a key role in lipopolysaccharide synthesis, specifically in the synthesis of the common polysaccharide antigen (CPA) also known as A-band LPS [37], (Figure S2). Most strains of P. aeruginosa produce two distinct LPS-types that differ in their O-antigen, but share the same core oligosaccharide. The CPA is predominantly a homopolymer of d-rhamnose and the O-specific antigen contains a heteropolymeric repeating unit that varies widely among strains [38]. Consistent with mutation of wbpZ, we found that production of CPA, as determined by immunoblotting with a CPA-specific monoclonal antibody [39], in both M4(E2) and M11(E2) was reduced to undetectable levels (Figure 1B). Visualisation of LPS from these strains was performed via silver staining and comparable quantities of LPS were shown to be present. These observations suggest that pyocin L1 may utilise CPA as a cellular receptor. To test this idea further, we obtained two transposon insertion mutants of P. aeruginosa PAO1, which is sensitive to pyocin L1, with insertions in the genes responsible for the transport of CPA to the periplasm [40]. These two genes, wzt and wzm, encode the ATP-binding component and membrane component of a CPA dedicated ABC transporter [38]. Pyocin L1, which shows good activity against PAO1 showed no activity against strains with insertions in wzm and wzt (Figure 1C) and immunoblotting with a CPA-specific antibody confirmed the absence of the CPA in these pyocin L1 resistant strains (Figure 1D). Thus, the presence of CPA on the cell surface is required for pyocin L1 killing.

In order to determine if the requirement for CPA is due to a direct interaction with pyocin L1 we purified LPS from wild-type PAO1 and from the pyocin L1 resistant, wzm and wzt mutants (which produce no CPA but do produce the O-specific antigen) and analysed the pyocin-CPA interaction by isothermal titration calorimetry (ITC). Titration of pyocin L1 into isolated LPS-derived polysaccharides (a mixture of CPA and the O-specific antigen containing polysaccharides) from PAO1 gave rise to strong saturable exothermic heats of binding (Figure 2A), whereas no binding was detected on titration of pyocin L1 into an equivalent concentration of LPS carbohydrates from PAO1 wzt, which produces the O-specific antigen but not the CPA (Figure 2B). These data show that pyocin L1 binds directly to the CPA and that this interaction is required for killing. The CPA is therefore likely to be the cellular receptor for pyocin L1.

Pyocin L1 binds the monosaccharide d-rhamnose
The evolutionary relationships between MMBL-like bacteriocins and the originally identified mannose-binding members of this protein family, led to the assumption that carbohydrate binding of polysaccharides by the lectin-like bacteriocins is primarily mediated through binding of d-mannose at one or more of their conserved QxDxNxVxY carbohydrate binding motifs. Indeed, the recent structures [23] of putidacin L1 bound to mannose-containing monosaccharides adds weight to this idea, although measured affinities between polysaccharides and putidacin L1 are weak (mM) and so may not be physiologically relevant. However, the strong interaction between pyocin L1 and CPA, is incompatible with this and suggests that d-rhamnose and not d-mannose is the likely physiological substrate for the QxDxNxVxY carbohydrate binding motifs.
To determine the affinity of pyocin L1 for d-rhamnose and d-mannose, isothermal titration calorimetry (ITC) was performed. Titration of pyocin L1 into d-rhamnose gave rise to weakly saturable heats of binding that are significantly larger than the heats observed on titration of pyocin L1 into an identical concentration of d-mannose (Figure 3). From this experiment an apparent Kd of 5–10 mM was estimated for the interaction of pyocin L1 with d-rhamnose with apparently weaker binding for d-mannose, Kd>50 mM. The interaction between pyocin L1 and these monosaccharides was also probed using NMR with 15N labelled pyocin L1, monitoring changes to its 15N-heteronuclear single quantum correlation (15N-HSQC) spectra on addition of d-rhamnose or d-mannose. In the absence of added monosaccharide 15N-HSQC spectra of pyocin L1, which should contain one crosspeak for each non-proline amide NH as well as peaks for the NH groups in various side chains, were well resolved and dispersed, indicative of a folded protein. Chemical shift perturbation monitored by 15N-HSQC allows the mapping of changes to a protein that occur on ligand binding. Addition of either d-rhamnose or d-mannose up to a concentration of 100 mM did not give rise to large or global changes in chemical shifts (Figure S3). On addition of d-rhamnose significant chemical shift changes were observed for a discrete subset of peaks including some in the amide side chain region of the spectra, while changes of a smaller magnitude were observed on the addition of equal concentrations of d-mannose (Figure S3). Fitting the chemical shift changes that occur on addition of d-rhamnose, for peaks showing strong shifts, to a single site binding model indicates a Kd for the pyocin L1 - d-rhamnose complex in the range of 5–20 mM (Figures 3C–F). These data correlated well with the ITC sugar binding data, with low mM binding of pyocin L1 to d-rhamnose and much weaker binding to d-mannose.
d-rhamnose and the CPA bind to the C-terminal QxDxNxVxY motifs of pyocin L1
In an attempt to determine the location of the pyocin L1 d-rhamnose binding site(s) and the structural basis of the d-rhamnose specificity of pyocin L1 we determined the X-ray structures of pyocin L1 with bound d-mannose, d-rhamnose and in the unbound form (Table 1). Pyocin L1, as predicted by sequence homology to MMBL proteins, consists of two tandem β-prism domains characteristic of MMBLs, connected by antiparallel strands propagating from the end of each MMBL domain and lending a strand to the reciprocal β-prism. The strands contain a tryptophan residue which forms π-stacking interactions with two other tryptophans in the β-prism to stabilise the structure (Figure 4A). This interaction is conserved throughout MMBLs, with most members of the class utilising it to form either homo - or hetero-dimers of single MMBL subunits. However, in pyocin L1, as with the recently described structure of putidacin L1, both domains are from a single polypeptide chain [23]. Other structural elements are also common between the two bacteriocins, namely a C-terminal extension of 30 amino acids and a two-turn α-helix insertion into loop 6 of the N-terminal MMBL domain (Figure 4B). The overall root mean square deviation (rmsd) of backbone atoms for pyocin L1 and putidacin L1 is 7.5 Å, which is relatively high due to a difference in the relative orientation of the two MMBL domains. In contrast, the relative orientation of the tandem MMBL domains of pyocin L1 matches those of the dimeric plant lectins very closely, with alignment of pyocin L1 with the snowdrop lectin homodimer (pdb ID: 1MSA) giving an rmsd of 4.81 Å. Comparison of the respective N - and C - terminal domains from pyocin L1 and putidacin L1 shows they possess very similar folds with rmsds of 2.77 Å and 2.02 Å, respectively (Figures 4C–D). The higher value for comparison of the N-terminal domains is due to the presence of a 2-strand extension to β-sheet two of the putidacin L1 N-terminal MMBL domain, which is absent from pyocin L1 and other MMBLs. In order to identify protein structures which share a similar fold to pyocin L1 we submitted the structure of the DALI server (http://ekhidna.biocenter.helsinki.fi/dali_server/start). The DALI server searches the protein data bank (PDB) to identify proteins structurally related to the query structure [41]. Significant structural homology was only identified for putidacin L1 and other proteins previously characterised as containing a MMBL fold such as the snowdrop lectin. MMBL dimers of plant origin often form higher order structures, however small angle X-ray scattering of pyocin L1 showed it to be monomeric in solution (Figure S4).


Electron density maps, derived from both d-mannose and d-rhamnose soaked crystals show clear density for sugar moieties in both sites, C1 and C2 (Figure 5). The sugars refined well in these densities at full occupancy, giving B-factors comparable to the surrounding protein side chains. The canonical MMBL hydrogen bonds observed for both d-mannose and d-rhamnose were the same: Gln to O3, Asp to O2, Asn to O2 and Tyr to O4. In addition, O6 of d-mannose forms a hydrogen bond with Tyr169 in C1 and His194 in C2. As d-rhamnose is C6 deoxy d-mannose, it lacks these interactions (Figure 6). The fact that d-mannose forms an additional hydrogen bond is counter-intuitive given that pyocin L1 has a significantly stronger affinity for d-rhamnose, however Val154, Val163 and Ala166 of C1 and Val184 and Ala191 of C2 form a hydrophobic pocket to accommodate the C6-methyl group of d-rhamnose (Figure S5).


Weak density was observed for both sugars at site N1, however given the high concentrations used in the soak and the overall low binding affinity of pyocin L1 for monomeric sugars, it is unlikely that N1 represents a primary binding site for d-rhamnose (Figure S5). The conserved residues in site N2 form interactions with the C-terminal extension of the protein and as such are inaccessible. Weak density was also observed adjacent to the binding site C1 of mol B in both the soaks and in mol A of the d-rhamnose form. This density may correspond to a peripheral binding site utilised in binding to the carbohydrate chain of LPS, as is observed in the structure of putidacin L1 bound to oligosaccharides [23].
To test the idea that the observed binding of d-rhamnose to sites C1 and C2 is reflective of CPA binding and that this binding is critical to pyocin L1 cytotoxicity, we created pyocin L1 variants in which the conserved aspartic acids of the QxDxNxVxY motifs of the C1 and C2 sugar binding sites were mutated to alanine and compared their cytotoxicity and ability to bind the CPA by ITC with the wild-type protein. Titrations with wild-type pyocin L1 and the D150A (C1) and D180A (C2) variants were performed by titrating protein at a concentration of 100 µM into a solution of LPS-derived polysaccharide (1 mg ml−1) from strain PAO1 (Figure 7). Under these conditions we were able to generate binding isotherms that enabled us to accurately determine an apparent Kd of 0.15 (±0.07) µM for the wild-type pyocin L1-CPA complex. For both the D150A (C1) and D180A (C2) variants, affinity for CPA was reduced. For the pyocin L1 D150A-CPA complex a Kd of 1.52 (±0.51) µM was determined, a 10-fold increase in Kd relative to the wild-type pyocin L1-CPA complex. However, CPA binding to the D180A variant was severely weakened and although heats of binding were still observed the Kd for this complex, which could not be accurately determined, is likely >500 µM. We also produced a double mutant in which both D150A and D180A mutations were present. For this double mutant, no binding to CPA was observed by ITC. These data show that both the C1 and C2 sugar binding motifs are required for full CPA binding, but that the C2 binding site is the major CPA binding determinant. The killing activity of these sugar binding motif variants showed a good correlation with their ability to bind the CPA. Both the D150A and D180A variants showed reduced cytotoxicity against PAO1 relative to pyocin L1, with the D150A showing a greater reduction in activity and for the D150A/D180A variant very low levels of cytotoxicity were observed (Figure 7).

Putidacin L1 binds to P. syringae LPS and d-rhamnose
Pyocin L1 targets sensitive strains of P. aeruginosa through binding to LPS and utilises this as a cell surface receptor. To determine if LPS binding is common to the homologous and previously characterised lectin-like bacteriocin putidacin L1, we purified this protein and determined if the susceptibility of a number of strains of P. syringae correlated with the ability of putidacin L1 to bind to LPS-derived carbohydrates from these strains.
From the five strains of P. syringae tested, LMG 5456 and LMG 2222 were found to be highly susceptible to putidacin L1 with killing down to concentrations of 0.3 and 7.6 nM respectively. DC3000 and NCPPB 2563 showed complete resistance and LMG 1247 was highly tolerant (killing down to 0.6 µM). Binding of putidacin L1 to the isolated LPS-derived polysaccharides of the above mentioned strains was tested by ITC. Large saturable heats of binding were observed for putidacin L1 and the LPS-derived polysaccharides from LMG 5456 and LMG 2222, while no binding was observed between putidacin L1 and the LPS-derived polysaccharides from LMG 1247, 2563 or DC3000 (Figure 8). Thus, there is excellent correlation between putidacin L1 cell killing and the binding of LPS-derived polysaccharide indicating that like pyocin L1, putidacin L1 utilises LPS as a surface receptor.

Although P. syringae O-antigens are diverse relative to CPA, the incorporation of d-rhamnose is widespread and seemingly almost universal in strains of this species [42], [43]. Interestingly, in cases where d-rhamnose is not a component of P. syringae LPS, l-rhamnose is present [42]. As with pyocin L1 we utilised ITC and NMR to characterise the binding affinity of putidacin L1 for d-rhamnose, in comparison with d-mannose and l-rhamnose. Putidacin L1 exhibited an affinity of 5–10 mM for d-rhamnose, which is comparable to that of pyocin L1, and approximately 10-fold stronger than its affinity for d-mannose (Figure S6). Interestingly, no binding of l-rhamnose to putidacin L1 or pyocin L1 was observed (Figure S7). It is interesting to note that in the strains of P. syringae we have tested, the killing spectrum (but not the potency) of pyocin L1 and putidacin L1 is identical. This observation combined with the specificity of putidacin L1 for d-rhamnose, strongly suggests that it also binds to a d-rhamnose containing O-antigen. Indeed branched d-rhamnose O-antigens are common in P. syringae [42], [43].
Our data for both pyocin L1 and putidacin L1 indicate that d-rhamnose containing O-antigens are utilised as surface receptors for lectin-like bacteriocins from Pseudomonas spp. This is an attractive hypothesis since the inclusion of d-rhamnose in the lipopolysaccharides from members of this genus is widespread and could form an important component of the genus specific activity of this group of bacteriocins.
Discussion
In this work we have shown that pyocin L1 targets susceptible cells through binding to the CPA component of LPS and that primary recognition of CPA occurs through binding of d-rhamnose at the conserved QxDxNxVxY sugar binding motifs of the C-terminal lectin domain. The ability of both pyocin L1 and putidacin L1 to recognise d-rhamnose containing carbohydrates is an important component of their ability to target sensitive strains of Pseudomonas spp. The use of the O-antigen as a primary receptor differentiates the lectin-like bacteriocins from other multidomain bacteriocins such as colicins and S-type pyocins (colicin-like bacteriocins) which utilise outer membrane proteins as their primary cell surface receptors [44]. The colicin-like bacteriocins also possess a flexible, or natively disordered N-terminal region that is thought to pass through the lumen of a coreceptor and interact with the periplasmic Tol or Ton complexes that mediate translocation of the bacteriocin across the outer membrane [11], [44]. The lack of such a flexible N-terminal region in the lectin-like bacteriocins suggests that either they do not need to cross the outer membrane in order to mediate their cytotoxicity or they do so by a mechanism that is fundamentally different to the diverse family of colicin-like bacteriocins. Given the extensive structural homology between the lectin-like bacteriocins and plant lectins it seems likely that these bacteriocins share a common ancestor with plant lectins and from an evolutionary perspective are unrelated to the colicin-like bacteriocins.
In addition to O-antigen recognition, additional factors, as yet to be determined, are clearly also important in strain and species specificity among the lectin-like bacteriocins. Indeed, recent work from Ghequire et al. has shown through domain swapping experiments that for putidacin L1 (LlpABW) and the homologous lectin-like bacteriocin LlpA1Pf-5 from Pseudomonas fluorescens, species specificity is governed by the identity of the N-terminal lectin domain [23]. Thus, in view of these data and our own data it seems likely that the C-terminal lectin domain of this class of bacteriocins plays a general role in the recognition of d-rhamnose containing O-antigens, with the N-terminal domain interacting with species-specific factors and thus determining the precise species and strain specificity of these bacteriocins. Although there are few clues as to how the lectin-like bacteriocins ultimately kill susceptible cells, we have established a clear role for the C-terminal MMBL domain of these proteins. The roles of the N-terminal MMBL domain and the C-terminal extension remain to be discovered [23]. However, from the previous work of Ghequire et al, it is clear that all three of these regions are required for killing of susceptible cells.
Interestingly, although rhamnose is frequently a component of plant and bacterial glycoconjugates, such as the rhamnolipids of P. aeruginosa [45] and pectic polysaccharides of plant cell walls [46], it is generally the l-form of this sugar that is found in nature. Although otherwise rare, d-rhamnose is found frequently as a component of the LPS of plant pathogens and plant associated bacteria such as P. syringae [42], [43], P. putida [47], Xanthomonas campestris [48] and Burkholderia spp. [49], but is a relatively rare component of the O-antigens of animal pathogens such as E. coli, Salmonella and Klebsiella. It is interesting to speculate that since d-rhamnose is a common component of the LPS of bacterial plant pathogens, that some of the many lectins produced by plants may have evolved to target d-rhamnose as part of plant defence to bacterial pathogens.
The specificity of lectin-like bacteriocins suggests that these protein antibiotics may be useful in combating plant pathogenic bacteria, either through the use of bacteriocin expressing biocontrol strains or by the production of transgenic plants engineered to express these proteins. The specific targeting mechanism described here, binding of d-rhamnose containing polymers, indicates that the lectin-like bacteriocins would not interact with either plant or animal cells, since these lack d-rhamnose containing glycoconjugates. In addition, these narrow spectrum antibiotics would leave the majority of the soil microbiome and the gut microbiome of plant-eating animals intact and so would be likely to have minimal environmental impact and minimal impact on animal health. This latter property and the potency of these protein antibiotics could also make the use of lectin-like bacteriocins in the treatment of chronic multidrug-resistant P. aeruginosa infections in humans an attractive proposition.
Materials and Methods
Bacterial strains, plasmids and growth conditions
Strains and plasmids utilised in this study are presented in Supplementary Table S1. Strains of P. aeruginosa were grown in LB at 37°C, P. syringae were grown in King's B Media (KB) (20 g peptone, 10 g glycerol, 1.5 g MgSO4, 1.5 g K2HPO4 per liter adjusted to pH 7.5) at 28°C.
Cloning and purification of lectin-like bacteriocins
Pyocin L1 was amplified from the genomic DNA of the producing strain P. aeruginosa C1433 [50] by PCR using primers designed to introduce an NdeI site at the start of the pyoL1 gene (ACA GAT CAT ATG AAG TCT CCA AAC AAA AGG AGG) and an XhoI site at the end of the gene (ACA GAT CTC GAG GAC CAC GGC GCG CCG TCG TGG ATA GTC GTG GGG CCA A). The PCR product was ligated into the corresponding sites of the E. coli expression vector pET21a to give pETPyoL1 which encodes pyocin L1 with a C-terminal His6 tag separated from the C-terminus of pyocin L1 by a 6 amino acid linker (RRRAVV). Pyocin L1 was overexpressed from E. coli BL21(DE3)pLysS carrying the plasmid pETPyoL1. Five litres of LB broth was inoculated (1∶100) from an overnight culture and cells were grown at 37°C in a shaking incubator to an OD600 = 0.6. Protein production was induced by the addition of 0.3 mM isopropyl β-d-1-thiogalactopyranoside (IPTG), the cells were grown at 22°C for a further 20 hand harvested by centrifugation. Cells were resuspended in 20 mM Tris-HCl, 500 mM NaCl, 5 mM imidazole (pH 7.5) and lysed using an MSE Soniprep 150 (Wolf Laboratories) and the cell debris was separated by centrifugation. The cell-free lysate was applied to a 5-ml His Trap HP column (GE Healthcare) equilibrated in 20 mM Tris-HCl, 500 mM NaCl, 5 mM imidazole (pH 7.5) and pyocin L1 was eluted over a 5–500 mM imidazole gradient. Pyocin L1 containing fractions were identified by SDS PAGE, pooled and dialyzed overnight into 50 mM Tris-HCl, 200 mM NaCl, pH 7.5 and remaining contaminants were removed by gel filtration chromatography on a Superdex S75 26/600 column (GE Healthcare) equilibrated in the same buffer. The protein was concentrated using a centrifugal concentrator (Vivaspin 20) with a molecular weight cut off of 5 kDa and stored at −80°C until required. The putidacin L1 open reading frame was synthesised (DNA 2.0) and cloned into pET21a via 5′ NdeI and 3′ XhoI restriction sites. The stop codon was removed in order to utilise the pET21a C-terminal His6 tag. Purification of putidacin L1 was performed as for pyocin L1. Constructs to express the pyocin L1 mutants D31A, D97A, D150A and D180A were created using the QuikChange Site Directed Mutagenesis Kit (Stratagene) utilising pETPyoL1 as a template. The primers used were CAA ATT GGT CAT GCA AGC GGC TGG CAA CTT GGT CCT TTA CG and CGT AAA GGA CCA AGT TGC CAG CCG CTT GCA TGA CCA ATT TG for D31A, GCG TAC CTG AAT CTT CAA GAT GCT GGG GAC TTC GGT ATA TTT TC and GAA AAT ATA CCG AAG TCC CCA GCA TCT TGA AGA TTC AGG TAC GC for D97A, CGC CTA GCG TTT CAG GGA GCT GGC AAC CTA GTG ATC TAT C and GAT AGA TCA CTA GGT TGC CAG CTC CCT GAA ACG CTA GGC G for D150A and GAT AGA GCA GTA GTG CAA GAG GCT GGA AAT TTT GTT ATC TAC AAA G and CTT TGT AGA TAA CAA AAT TTC CAG CCT CTT GCA CTA CTG CTC TAT C for D180A. Mutant proteins were purified as described above for wild-type pyocin L1.
Pyocin sensitivity assays: Overlay spot plate method
Soft agar overlay spot plates were performed using the method of [35]. 150 µl of test strain culture at OD600 = 0.6 was added to 6 ml of 0.8% soft agar and poured over an LB or KB agar plate. 5 µl of bacteriocin at varying concentrations was spotted onto the plates and incubated for 20 h at 37 or 28°C.
Isolation of pyocin L1 tolerant mutants
1.5 ml of a culture of P. aeruginosa E2 (OD600 = 0.6) was centrifuged and resuspended in 100 µl of LB, to which 100 µl (8 mg ml−1) of purified pyocin L1 was added. The culture was grown for 1 h, plated onto a LB agar plate and incubated for 20 h at 37°C. Isolated colonies were identified as P. aeruginosa using 16S PCR as described previously [51].
Whole genome sequencing
The genomes of P. aeruginosa E2 and derived pyocin L1 tolerant mutants were sequenced at the Glasgow Polyomics Facility, generating paired-end reads on an Illumina MiSeq Personal Sequencer. Reads were mapped to the previously sequenced parent genomes of P. aeruginosa E2 using the CLC genomics workbench, MAUVE and RAST to create an ordered annotated genome. The CLC genomics workbench was used for genome comparisons and the identification of SNPs/INDELs.
LPS purification and isolation of LPS-derived polysaccharide
LPS was purified from 1 litre cultures of P. aeruginosa and P. syringae strains as described previously, with modifications including the omission of the final trifluoroacetic acid hydrolysis and chromatography steps [52]. Cells were grown for 20 h at 37°C and 28°C for P. aeruginosa and P. syringae respectively, pelleted by centrifugation at 6000 g for 20 min, and resuspended in 50 mM Tris, pH 7.5 containing lysozyme (2 mg ml−1) and DNase I (0.5 mg ml−1). Cells were lysed by sonication and the cell lysate was incubated at 20°C for 30 min before EDTA was added to a final concentration of 2 mM. An equal volume of aqueous phenol was added and the solution was heated at 70°C for 20 min, with vigorous mixing. The solution was then cooled on ice for 30 min, centrifuged at 7000 g for 20 min and the aqueous phase extracted. Proteinase K was added to a final concentration of 0.05 mg ml−1 and dialysed for 12 h against 2×5 L H2O. LPS was pelleted by ultracentrifugation at 100,000 g for 1 h, resuspended in H2O and heated to 60°C for 30 min to remove residual proteinase K activity. LPS-derived carbohydrates were isolated by heating LPS in 2% acetic acid for 1.5 h at 96°C. Lipid A was removed by centrifugation at 13,500 g for 3 min followed by extraction with an equal volume of chloroform. The aqueous phase was then lyophilised.
SDS-PAGE, silver staining and immunoblotting
Purified LPS from wild-type and mutant samples were resolved by electrophoresis on 12% SDS-polyacrylamide gels. The LPS banding patterns were visualised by the Invitrogen ultrafast silver staining method. For immunoblotting LPS was transferred onto nitrocellulose membranes and western immunoblotting was performed as previously described using the CPA-specific monoclonal antibody N1F10 and alkaline phosphatase-conjugated goat anti-mouse Fab2 as the secondary antibody [39]. The blots were developed using SIGMAFAS BCIP/NBT tablets.
Isothermal titration calorimetry
ITC experiments were performed on a VP-ITC microcalorimeter (MicroCal LLC). For monosaccharide binding, titrations were carried out at 299 K with regular 15 µl injections of ligands into 60–100 µM pyocin L1 or putidacin L1 at 300 s intervals. 50 mM d-rhamnose, d-mannose or l-rhamnose were used as titrants and reactions were performed in 0.2 M sodium phosphate buffer, pH 7.5. d-rhamnose (>97%) was obtained from Carbosynth Limited (UK) and d-mannose and l-rhamnose (>99%) from Sigma-Aldrich (UK). For O-antigen-pyocin L1 binding reactions, pyocin L1 or pyocin L1 variants were used as titrant at 100 or 150 µM with cleaved O-antigen sugars dissolved at 1 mg ml−1 in the chamber. For curve fitting we estimated the molar concentration of LPS-derived CPA containing carbohydrate chains at 20 µM based on an estimated average molecular weight of 10 kDa for CPA containing polysaccharides and estimating the percentage of total LPS represented by CPA containing carbohydrates as 20% of the total by weight [53]. This value may not be accurate and as such the stoichiometry implied by the fit is likely to be unreliable. However, the use of this estimated value has no impact on the reported parameters of ΔH, ΔS and Kd. For O-antigen-putidacin L1 binding reactions, O-antigen was used as the titrant at 3 mg ml−1 with 60 µM putidacin L1 in the chamber. Reactions were performed in 20 mM HEPES buffer pH 7.5. All samples were degassed extensively prior to the experiments. Calorimetric data were calculated by integrating the area under each peak and fitted with a single-site binding model with Microcal LLC Origin software. The heats of dilution for each titration were obtained and subtracted from the raw data.
NMR titration experiments
NMR chemical shift perturbation analysis of sugar binding by pyocin L1 and putidacin L1 was carried out at 305 K and 300 K respectively. Fast-HSQC spectra [54] were recorded using 15N labelled proteins (0.1–0.2 mM) and unlabelled ligands, d-rhamnose and d-mannose (100 mM), on a Bruker AVANCE 600 MHz spectrometer. Protein samples were prepared with and without the sugars present and volumes were exchanged at fixed ratios, making sure the protein concentration remained unchanged. The spectra were processed with Topspin and analysed with CCPNmr analysis [55].
Crystallisation and data collection for pyocin L1
Purified pyocin L1 at a concentration of 15 mg ml−1 was screened for crystallisation conditions using the Morpheus and PGA crystallisation screens (Molecular Dimensions) [56]. Screens were prepared using a Cartesian Honeybee 8+1 dispensing robot, into 96-well, MRC-format, sitting drop plates (reservoir volume of 80 µl; drop size of 0.5 µl of protein and 0.5 µl of reservoir solution). Clusters of needle shaped crystals grew in a number of conditions in each screen over 3 to 7 days. Two of these conditions, condition 1 (20% v/v ethylene glycol, 10% w/v PEG 8000, 0.03 M CaCl2, 0.03 M MgCl2, 0.1 M Tris/Bicine, pH 8.5) and condition 2 (20% PEG 550 MME, 20% PEG 20 K, 0.03 M CaCl2, 0.03 M MgCl2 0.1 M MOPS/HEPES, pH 7.5) from the Morpheus screen were selected for optimisation by vapour diffusion in 24 well plates (reservoir volume 500 µl, drop size 1 µl protein and 1 µl reservoir solution). Clusters of needles from these trays grew after 3–7 days and were mechanically separated. The un-soaked crystals were from condition 1, while soaked crystals were from condition 2. Un-soaked crystals were looped and directly cryo-cooled to 110 K in liquid nitrogen; D-mannose and D-rhamnose soaked crystals were soaked for 2–12 min in artificial mother liquor containing 4 M d-mannose or 2 M d-rhamnose, before cryo-cooling to 110 K. X-ray diffraction data were collected at the Diamond Light Source, Oxfordshire, UK at beam lines I04, I04-1 and I24. Automatic data processing was performed with Xia2 within the EDNA package [57].
Structure solution and refinement for pyocin L1
A dataset from an un-soaked pyocin L1 crystal was submitted to the Balbes pipeline along with the amino acid sequence for pyocin L1 [58]. Balbes produced a partial molecular replacement solution based on the structure of Galanthus nivalis agglutinin (PDB ID: 1MSA). Initial phases from Balbes were improved via density modification and an initial model was built using Phase and Build from the Phenix package [59]. The model was then built and refined using REFMAC5 and Coot 0.7 [60], [61]. Validation of all models was performed using the Molprobity web server and Procheck from CCP4-I [62], [63]. Two structures of sugar soaked pyocin L1 were solved by molecular replacement using Phaser [64], with the sugar-free pyocin L1 as the search model. Additional electron density corresponding to bound sugars, was observed in both 2Fo-2Fc and Fo-Fc maps [65]. Sugars were fitted and structures refined using Coot 0.7 and REFMAC5. β-d-mannose (PDB ID: BMA) corresponded best to the density of bound d-mannose. The density in the d-rhamnose complex best corresponded to α-d-rhamnose, for which no PDB ligand exists; a model for α-d-rhamnose was prepared by removing the oxygen from carbon 6 of α-d-mannose and submitting these PDB coordinates to the Prodrg server, which generated the model and modeling restraints [65]. The resultant α-d-rhamnose was designated with the PDB ID: XXR.
Small angle X-ray scattering
SAXS was carried out on the X33 beamline at the Deutsches Elektronen Synchrotron (DESY, Hamburg, Germany). Data were collected on samples of Pyocin L1 in the range of 0.5–5 mg ml−1. Buffer was read before and after each sample and an average of the buffer scattering was subtracted from the sample scattering. The data obtained for each sample were analysed using PRIMUS [66], merging scattering data at low angles with high angle data. The distance distribution function, p(r), was obtained by indirect Fourier transform of the scattering intensity using GNOM [67]. A Guinier plot (ln I(s) vs s2) was used to calculate the molecular weight at I(0) and radius of gyration, Rg, of PyoL1. Ab initio models of the protein in solution were built using DAMMIF [68], averaged with DAMAVER [69] and overlaid with the available crystal structure using SUPCOMB [70].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. GorkiewiczG (2009) Nosocomial and antibiotic-associated diarrhoea caused by organisms other than Clostridium difficile. International Journal of Antimicrobial Agents 33: S37–S41.
2. CarrollKC, BartlettJG (2011) Biology of Clostridium difficile: Implications for Epidemiology and Diagnosis. Annual Review of Microbiology 65 : 501–521.
3. ManichanhC, BorruelN, CasellasF, GuarnerF (2012) The gut microbiota in IBD. Nature Reviews Gastroenterology & Hepatology 9 : 599–608.
4. QinJ, LiY, CaiZ, LiS, ZhuJ, et al. (2012) A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490 : 55–60.
5. Henao-MejiaJ, ElinavE, JinC, HaoL, MehalWZ, et al. (2012) Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482 : 179–U167.
6. ScherJU, AbramsonSB (2011) The microbiome and rheumatoid arthritis. Nature Reviews Rheumatology 7 : 569–578.
7. HviidA, SvanstromH, FrischM (2011) Antibiotic use and inflammatory bowel diseases in childhood. Gut 60 : 49–54.
8. ShawSY, BlanchardJF, BernsteinCN (2011) Association Between the Use of Antibiotics and New Diagnoses of Crohn's Disease and Ulcerative Colitis. American Journal of Gastroenterology 106 : 2133–2142.
9. SpehlmannME, BegunAZ, SaroglouE, HinrichsF, TiemannU, et al. (2012) Risk factors in German twins with inflammatory bowel disease: Results of a questionnaire-based survey. Journal of Crohns & Colitis 6 : 29–42.
10. GrinterR, MilnerJ, WalkerD (2012) Ferredoxin containing bacteriocins suggest a novel mechanism of iron uptake in Pectobacterium spp. PLoS ONE 7: e33033.
11. GrinterR, RoszakAW, CogdellRJ, MilnerJJ, WalkerD (2012) The Crystal Structure of the Lipid II-degrading Bacteriocin Syringacin M Suggests Unexpected Evolutionary Relationships between Colicin M-like Bacteriocins. Journal of Biological Chemistry 287 : 38876–38888.
12. CascalesE, BuchananSK, DuchéD, KleanthousC, LloubèsR, et al. (2007) Colicin biology. Microbiology and Molecular Biology Reviews 71 : 158–229.
13. Michel-BriandY, BaysseC (2002) The pyocins of Pseudomonas aeruginosa. Biochimie 84 : 499–510.
14. WalkerD, MoshbahiK, VankemmelbekeM, JamesR, KleanthousC (2007) The role of electrostatics in colicin nuclease domain translocation into bacterial cells. Journal of Biological Chemistry 282 : 31389–31397.
15. OgawaT, TomitaK, UedaT, WatanabeK, UozumiT, et al. (1999) A cytotoxic ribonuclease targeting specific transfer RNA anticodons. Science 283 : 2097–2100.
16. NgCL, LangK, MeenanNAG, SharmaA, KelleyAC, et al. (2010) Structural basis for 16S ribosomal RNA cleavage by the cytotoxic domain of colicin E3. Nature Structural & Molecular Biology 17 : 1241–+.
17. ZethK, RoemerC, PatzerSI, BraunV (2008) Crystal structure of colicin M, a novel phosphatase specifically imported by Escherichia coli. Journal of Biological Chemistry 283 : 25324–25331.
18. GrahamAC, StockerBAD (1977) GENETICS OF SENSITIVITY OF SALMONELLA SPECIES TO COLICIN-M AND BACTERIOPHAGES T5 T1, AND ES18. Journal of Bacteriology 130 : 1214–1223.
19. KurisuG, ZakharovSD, ZhalninaMV, BanoS, EroukovaVY, et al. (2003) The structure of BtuB with bound colicin E3 R-domain implies a translocon. Nature Structural Biology 10 : 948–954.
20. SmithK, MartinL, RinaldiA, RajendranR, RamageG, et al. (2012) Activity of Pyocin S2 against Pseudomonas aeruginosa Biofilms. Antimicrobial Agents and Chemotherapy 56 : 1599–1601.
21. BrownCL, SmithK, McCaugheyL, WalkerD (2012) Colicin-like bacteriocins as novel therapeutic agents for the treatment of chronic biofilm-mediated infection. Biochemical Society Transactions 40 : 1549–1552.
22. LyczakJB, CannonCL, PierGB (2002) Lung Infections Associated with Cystic Fibrosis. Clinical Microbiology Reviews 15 : 194–222.
23. GhequireMGK, Garcia-PinoA, LebbeEKM, SpaepenS, LorisR, et al. (2013) Structural Determinants for Activity and Specificity of the Bacterial Toxin LlpA. PLoS pathogens 9: e1003199–e1003199.
24. GhequireMGK, LiW, ProostP, LorisR, De MotR (2012) Plant lectin-like antibacterial proteins from phytopathogens Pseudomonas syringae and Xanthomonas citri. Environmental Microbiology Reports 4 : 373–380.
25. GhequireMGK, LorisR, De MotR (2012) MMBL proteins: from lectin to bacteriocin. Biochemical Society Transactions 40 : 1553–U1433.
26. ParretAHA, SchoofsG, ProostP, De MotR (2003) Plant lectin-like bacteriocin from a rhizosphere-colonizing Pseudomonas isolate. Journal of Bacteriology 185 : 897–908.
27. ParretAHA, TemmermanK, De MotR (2005) Novel lectin-like bacteriocins of biocontrol strain Pseudomonas fluorescens Pf-5. Applied and Environmental Microbiology 71 : 5197–5207.
28. Sharon N (2001) Lectins. eLS: John Wiley & Sons, Ltd.
29. SharonN, LisH (2004) History of lectins: from hemagglutinins to biological recognition molecules. Glycobiology 14 : 53R–62R.
30. Van DammeEJM, Nakamura-TsurutaS, SmithDF, OngenaertM, WinterHC, et al. (2007) Phylogenetic and specificity studies of two-domain GNA-related lectins: generation of multispecificity through domain duplication and divergent evolution. Biochemical Journal 404 : 51–61.
31. ChandraNR, RamachandraiahG, BachhawatK, DamTK, SuroliaA, et al. (1999) Crystal structure of a dimeric mannose-specific agglutinin from garlic: Quaternary association and carbohydrate specificity. Journal of Molecular Biology 285 : 1157–1168.
32. VastaGR, Nita-LazarM, GiomarelliB, AhmedH, DuS, et al. (2011) Structural and functional diversity of the lectin repertoire in teleost fish: Relevance to innate and adaptive immunity. Developmental and Comparative Immunology 35 : 1388–1399.
33. KurimotoE, SuzukiM, AmemiyaE, YamaguchiY, NirasawaS, et al. (2007) Curculin Exhibits Sweet-tasting and Taste-modifying Activities through Its Distinct Molecular Surfaces. Journal of Biological Chemistry 282 : 33252–33256.
34. ShimokawaM, FukudomeA, YamashitaR, MinamiY, YagiF, et al. (2012) Characterization and cloning of GNA-like lectin from the mushroom Marasmius oreades. Glycoconjugate Journal 29 : 457–465.
35. HesterG, WrightCS (1996) The Mannose-specific bulb lectin from Galanthus nivalis (snowdrop) binds mono - and dimannosides at distinct sites. Structure analysis of refined complexes at 2.3 angstrom and 3.0 angstrom resolution. Journal of Molecular Biology 262 : 516–531.
36. FyfeJAM, HarrisG, GovanJRW (1984) Revised Pyocin Typing Method For Pseudomonas-Aeruginosa. Journal of Clinical Microbiology 20 : 47–50.
37. RocchettaHL, BurrowsLL, PacanJC, LamJS (1998) Three rhamnosyltransferases responsible for assembly of the A-band D-rhamnan polysaccharide in Pseudomonas aeruginosa: a fourth transferase, WbpL, is required for the initiation of both A-band and B-band lipopolysaccharide synthesis. Molecular Microbiology 30 : 1131–1131.
38. LamJS, TaylorVL, IslamST, HaoY, KocincovaD (2011) Genetic and Functional Diversity of Pseudomonas aeruginosa Lipopolysaccharide. Frontiers in microbiology 2 : 118–118.
39. HaoY, KingJD, HuszczynskiS, KocincovaD, LamJS (2013) Five New Genes Are Important for Common Polysaccharide Antigen Biosynthesis in Pseudomonas aeruginosa. Mbio 4.
40. JacobsMA, AlwoodA, ThaipisuttikulI, SpencerD, HaugenE, et al. (2003) Comprehensive transposon mutant library of Pseudomonas aeruginosa. Proceedings of the National Academy of Sciences of the United States of America 100 : 14339–14344.
41. HolmL, RosenströmP (2010) Dali server: conservation mapping in 3D. Nucleic Acids Research 38: W545–W549.
42. OvodV, RudolphK, KnirelY, KrohnK (1996) Immunochemical characterization of O polysaccharides composing the alpha-D-rhamnose backbone of lipopolysaccharide of Pseudomonas syringae and classification of bacteria into serogroups O1 and O2 with monoclonal antibodies. Journal of Bacteriology 178 : 6459–6465.
43. OvodVV, KnirelYA, SamsonR, KrohnKJ (1999) Immunochemical characterization and taxonomic evaluation of the O polysaccharides of the lipopolysaccharides of Pseudomonas syringae serogroup O1 strains. Journal of Bacteriology 181 : 6937–6947.
44. KleanthousC (2010) Swimming against the tide: progress and challenges in our understanding of colicin translocation. Nature Reviews Microbiology 8 : 843–848.
45. Abdel-MawgoudAM, LepineF, DezielE (2010) Rhamnolipids: diversity of structures, microbial origins and roles. Applied Microbiology and Biotechnology 86 : 1323–1336.
46. CaffallKH, MohnenD (2009) The structure, function, and biosynthesis of plant cell wall pectic polysaccharides. Carbohydrate Research 344 : 1879–1900.
47. KnirelYA, ShashkovAS, SenchenkovaS, AjikiY, FukuokaS (2002) Structure of the O-polysaccharide of Pseudomonas putida FERM p-18867. Carbohydrate Research 337 : 1589–1591.
48. MolinaroA, SilipoA, LanzettaR, NewmanMA, DowJM, et al. (2003) Structural elucidation of the O-chain of the lipopolysaccharide from Xanthomonas campestris strain 8004. Carbohydrate Research 338 : 277–281.
49. Vinion-DubielAD, GoldbergJB (2003) Lipopolysaccharide of Burkholderia cepacia complex. Journal of Endotoxin Research 9 : 201–213.
50. StewartL, FordA, SangalV, JeukensJ, BoyleB, et al. (2013) Draft genomes of twelve host adapted and environmental isolates of Pseudomonas aeruginosa and their position in the core genome phylogeny. Pathogens and Disease [epub ahead of print].
51. ClaessonMJ, WangQ, O'SullivanO, Greene-DinizR, ColeJR, et al. (2010) Comparison of two next-generation sequencing technologies for resolving highly complex microbiota composition using tandem variable 16S rRNA gene regions. Nucleic Acids Research 38.
52. RammM, LobeM, HamburgerM (2003) A simple method for preparation of D-rhamnose. Carbohydrate Research 338 : 109–112.
53. RiveraM, BryanLE, HancockREW, McGroartyEJ (1988) Heterogeneity Of Lipopolysaccharides From Pseudomonas-Aeruginosa - Analysis Of Lipopolysaccharide Chain-Length. Journal of Bacteriology 170 : 512–521.
54. MoriS, AbeygunawardanaC, JohnsonMO, vanZijlP (1996) Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation (vol 108, pg 94, 1995). Journal of Magnetic Resonance Series B 110 : 321–321.
55. VrankenWF, BoucherW, StevensTJ, FoghRH, PajonA, et al. (2005) The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins-Structure Function and Bioinformatics 59 : 687–696.
56. GorrecF (2009) The MORPHEUS protein crystallization screen. Journal of Applied Crystallography 42 : 1035–1042.
57. IncardonaM-F, BourenkovGP, LevikK, PieritzRA, PopovAN, et al. (2009) EDNA: a framework for plugin-based applications applied to X-ray experiment online data analysis. Journal of Synchrotron Radiation 16 : 872–879.
58. LongF, VaginAA, YoungP, MurshudovGN (2008) BALBES: a molecular-replacement pipeline. Acta Crystallographica Section D-Biological Crystallography 64 : 125–132.
59. AdamsPD, AfoninePV, BunkocziG, ChenVB, DavisIW, et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallographica Section D-Biological Crystallography 66 : 213–221.
60. EmsleyP, LohkampB, ScottWG, CowtanK (2010) Features and development of Coot. Acta Crystallographica Section D-Biological Crystallography 66 : 486–501.
61. MurshudovGN, SkubakP, LebedevAA, PannuNS, SteinerRA, et al. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallographica Section D-Biological Crystallography 67 : 355–367.
62. ChenVB, ArendallWBIII, HeaddJJ, KeedyDA, ImmorminoRM, et al. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallographica Section D-Biological Crystallography 66 : 12–21.
63. LaskowskiRA, MacarthurMW, MossDS, ThorntonJM (1993) PROCHECK - A program to check the stereochemical quality of protein structures. Journal of Applied Crystallography 26 : 283–291.
64. McCoyAJ, Grosse-KunstleveRW, AdamsPD, WinnMD, StoroniLC, et al. (2007) Phaser crystallographic software. Journal of Applied Crystallography 40 : 658–674.
65. SchuttelkopfAW, van AaltenDMF (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallographica Section D-Biological Crystallography 60 : 1355–1363.
66. KonarevPV, VolkovVV, SokolovaAV, KochMHJ, SvergunDI (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. Journal of Applied Crystallography 36 : 1277–1282.
67. SvergunDI (1992) Determination Of The Regularization Parameter In Indirect-Transform Methods Using Perceptual Criteria. Journal of Applied Crystallography 25 : 495–503.
68. FrankeD, SvergunDI (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. Journal of Applied Crystallography 42 : 342–346.
69. VolkovVV, SvergunDI (2003) Uniqueness of ab initio shape determination in small-angle scattering. Journal of Applied Crystallography 36 : 860–864.
70. KozinMB, SvergunDI (2001) Automated matching of high - and low-resolution structural models. Journal of Applied Crystallography 34 : 33–41.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 2
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Suppression of RNA Silencing by a Plant DNA Virus Satellite Requires a Host Calmodulin-Like Protein to Repress Expression
- Reversible Silencing of Cytomegalovirus Genomes by Type I Interferon Governs Virus Latency
- Identification of Host-Targeted Small Molecules That Restrict Intracellular Growth
- Implication of PMLIV in Both Intrinsic and Innate Immunity
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy