
Genome-Wide RNAi Screen Identifies Broadly-Acting Host Factors That Inhibit Arbovirus Infection
Vector-borne viruses are an important class of emerging and re-emerging pathogens; thus, an improved understanding of the cellular factors that modulate infection in their respective vertebrate and insect hosts may aid control efforts. In particular, cell-intrinsic antiviral pathways restrict vector-borne viruses including the type I interferon response in vertebrates and the RNA interference (RNAi) pathway in insects. However, it is likely that additional cell-intrinsic mechanisms exist to limit these viruses. Since insects rely on innate immune mechanisms to inhibit virus infections, we used Drosophila as a model insect to identify cellular factors that restrict West Nile virus (WNV), a flavivirus with a broad and expanding geographical host range. Our genome-wide RNAi screen identified 50 genes that inhibited WNV infection. Further screening revealed that 17 of these genes were antiviral against additional flaviviruses, and seven of these were antiviral against other vector-borne viruses, expanding our knowledge of invertebrate cell-intrinsic immunity. Investigation of two newly identified factors that restrict diverse viruses, dXPO1 and dRUVBL1, in the Tip60 complex, demonstrated they contributed to antiviral defense at the organismal level in adult flies, in mosquito cells, and in mammalian cells. These data suggest the existence of broadly acting and functionally conserved antiviral genes and pathways that restrict virus infections in evolutionarily divergent hosts.
Published in the journal:
. PLoS Pathog 10(2): e32767. doi:10.1371/journal.ppat.1003914
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003914
Summary
Vector-borne viruses are an important class of emerging and re-emerging pathogens; thus, an improved understanding of the cellular factors that modulate infection in their respective vertebrate and insect hosts may aid control efforts. In particular, cell-intrinsic antiviral pathways restrict vector-borne viruses including the type I interferon response in vertebrates and the RNA interference (RNAi) pathway in insects. However, it is likely that additional cell-intrinsic mechanisms exist to limit these viruses. Since insects rely on innate immune mechanisms to inhibit virus infections, we used Drosophila as a model insect to identify cellular factors that restrict West Nile virus (WNV), a flavivirus with a broad and expanding geographical host range. Our genome-wide RNAi screen identified 50 genes that inhibited WNV infection. Further screening revealed that 17 of these genes were antiviral against additional flaviviruses, and seven of these were antiviral against other vector-borne viruses, expanding our knowledge of invertebrate cell-intrinsic immunity. Investigation of two newly identified factors that restrict diverse viruses, dXPO1 and dRUVBL1, in the Tip60 complex, demonstrated they contributed to antiviral defense at the organismal level in adult flies, in mosquito cells, and in mammalian cells. These data suggest the existence of broadly acting and functionally conserved antiviral genes and pathways that restrict virus infections in evolutionarily divergent hosts.
Introduction
Historically, West Nile virus (WNV) epidemics were observed in Africa, the Middle East, Europe, India, Australia, and parts of Asia, In 1999, WNV entered into the North America as part of an outbreak of neuroinvasive disease in New York City [1], and since then has become endemic in the United States with large numbers of cases occurring annually in different regions of the country. Indeed, the occurrence, size, and severity of outbreaks in humans overall have increased worldwide since the mid 1990s [2], with a large outbreak in Texas in 2012 leading to many fatalities [3], [4]. Different strains of WNV, with variable worldwide distributions, exhibit significant differences in pathogenesis. In humans infected with North American WNV strains, approximately 80% of infections are asymptomatic, with 20% developing WNV fever and other relatively mild symptoms, and 1% progressing to encephalitis, meningitis, or flaccid paralysis [2]. In contrast, WNV-Kunjin, endemic in Australia, has not been associated with any human fatalities or severe disease [5]. The natural transmission cycle of WNV is between mosquitoes and birds, with humans, horses, and other vertebrates being incidental dead-end hosts [2]. WNV is a member of the Flavivirus genus, which includes many globally important vector-borne pathogens, such as Dengue (DENV), yellow fever (YFV), tick-borne encephalitis (TBEV), and Japanese encephalitis viruses (JEV) [6]. DENV is endemic in more than 110 countries with 3.6 billion people at risk, and 390 million people infected yearly [7], [8]. At present, there are no specific antiviral therapies against any flavivirus, and only three insect-borne flaviviruses have approved vaccines for humans (YFV, TBEV, and JEV) [9].
Flaviviruses are small (∼50 nm diameter) enveloped viruses that contain a single-stranded, positive-sense RNA genome of ∼11-kb with a 5′ cap, but unlike mRNA, lack a 3′ polyadenylated tail [10]. WNV enters both vertebrate and invertebrate cells through clathrin-mediated endocytosis [11], and then traffics to an acidic compartment that facilitates viral fusion with endosomal membranes and release of the nucleocapsid into the cytoplasm [12]. The viral genome encodes one open reading frame and is translated as a single polyprotein at the rough endoplasmic reticulum (ER), which is subsequently processed by both viral and cellular proteases into 3 structural and 7 non-structural viral proteins [13]. Viral RNA replication occurs within cytoplasmic complexes associated with perinuclear membranes requiring lipid rearrangements [14], [15], [16], [17], and progeny viruses bud into the ER and traffic through the Golgi network where virions are processed into mature particles prior to exocytosis [18].
While there has been extensive study into the cellular pathways that are hijacked to facilitate WNV infection in mammalian cells, less is known about the cell-intrinsic pathways that restrict WNV in insects and whether these pathways have conserved roles in vertebrates. Furthermore, Flaviviruses belong to a larger group of vector-borne RNA viruses (including Togaviruses and Bunyaviruses), raising the possibility that these viruses as a group may be restricted using shared host defense pathways. Indeed, RNA interference (RNAi) is recognized as a major antiviral mechanism in insects and is active against all human arthropod-borne viruses tested including the flaviviruses WNV and DENV [19], [20]. The Jak-STAT and Toll pathways also are active in diverse insect hosts and restrict flavivirus infection in mosquitoes [19], [20]. Indeed, many antiviral pathways active in vector insects were first shown to restrict viral infection in the fruit fly (Drosophila melanogaster) model. This is in part due to the depth of Drosophila genome annotation, powerful genetic tools, potent gene silencing by RNAi, limited genetic redundancy, a high percentage of identifiable functional orthologs in both mosquitoes and vertebrates, lack of an acquired immune system, and that Drosophila can be experimentally infected by a large number of human arthropod-transmitted viruses. Furthermore, RNAi screening is robust in Drosophila cells and has been used effectively to analyze host-pathogen interactions and identify genes involved in antiviral defense including components of the RNAi silencing machinery [21], [22], [23], [24]. Additionally, findings in Drosophila have been extended to mosquitoes and mammals further validating this approach [23], [24], [25], [26], [27], [28], [29], [30], [31].
In this study, we used Drosophila to identify cell-intrinsic antiviral genes that restrict WNV and hypothesized that a number of these would restrict other insect-borne viruses, and some might have conserved roles in vector insects such as mosquitoes. Since many antiviral pathways (e.g., autophagy, Jak/Stat and Toll pathways) are active both in mammals and insects, we speculated that some of these newly identified factors also would confer antiviral activity in mammalian cells. To identify such genes using an unbiased approach we performed a genome-wide high-content RNAi screen in Drosophila cells to identify cellular factors that limited WNV infection. To date, RNAi screens have mainly focused on cellular factors usurped by pathogens to promote infection. While 22 restriction factors have been identified as anti-flaviviral in genome-wide RNAi screens [24], [32], only 2 of these are conserved between humans and insects. We optimized the assay for the discovery of restriction factors and identified 50 genes that when silenced resulted in enhanced WNV infection in Drosophila cells. All 50 are conserved in mosquitoes and 86% have clearly defined human orthologs. Furthermore, 17 of these genes had antiviral activity against multiple flaviviruses, and 7 genes were antiviral against a diverse panel of additional vector-borne RNA viruses. We focused on two broadly acting conserved genes, dRUVBL1 (pontin) and dXPO1 (embargoed), and found both restricted viral infection in adult flies, were antiviral in mosquito Aedes aegypti cell culture as well as in human cells. Furthermore, since WNV is neurotropic we tested whether RUVBL1 contributes to control of WNV in neurons and found it to be antiviral in these cells. Mechanistically, our studies establish that dRUVBL1 along with other members of the Tip60 histone acetylase complex are antiviral suggesting a role for this complex in virus restriction. Furthermore, we found that dXPO1 controls the nuclear export of specific host mRNAs, including the mRNA encoding Aldolase, which we identified as antiviral. Collectively, we identified additional novel, broadly acting cell-intrinsic antiviral genes in Drosophila at least some of which function in mosquito and vertebrate cells.
Results
RNAi screening of WNV infection of Drosophila cells
To identify cellular factors that restrict WNV infection, we first characterized the infection of a pathogenic North American WNV isolate (New York 2000) (referred to as WNV) [33], in Drosophila DL1 cells. WNV successfully infected and produced infectious virions from DL1 cells, although infection levels were substantially lower than that observed in human cells (Figure S1A and B in Text S1). Kinetic experiments revealed that peak immunofluorescence signal of virally produced NS1 protein was 48 hours post infection (hpi), a time point prior to substantial virus spread (Figure S1B and C in Text S1). We next tested whether WNV infection of Drosophila cells was dependent on similar entry and replication pathways as in mammalian and mosquito cells. Chlorpromazine, an inhibitor of clathrin-mediated endocytosis, blocks entry of WNV in both mammalian and mosquito cells [34], [35], and also effectively inhibited WNV infection of Drosophila DL1 cells (Figure S1D in Text S1). Ribavirin, a nucleoside analog and a inhibitor of Flavivirus replication in many mammalian cell types [36], also inhibited WNV infection of Drosophila cells (Figure S1E in Text S1).
Next, we optimized RNAi in a 384-well format using dsRNAs against β-galactosidase (βgal) as a negative control, and dsRNA against the WNV genome as a positive control (Figure 1A and B). We also included dsRNA targeting Ars2, a gene that we previously established as antiviral in Drosophila against many unrelated RNA viruses [21]. By selecting the infection level at ∼7%, this maximized the fold-change in infection upon loss of Ars2, allowing us to focus the assay on genes which restrict infection. This approach contrasts with previous screens that used a higher infection level and focused on genes that promote infection [24], [32]. Briefly, DL1 cells were seeded onto 384 well plates pre-arrayed with dsRNAs, incubated for 3 days for effective knockdown of target genes, and infected with WNV (Multiplicity of infection (MOI) of 10) for 48 hours. Cells were fixed, permeabilized and stained for the viral protein NS1 [37] and counterstained for nuclei. Automated microscopy and image analysis calculated the cell number per well (nuclei) and number of infected cells (WNV NS1) to measure the percent infection. As expected, we observed a decrease in infection after treatment with dsRNA against WNV. Importantly, we also observed a robust increase in WNV infection upon loss of Ars2 (Figure 1A and B); thus these optimized conditions were used for RNAi screening.
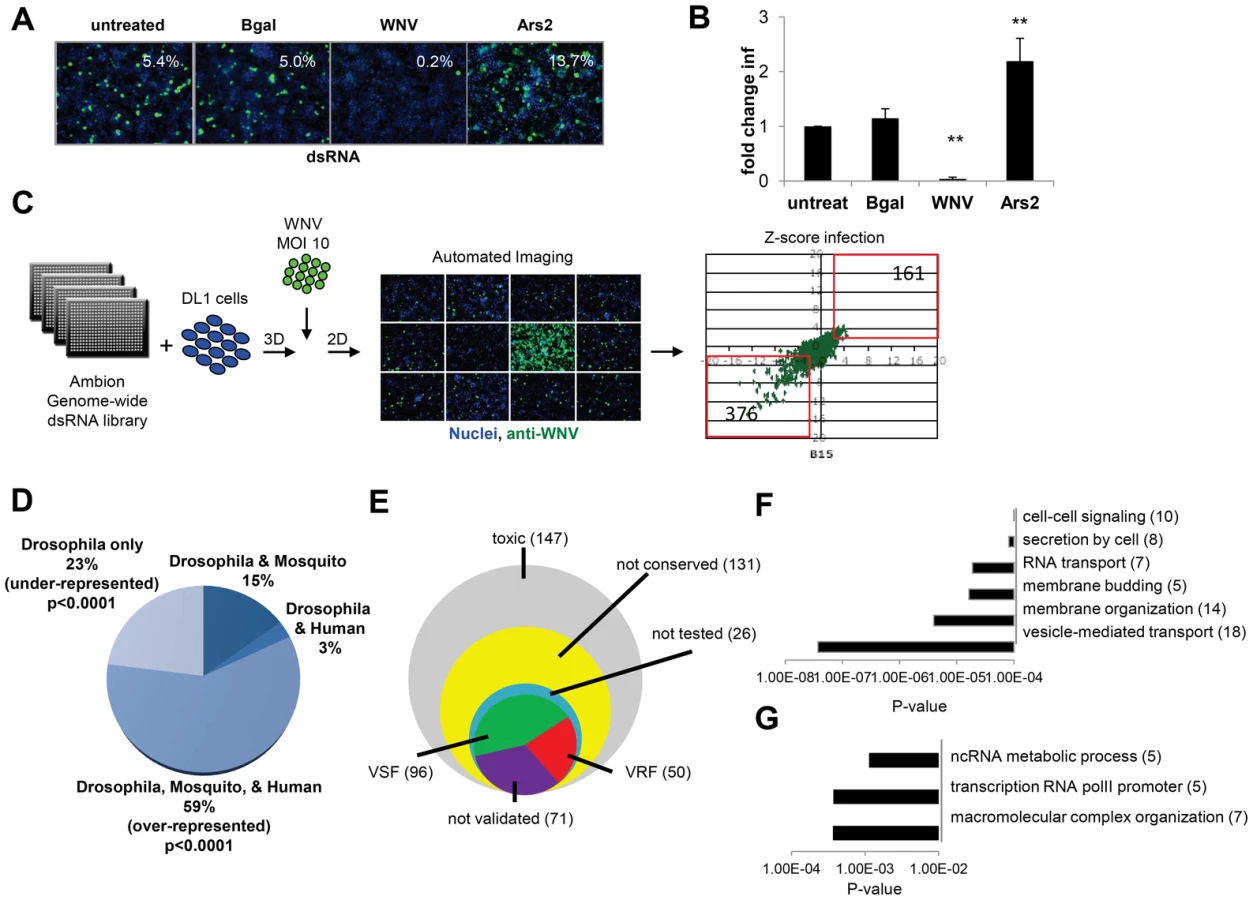
A genome-wide RNAi screen was performed in duplicate and statistical analysis identified 537 genes (3.6% of the Drosophila genome) that when silenced had a significant effect on the percentage of WNV infected cells, with a robust Z score of ≥2 or ≤−2 in both replicates (p<0.001; ∼40% change; Figure 1C). None of the non-targeting controls spotted on each plate were identified whereas 100% of the positive control dsRNAs spotted on each plate against WNV genome and Ars2 were identified. Silencing of 376 of these 537 genes resulted in decreased viral infection, indicating WNV was dependent on these genes for replication (viral sensitivity factors (VSF)). Silencing of 161 genes resulted in increased WNV infection suggesting they normally restrict replication (viral resistance factors (VRF)). As WNV infects mosquitoes, birds, and vertebrates we were interested in those genes having orthologs in hosts that normally encounter the virus, rather than genes annotated as Drosophila specific, as flies are not natural hosts. Analysis of this candidate gene list revealed that ∼59% of the genes have orthologs in both humans and mosquitoes (p<0.0001), with Drosophila-specific genes being greatly under-represented (∼23% of the total; p<0.0001) (Figure 1D). Of the 537 genes identified in the primary screen, 147 were cytotoxic (robust Z score<−2 in duplicate; ∼15% decrease in cell number) and were excluded from further analysis. Only one gene had a robust Z score>2 in duplicate but did not validate subsequently. Additionally, 131 genes were not clearly conserved in mosquitoes or humans (as determined by Homologene) and also were excluded from further analysis. Of the 280 remaining genes, we set out to validate all of the genes except for a handful that were members of complexes in which we identified >2 components. In those cases, we chose to validate representative genes from these complexes (Table S1). To do this, we generated independent dsRNA reagents that targeted 217 genes and screened this secondary gene set under two conditions: we infected cells at a low level of infection (4%) to maximize identification of genes that restricted infection, and at a higher level of infection (18%) to maximize validation of genes that promote infection. Of the 217 genes, 121 validated (56%): 82 genes (68%) facilitated WNV infection (VSFs) and 39 genes (32%) restricted infection (VRFs). We also validated a total of 23 genes from larger complexes (Table S1, Table S2, and Figure S1F in Text S1). If we include the remaining 17 genes in the complex, the screen identified 96 VSFs and 50 VRFs in total (Figure 1E; Table S1 and S2).
Bioinformatics analysis was used to identify processes or pathways that impact WNV infection. First, we performed Gene Ontology enrichment analysis on the VSF and VRF gene sets independently (Figure 1F and G) and found biological pathways including vesicle-mediated transport and membrane modifications were enriched within the VSF data set, consistent with the important role of vesicular trafficking and membrane modifications in WNV entry and replication [6]. Second, we used several functional annotation metrics to place these validated genes into cellular pathways and sub-cellular compartments most likely relevant to WNV infection (red genes, VRF; green genes, VSF; black genes not tested but in validated complexes; Figure S1G in Text S1). We identified 29 genes involved in endocytosis and endosomal acidification, a known entry pathway for WNV. Furthermore, although we tested and validated only 4 of the components in the signal recognition particle complex, we identified 6 subunits of this complex in our primary screen, supporting the importance of targeting the WNV polyprotein to the ER for proper translation and processing. These findings suggest that this screen was robust and identified important host factors that promote infection.
The VRFs were highly conserved (86% have human orthologs) and fell into distinct enriched groups. Two of the three categories involved RNA metabolism, including RNA transcription, which may be involved in an antiviral transcriptional program [31]. In fact, 28% of the WNV VRFs (p<0.00012) have a function within the nucleus suggesting a complex host response to infection since WNV replicates exclusively in the cytoplasm (Figure S1F in Text S1).
Identification of broadly antiviral factors in vector-borne virus infections
While few antiviral pathways have been described in Drosophila, the well characterized ones (e.g., RNA silencing machinery) appear to inhibit infection of diverse viruses [38], [39]. Given this, we explored whether the anti-WNV factors identified also restricted other viral pathogens. We tested two additional flaviviruses: the WNV strain Kunjin (CH 16532; WNV-KUN) and Dengue virus (Drosophila adapted Dengue-2 (DENV)). In addition we tested three additional human vector-borne viruses: Sindbis virus (HRsp; SINV), Rift Valley Fever virus (MP12; RVFV), and vesicular stomatitis virus (Indiana; VSV) (Figure 2A). All of these are enveloped RNA viruses transmitted to vertebrates by an insect vector. While mosquitoes are the natural vector for WNV, WNV-KUN, DENV, SINV and RVFV, sandflies are the primary vector for VSV. The flaviviruses and SINV are positive sense RNA viruses, whereas RVFV and VSV are negative sense. RVFV has a tri-segmented genome, while the other viruses encode a non-segmented genome. Thus, these viruses represent divergent families and genomic architectures.

We and others have previously infected Drosophila with WNV, DENV, SINV, RVFV and VSV [24], [25], [40], [41], [42]. However, WNV-KUN infection of Drosophila has not been characterized. WNV-KUN is a less pathogenic strain of WNV endemic to Oceania [5]. We found that WNV-KUN, analogous to WNV New York, productively infected Drosophila cells (Figure S2A and B in Text S1). Next, we optimized conditions for RNAi screening in 384 well plates using both negative and positive control dsRNAs based upon our previous studies selecting conditions to identify restriction factors for WNV-KUN, DENV, SINV, RVFV and VSV (Figure S2C–G in Text S1) [25], [40], [41]. We screened the validated WNV gene set in duplicate against each virus, and Z-scores were calculated (Table S3). We used hierarchical clustering to compare the VRF gene dependencies of all six viruses (Figure 2B). The four positive sense viruses clustered together (flaviviruses WNV, WNV-KUN, and DENV, and alphavirus SINV), while RVFV and VSV, the two negative sense viruses clustered together. This suggests the gene signature of restriction is related to a fundamental aspect of viral structure.
The WNV VRFs had a high propensity to impact infection by multiple different viruses. There was a high concordance of gene dependencies across the three flaviviruses; 31 genes (86%) restricted WNV-KUN and 22 genes (61%) restricted DENV (Figure 2C and Table S3). There also was a large overlap between WNV and SINV VRFs (64%), while less so with RVFV (38%) and VSV (25%). Thus, many anti-WNV factors appear broadly antiviral against other flaviviruses and an unrelated positive strand RNA virus in insect cells. The degree of VRF overlap diminished as the viruses became more disparate (Figure 2C). Nonetheless, we identified 7 host factors that significantly restricted infection by all six vector-borne viruses tested (p<0.05): dXPO1 (emb), dRUVBL1 (pont), dYARS (Aats-tyr), dEIF1B (CG17737), dPPM1L (CG7115), dCTNS (CG17119) and dICT1 (CG6094). All seven of these VRF genes have human and mosquito orthologs (Figure 2D).
dRUVBL1 and the Tip60 complex restrict vector-borne virus infection in Drosophila
Among the validated WNV VRFs, genes with putative nuclear roles were enriched (p<0.00012) and included dRUVBL1 (pontin, also known as Tip49), which was antiviral against all six viruses. RUVBL1 is an ATP-binding protein belonging to the AAA+ (ATPase associated with diverse cellular activities) family of ATPases implicated in diverse cellular pathways in the nucleus and cytoplasm [43], [44], [45], [46], [47], [48], [49]. First, we validated the antiviral activity of dRUVBL1 using independent dsRNA targeting dRUVBL1 outside of the screening format and observed a significant increase (p<0.05) in infection by WNV, WNV-KUN, DENV, SINV, RVFV and VSV compared to control (Figure 3A and B). There was no impact on cell number upon depletion of dRUVBL1 (Figure S3A in Text S1). Next, using quantitative RT-PCR (RT-qPCR) as an independent assay, we found that both WNV and VSV RNA levels were increased upon dRUVBL1-depletion compared to the control (Figure 3C and D).

One advantage of the Drosophila system is the powerful genetic tools including the availability of genome-wide in vivo RNAi transgenic flies. Furthermore, Drosophila are not hematophagous, so they can be challenged easily and safely with highly pathogenic human viruses. We took advantage of WNV-KUN as it is a BSL2 agent in comparison to the more virulent North American WNV strains, which require a BSL3 facility [50]. Wild-type flies were permissive to WNV-KUN infection as measured by plaque assay and exhibited no increase in mortality compared to control flies (Figure S3B and C in Text S1). This is consistent with the natural infection of mosquitoes where limited pathogenesis is observed, and similar to our observations with other vector-borne viruses (VSV, SINV, RVFV) that display limited pathology upon viral infection [25], [40], [41]. However, loss of innate immune defenses in Drosophila or mosquitoes can render insects more susceptible to infection and result in increased viral replication and mortality [19], [20], [21], [40], [51], [52], [53]. Because null mutants in dRUVBL1 are lethal, we took advantage of inducible RNAi transgenic flies [54]. We used the GAL4/UAS system to promote expression of a UAS - inverted repeat (IR) transgene that bears long hairpin dsRNA against dRUVBL1 to target the endogenous transcript in vivo. We induced expression of the transgene using a heat shock (hs) promoter in adult flies allowing us to bypass any developmental requirements. Indeed, expression of the hairpin during development was lethal (data not shown). Importantly, heat shock driven dRUVBL1 RNAi flies had decreased dRUVBL1 mRNA (Figure S3D in Text S1). Next, dRUVBL1-depleted (hs-GAL4<dRUVBL1 IR) and control flies (hs-GAL4<+) were challenged with WNV-KUN and survival was monitored. Unchallenged dRUVBL1-depleted flies exhibited no increase in mortality nor did control flies challenged with WNV-KUN. Notably, the majority of WNV-KUN infected dRUVBL1-depleted flies succumbed to infection (p<0.01, Figure 3E). We next tested if there was an impact on viral load. Groups of 15 flies were challenged, and whole animals were crushed, and assayed for WNV-KUN by plaque assay in four independent experiments (shown as individual dots). We observed modest, but increased viral loads in dRUVBL1-depleted animals compared to controls (set to 1) at day 6 post infection; similar results were observed at day 9 post infection (Figure 3F, not shown).
We subsequently explored the requirement of dRUVBL1 during VSV infection, the best-studied human arbovirus in flies, and most divergent from WNV of the vector-borne viruses tested (Figure 2C). Again, while uninfected flies or wild type control flies challenged with VSV exhibited little mortality, flies depleted for dRUVBL1 and challenged with VSV showed an increase in mortality after infection (p<0.01, Figure 3G). Groups of 15 flies were challenged, and whole animals were crushed, and assayed for VSV by plaque assay in seven independent experiments (shown as individual dots). We observed modest, but increased viral loads in dRUVBL1-depleted animals compared to controls (set to 1) at day 6 post infection (Figure 3H). Together, these results demonstrate the important and broad-spectrum antiviral requirement for dRUVBL1 both in vitro and in vivo in Drosophila.
dRUVBL1 has been shown to function in many complexes, most often in conjunction with another AAA+ ATPase, dRUVBL2 (reptin, also known as Tip48) (depicted in Figure 4A) [43], [54]. Indeed, structural and functional analysis of human and yeast RUVBL1 and RUVBL2 suggest these proteins work as a scaffold in addition to functioning as ATPases, potentially explaining their association with a diverse set of cellular complexes [44]. dRUVBL1, along with dRUVBL2, is involved in chromatin remodeling, most notably in the Ino80 and Tip60/Swr1 complexes [55], [56]. Furthermore, roles in transcriptional regulation facilitating the activity of c-Myc and β-catenin also have been reported in Drosophila and human cells [57], [58]. Additional roles for dRUVBL1 and dRUVBL2 have been described in snoRNA maturation, nonsense mediated mRNA decay, and telomere maintenance. Lastly, dRUVBL1 also has been implicated in chromatin remodeling with the Drosophila Trithorax complex, although this is thought to be independent of dRUVBL2 [46]. Based on these possible functions, we tested components of these complexes for their impact on viral infection to identify which of the putative dRUVBL1 containing complex(es) mediated the antiviral activity. We designed dsRNAs against dRUVBL2 along with the indicated genes in each of the complexes in Figure 4A. Cells were treated with these dsRNAs, along with β-gal (negative) and dRUVBL1 (positive) controls. Importantly, no impact on cell viability was observed (Figure S4A in Text S1). Next, the dsRNA treated cells were infected with either WNV or VSV. Depletion of c-Myc, arm (Drosophila β–catenin), Smg1, Fib and Nop60B did not impact WNV or VSV infection levels (Figure 4B and C). In contrast, dRUVBL2 was antiviral against both VSV and WNV (Figure 4B and C). Depletion of both dRUVBL1 and dRUVBL2 together did not increase infection beyond that observed with silencing of either gene, indicating their effect was not additive (data not shown). Increased WNV and VSV infection also was observed when dTIP60 (Tip60), dEP400 (domino (dom)) and dSMARCA4 (Brahma (brm)) were depleted. Since dRUVBL1, dRUVBL2, dEP400 and dTIP60 are all antiviral, and members of the Tip60 complex, these data suggest that a major antiviral role of dRUVBL1 is through its function in the Tip60 complex.

Next, we tested whether Tip60 also restricted infection of adult flies. Indeed, we depletion of Tip60 using in vivo RNAi led to decreased survival of flies challenged with WNV-KUN and VSV but did not impact survival of unchallenged animals (Figure S4E–G in Text S1). Thus, the Tip60 complex also has antiviral roles in vivo.
Tip60 complex is antiviral in mosquito cells
Mosquitoes are the natural vectors for WNV, WNV-KUN, DENV, RVFV and SINV although the particular mosquito species that transmit each of these viruses varies [59], [60]. In contrast, the primary vector for VSV is the sandfly, although VSV has been isolated from mosquitoes [61]. Aedes aegypti is the primary vector species for DENV transmission, and can be infected by RVFV, WNV, WNV-KUN, and SINV [62]. Furthermore, the Aedes aegypti genome has been sequenced [63] and the Aedes aegypti cell line Aag2 is amenable to RNAi and routinely used as a model for mosquito cell studies [64].
We designed dsRNAs against Aedes aegypti RUVBL1 (AAEL004686), RUVBL2 (AAEL010341), and TIP60 (AAEL014072) orthologs. Prior to infection, Aag2 cells were treated with these dsRNAs or with dsRNAs against Bgal or the viral genome as negative and positive controls, respectively. Loss of RUVBL1, RUVBL2, or TIP60 mosquito orthologs did not affect cell number (Figure S4B in Text S1) but led to a significant increase in WNV-KUN infection (p<0.05, Figure S4C in Text S1 and Figure 4D). Similarly, each of these genes had antiviral effects against VSV, as silencing resulted in increased infection (p<0.05, Figure S4D in Text S1 and Figure 4E). These data indicate that members of the Tip60 complex also have antiviral activity in cells from a mosquito vector.
Export receptor, dXPO1, restricts virus infection in Drosophila
dXPO1 (embargoed (emb), also known as CRM1), another broadly antiviral gene identified in our screen, is a nuclear export receptor conserved from yeast to humans. XPO1 shuttles proteins and RNAs from the nucleus to the cytoplasm [65], [66]. To validate the role of dXPO1 in viral infection we tested whether an independent dsRNA against dXPO1 modulated infection. Silencing of XPO1 with an independent dsRNA did not impact cell number (Figure S5A in Text S1) but resulted in to 2 to 4-fold increases in the percentage of cells infected with WNV, WNV-KUN, DENV, SINV, RVFV or VSV as measured by microscopy (p<0.05, Figure 5A and B). Consistent with this, loss of dXPO1 led to a ≥6-fold increase in both WNV and VSV RNA, as measured by RT-qPCR (p<0.05, Figure 5C and D). Thus, a loss of dXPO1 expression leads to increased viral replication in Drosophila cells.

Next, we assessed whether dXPO1 was antiviral in vivo in adult flies. Null mutants of dXPO1 are lethal [67] so we again used an inducible RNAi and observed in vivo silencing of the mRNA (Figure S5B in Text S1). We then challenged control (hs-GAL4>+) or dXPO1-depleted (hs-GAL4>dXPO1 IR) flies with vehicle, WNV-KUN or VSV. While unchallenged flies or control challenged flies did not exhibit increased mortality, dXPO1-depleted flies challenged with either WNV-KUN or VSV had increased mortality (p<0.01, Figure 5E and F). Furthermore, dXPO1-depleted flies had modestly increased WNV-KUN viral loads, as measured by plaque assay of whole flies in four independent experiments (individual dots) relative to control (set to 1) (Figure 5G). And increased VSV loads, as measured by plaque assay of whole flies in three independent experiments (individual dots) relative to control (set to 1) (Figure 5H). These results establish that dXPO1 is required for antiviral defense both in cells and at the organismal level in adult flies.
XPO1 is antiviral in mosquito cells
To assess whether XPO1 also had antiviral activity in the vector mosquito cells, we treated Aag2 cells with dsRNAs against the Aedes aegypti XPO1 ortholog (AAEL001484) or against Bgal or the viral genome as negative and positive controls, respectively. These cells were subsequently challenged with WNV-KUN or VSV. While depletion of XPO1 did not affect cell number (Figure S5B in Text S1), we observed a significant increase in the percentage of Aag2 cells infected with WNV-KUN or VSV (p<0.05, Figure S5C and D in Text S1 and Figure 5I and J).
Aldolase, a target of dXPO1 nuclear export, is antiviral
Since dXPO1 is as a nuclear export receptor, we speculated that dXPO1-dependent regulation of either host genes required for infection or virus-induced antiviral genes may account for the antiviral activity. Indeed, antiviral transcriptional programs have been shown to restrict viral infections in Drosophila [19], [31], [52], [68], [69]. Leptomycin B (LMB) is a potent and specific inhibitor of dXPO1 mediated nuclear export [70]. Previous work demonstrated that LMB treatment of Drosophila cells altered the nuclear export of only 85 mRNAs (<2% of the transcripts surveyed) [71]. One gene, bsg, was XPO1-dependent and required for WNV infection. However, this cannot explain the phenotype of XPO1 because bsg was required for WNV infection and not the other viruses that are sensitive to XPO1 restriction (Table S3). Moreover, 2 XPO1-dependent genes also were transcriptionally induced by VSV infection (CG4294, CG30389) [31]. We generated dsRNAs targeting CG4294 and CG30389 but observed no impact on WNV-KUN or VSV infection (Figure 6C and D). None of the 50 VRFs from our screen were within this set; however, data mining of an RNAi screen with VSV in DL1 cells (S. Cherry unpublished data) identified one additional gene (Aldolase, dALDOA) from this LMB-dependent gene set that showed antiviral activity against VSV (Figure 6A). We generated an independent dsRNA targeting dALDOA and observed that depletion did not affect cell number (Figure S6A in Text S1) but resulted in a 1.5 to 2.5-fold increase (p<0.05) in the percentage of cells infected with WNV-KUN and VSV, respectively (Figure 6C and D). This suggests that dXPO1-dependent mRNA export of dALDOA contributes to the defense against multiple virus families.

Aldolase is a critical enzyme in glycolysis, catalyzing the conversion of fructose 1,6-biphosphate to glyceraldehyde-3-phosphate (G3P) and dihydroxyacetone phosphate (DHAP) (Figure 6B). However, Aldolase may have functions apart from glycolysis, as its expression but not all core glycolytic enzymes are increased in response to LPS treatment [72]. To define whether the antiviral activity of Aldolase was related to glycolysis we performed two complementary experiments. First, we depleted Drosophila cells of additional enzymes essential for glycolysis (Phosphoglucose isomerase (Pgi), Phosphofructokinase (Pfk), Phosphoglycerate kinase (Pgk), and Phosphoglycerate mutase (Pglym87)) (Figure 6B). Depletion of these canonical glycolysis enzymes had no impact on cell number (Figure S6A in Text S1) or WNV-KUN and VSV infection (Figure 6C and D). Second, to overcome the fact that RNAi is incomplete, and that these are enzymes which may be fully active at low levels, we took advantage of two specific and potent glycolysis pathway inhibitors, Dichloracetic Acid (DCA), which inhibits the enzyme pyruvate dehydrogenase kinase, and a hexokinase inhibitor (3Br) [73]. Neither of these treatments impacted cell number (Figure S6B–E in Text S1) or WNV-KUN and VSV infection of Drosophila cells (Figure 6B–D). Together, these data suggest the antiviral effect of Aldolase is not mediated through the glycolysis pathway.
XPO1 and RUVBL1 have antiviral activity in mammalian cells
As dRUBVL1 and dXPO1 are conserved from insects to mammals, we tested whether silencing of these genes in human cells impacted infection. For these studies, we transfected human osteosarcoma U2OS cells with siRNAs against a non-targeting control, hRUVBL1 or hXPO1. Three days later, we confirmed silencing of these genes by RT-qPCR (Figure S7A and B in Text S1) with no impact on cell number (Figure S7C in Text S1). Next, the cells were infected with WNV-KUN (MOI of 0.5), and infection levels were monitored using immunofluorescence 20 hpi. Loss of either hRUVBL1 or hXPO1 resulted in a 2 to 3-fold increase in the percentage of WNV-KUN-infected cells, as measured by microscopy (p<0.05, Figure 7A). Consistent with this, we observed an increase in viral RNA levels in cells depleted of hRUVBL1 or hXPO1 as measured by Northern blot and quantified (p<0.05, Figure 7B). Similarly, depletion of hRUVBL1 or hXPO1 enhanced VSV infection, as measured by the percentage of infected cells (p<0.05, Figure 7C) or levels of viral RNA (p<0.05, Figure 7D). Furthermore, we tested whether RUVBL1 likely acted through the same Tip60 complex as we found in Drosophila. To this end, we obtained independent siRNAs against hTIP60 (KAT5) and confirmed they reduced TIP60 expression in human 293T cells as measured by RT-qPCR (Figure S7D in Text S1). Furthermore, we observed significantly increased WNV infection in the depleted cells (Figure 7E). Together, these data suggest that the Tip60 complex is antiviral against multiple viruses and in disparate hosts ranging from insects to vertebrates.

Our initial studies in Drosophila were performed in a single round of infection suggesting that the requirements for the genes in the viral lifecycle included: entry, uncoating, translation, polyprotein processing, and RNA replication. To study the step in the viral lifecycle impacted by the Tip60 complex in mammalian cells we took advantage of a human cell line (293T) that stably maintains a subgenomic WNV replicon expressing GFP [74], [75]. If these genes restricted infection downstream of entry, but upstream of assembly, they should restrict the replication of this WNV replicon. Indeed, siRNA depletion of hRUVBL1 or hTIP60 led to increased levels of WNV replicon replication as measured by immunoblot (Figure S7E in Text S1). Therefore, the action of these genes is at the step of translation, polyprotein processing, or RNA replication.
Since WNV is a neurotropic virus we tested whether RUVBL1 restricted infection in primary neuronal cultures. We prepared cerebellar granule cell neurons from wild-type C57BL/6 mice and transduced them with lentiviruses expressing either a control shRNA, or 4 independent shRNAs against RUVBL1. Three days later, we challenged the cells with WNV (MOI = 0.1), and harvested virus in the supernatant 24 hours later. Notably, all four independent shRNA depleted RUVBL1 to varying extents (Figure S7F in Text S1), and the level of depletion correlated with a significant increase in viral titers (p<0.05, Figure 7F). These data demonstrate that RUVBL1 restricts WNV infection in primary neurons.
To confirm a role for hXPO1 in antiviral defense in human cells using a small molecule inhibitor to complement our RNAi studies, we treated U2OS cells with the XPO1 export inhibitor LMB and monitored WNV-KUN or VSV infection. Treatment with LMB significantly enhanced (2–3 fold) viral replication by both viruses (p<0.05, Figure 7G and H), as measured by an increase in the percentage of infected cells. LMB treatment did not impact cell number (Figure S7G in Text S1). Furthermore, siRNA-mediated depletion of hXPO1 or LMB treatment of 293T cells carrying a WNV replicon revealed that the dependence was again downstream of entry and upstream of assembly since both perturbations led to increased levels of replication (Figure S7E and S7H in Text S1). These data suggest that the hXPO1 has antiviral activity through the regulation of XPO1-dependent cargo export downstream of entry in evolutionarily diverse cell types from insects to mammals.
Discussion
Genome-wide RNAi screens have been employed to identify cellular factors required by viruses to successfully infect cells as well as factors that, if left unmodulated by the virus, serve to suppress infection. In addition, this screening approach can identify pathways that regulate the expression and activity of direct antiviral factors, orchestrating a robust antiviral response. Since our goal was to identify conserved inhibitory pathways that span insects and mammals with a particular interest in those having broad antiviral activity against disparate viruses, we performed a genome-wide RNAi screen in Drosophila in which we deliberately set a low infection rate, thereby sensitizing our assay to detect factors that when suppressed result in higher levels of infection. This is in contrast to previous genome-wide flavivirus RNAi screens, which targeted a higher level of infection and so led to the identification of a larger number of genes that promote infection [24], [32]. Nonetheless, our screen was sufficiently sensitive and robust to enable us to identify 96 genes that promoted WNV infection. Enriched gene ontology categories included pathways such as clathrin-mediated endocytosis and endosomal acidification that are required for flavivirus entry and were identified by earlier RNAi screens.
We identified 50 restriction factors, greatly expanding the number of cell intrinsic anti-WNV factors known [32], [76], [77], [78], [79], [80]. We compared our restriction factors with previous studies (Table S4). A genome-wide siRNA screen against WNV in human cells identified 22 genes that were antiviral of which 6 had Drosophila homologs; none of which were within our validated antiviral genes [32]. A genome wide screen against hepatitis C virus, a distantly related Flaviviridae family member, in human cells identified 25 antiviral genes of which 12 had Drosophila orthologs; again, none of which were within our gene set [81]. Two screens querying the antiviral role of interferon stimulated genes (ISGs) against flaviviruses were recently published [80], [82]; however, none of our antiviral genes are known ISGs. The Schoggins screen identified 47 ISGs that when ectopically expressed restricted a flavivirus amongst which there were 12 homologs in Drosophila; none of which we identified as antiviral in our screen. The Li screen identified 47 ISGs that when depleted by RNAi restricted infection amongst which 13 had homologs in Drosophila; none of which were identified in our screen. None of the Drosophila homologs from any of these screens were within any other screen making conclusions difficult. Additional screens performed at low levels of infection may reveal additional intrinsic restriction factors.
Unexpectedly, our antiviral gene set was enriched for nuclear functions such as RNA metabolism and transcription even though WNV replicates exclusively in the cytoplasm. This observation suggested that we had uncovered pathways and processes that orchestrate an antiviral response rather than factors that interact directly with the virus. If this were the case, as is seen with antiviral interferon (IFN) responses in mammals, we reasoned that many of the VRFs might have antiviral activities against additional viruses. This in fact proved to be the case - not only did we discover a high degree of concordance between the WNV, WNV-KUN and DEN VRFs (WNV-KUN 86%, 31 genes; DENV 61%, 22 genes), we identified seven genes that restricted infection of all six different arboviruses tested, which included both positive and negative-sense RNA genomes: dYARS (Aats-tyr), dEIF1(CG17737), dPPM1L (CG7115), dCTNS (CG17119), dICT1 (CG6094), dXPO1 (emb), and dRUVBL1 (pont). Since none of these genes have been suggested previously to have an antiviral role in insects, we chose two genes for more detailed analysis: dRUVBL1 and dXPO1.
RUVBL1 had antiviral activity in Drosophila and mosquito cells. Depletion of dRUVBL1 in adult flies converted a non-pathogenic infection by WNV-KUN or VSV into a pathogenic infection with increased mortality and viral replication. These data suggest that RUVBL1 has a highly conserved role in antiviral defense in insects, including mosquito vectors. RUVBL1 is an AAA+ ATPase implicated in many cellular pathways [43] and that interacts with a number of other molecules that impact its function. By methodically suppressing each of its known interacting partners, we found that components of the Tip60 chromatin-remodeling complex (TIP60, EP400 and RUVBL2) that regulates transcription [43] were antiviral against multiple viruses in Drosophila and mosquito cells. dTip60 also was antiviral in adult flies. Furthermore, silencing of RUVBL1 led to increased viral infection in human cultured cells and primary mouse neurons. Silencing of TIP60 in human cells also rendered them more susceptible to WNV infection. Together, these results suggest a conserved role for the Tip60 complex in antiviral defense across phylogeny. WNV subgenomic replicons were used to show that the requirement for these genes in restriction is downstream of entry and upstream of viral assembly, suggesting a restriction of translation, polyprotein processing and/or RNA replication. While further investigation is required to determine the Tip60 targets that are responsible for the antiviral activity and the precise step of the lifecycle impacted, the identification of a chromatin remodeling complex as broadly antiviral is intriguing. Innate immunity is controlled, in large part, through the tight regulation of sequential gene expression programs that have effector function to restrict pathogen replication. We recently characterized a complex and rapid transcriptional antiviral host program active in insects that includes both primary responses which are translation-independent and secondary responses that are translation-dependent [31]. Half of this response was controlled at the level of transcriptional pausing, which also plays a role in innate immune responses in mammals [31], [83], [84], [85]. This antiviral transcriptional program was active against a broad panel of viruses, as we found with the Tip60 complex here. Thus, we hypothesize that the Tip60 chromatin remodeling complex may contribute to the orchestration of this sophisticated antiviral transcriptional response.
A recent study found that RUVBL2 was antiviral against influenza virus by interfering with nucleoprotein (NP) oligomerization that drives viral RNA polymerase activity in the nucleus; however, in contrast to our findings, this effect was independent of RUVBL1 function [86]. This may be a distinct and direct role for RUVBL2 in influenza replication independent of the role for the Tip60 complex in antiviral defense.
Many viruses, including those used in our studies inhibit host transcriptional responses to prevent the induction of antiviral mRNAs including IFN genes. Whether viruses target this Tip60 complex to block an antiviral transcriptional program is unknown. Tip60 is degraded by a number of nuclear viruses including HIV, adenovirus, papilloma virus and cytomegalovirus to promote viral replication [87], [88], [89], [90]. This is thought to alleviate its repression of early gene transcription. Whether these virus interactions alter the activity of the Tip60 complex on antiviral gene expression remains unknown.
The second broadly-acting VRF we investigated was XPO1. At the organismal level depletion of dXPO1 enhanced viral replication and mortality by both WNV-KUN and VSV. Data from yeast and humans suggest that XPO1 is a nuclear export receptor responsible for the translocation of RNAs and proteins from the nucleus to the cytoplasm [91], [92]. However, more recent studies have suggested that the mRNA cargo dependent on XPO1 is limited [71], [93]. Inhibition of XPO1 either by RNAi or using the specific inhibitor LMB, which blocks the nuclear export function of XPO1, resulted in increased viral infection, which suggests the antiviral role of XPO1 is at the step of nuclear export. As the vector-borne RNA viruses studied here replicate exclusively in the cytoplasm, we hypothesize that XPO1 transports cellular mRNAs critical for an antiviral response. Indeed, viruses including VSV (used in our study), HIV, VEEV, ebolavirus and picornaviruses inhibit nuclear export of antiviral genes including ISG mRNAs required for defense in mammalian cells [94], [95], [96], [97], [98], [99], [100], [101]. Consistent with this, LMB inhibition of XPO1 mediated export in human cells suppressed the export of IFNα1 mRNA [99].
While a role for nuclear export in antiviral immunity has been described in mammalian cells, its function in insect immunity was unknown. To identify the particular mRNAs responsible for the antiviral effects of XPO1 in Drosophila we mined a microarray study of Drosophila cells that found less than 2% of the mRNAs tested (85 mRNAs) exhibited nuclear export dysregulation upon LMB treatment [71]. Depletion of one XPO1-dependent mRNA, dALDOA (Aldolase A), resulted in enhanced virus infection in Drosophila cells. This suggests that the transport of dALDOA mRNA plays a role in the innate immune response to vector-borne viral infections. As part of the glycolysis pathway, ALDOA enzymatically cleaves fructose 1,6-bisphosphate (F-1,6-BP) into glyceraldehyde 3-phosphate (G3P) and dihydroxyacetone phosphate (DHAP). Our RNAi and pharmacological experiments suggested the mechanism of viral suppression by ALDOA was independent of its effects on glycolysis. Future studies will be required to define mechanistically how ALDOA acts to inhibit viral infections in insect cells.
Collectively, we have begun to describe a series of conserved pathways, including transcriptional pausing, chromatin remodeling and RNA export that likely regulate the expression of gene sets whose products are antiviral, perhaps in a direct way. For the most part, virus-host interaction studies have often concentrated on proteins that interact directly with the virus. Our work has revealed pathways that orchestrate larger responses, and this confers potent antiviral activity against a broad range of divergent viruses. Clearly, there is still much to be learned about the cellular factors critical for an innate immune response to vector-borne viruses in both vertebrate and invertebrate hosts. Our identification of these cell-intrinsic antiviral genes restricting WNV, and in many cases additional viruses, provides new opportunities for understanding the control mechanisms and larger antiviral programs active against globally relevant classes of emerging viral pathogens.
Materials and Methods
Ethics statement
This animal studies were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee at the Washington University School of Medicine (Assurance Number: A3381-01).
Cells, antibodies, and reagents
DL1 and Aag-2 cells were grown as previously described [31]. BHK, U2OS, and 293T cells were maintained as previously described [102]. 293T cells harboring WNV subgenomic replicon were maintained as previously described [74], [75]. Cerebellar granule cell neurons from neonatal (E6) wild-type C57BL/6 mice were generated from cerebella dissected in HBSS and dissociated in 1 mg/ml trypsin with 125 U/ml DNAse (Sigma, St. Louis, MO) for 20 min. Enzymatic digestion was quenched with DMEM/10% FCS and the tissue was pelleted, washed in HBSS, dissociated by trituration through a P-200 pipette tip and layered on a Percoll gradient. Cells were plated in neurobasal media (Gibco) supplemented with B-27 serum-free supplement (Gibco) on poly-D-lysine (PDL)-treated dishes for 1 hour to remove adherent glial cells. Nonadherent cells were then washed in HBSS, counted plated on PDL-coated wells in serum-free DMEM (supplemented with N2 growth medium (Gibco, Grand Island, NY) and 20 mM KCl. Cultures were >95% pure and were used 3 to 4 days later for lentivirus infections [103]. Antibodies were obtained from the following sources, anti-WNV NS1 (9-NS1; [37]), anti-RVFV N (1D8 – gift from C. Schmaljohn), anti-hTIP60 (abcam, ab23886) and Alexa-488 donkey anti-mouse secondary (Jackson Immunochemicals). The following inhibitors were used: Leptomycin B (SIGMA) 50 ng/ml; Dichloracetic Acid (SIGMA) 60 µM; Hexokinase II inhibitor II (Calbiochem) 0.1 mM.
Virus stocks
West Nile virus (WNV lineage I strain 3000.0259 New York 2000) was generated in BHK cells, concentrated using Centricon Plus-70 (Millipore), and ultracentrifuged through a sucrose cushion as described previously [104]. The WNV-KUNV isolate (CH16532) was a generous gift of R. Tesh (World Reference Center of Emerging Viruses and Arboviruses, Galveston, TX) was propagated using the same protocol as WNV. DENV (gift from M. Garcia-Blanco) was grown as previously described [24]. SINV was propagated as previously described [25]. RVFV strain MP12 was propagated as described [41]. VSV was grown as described [40]. All MOIs were determined on BHK cells.
Drosophila RNAi screen
For the primary WNV screen dsRNAs targeting 13,071 genes were pre-arrayed in thirty-two 384-well plates at 250 ng per well (Ambion). 16,000 DL1 cells were seeded in serum-free Schneider's media (10 uL/well). One hour later complete media was added (20 uL/well). Three days post plating, cells were infected with WNV at an MOI of 10 (10 uL/well). 48 hours post infection cells were fixed (4% formaldehyde), and a mAb against WNV NS1 (9-NS1) was used to identify infected cells (anti-mouse Alexa-fluor488 (Jackson Immunochemicals)) and counterstained with Hoechst 33342 to monitor nuclei. 3 images per well were captured at 20× using an automated microscope (ImageXpressMicro) and analyzed using MetaXpress software. Average infection and nuclei number were calculated for each site and averaged for each well. The percent infection was log-transformed, and the median and interquartile range were used to calculate a z-score: (log10(%infection)-log10(median))/(IQR*0.74) for each plate. The entire screen was performed in duplicate and those wells with Robust Z-scores≥to 2.0 or ≤to −2.0 in both replicates were considered ‘hits’.
Similar to the primary screen, secondary screen plates were arrayed with dsRNA (250 ng) targeting a different region of the genes identified in the primary screen (DRSC). WNV infections were performed in duplicate at a higher (20) and lower (5) MOI (18% and 4% respectively). Infections with other viruses used the same protocol as WNV and were fixed at the following hours post-infection: WNV-KUN - 48 hrs (MOI = 10), DENV - 72 hrs (MOI = 10), SINV – 40 hrs (MOI = 5), VSV – 24 hrs (MOI = 1), RVFV MP12 – 30 hrs (MOI = 1). Robust Z-scores in each duplicate viral infection set of ≥1.5 or ≤1.5 in duplicate (∼40% change) were considered ‘hits’ (p<0.009); none of the negative controls (non-targeting) were identified as positive, and all of the positive controls (dsRNA against virus genome) were identified.
Bioinfomatic analysis
The functional annotation and clustering of WNV ‘hits’ was performed using the DAVID Bioinfomatics resource. Homologene (NCBI) was used to identify orthologs. Gene Cluster and TreeView were used to generate heat maps.
Adult infections
All flies were maintained on standard medium at room temperature. Flies carrying UAS-dXPO1 IR (VDRC v3347) or UAS-dRUVBL1 IR (VDRC v105408) were crossed to heat shock (HS)-GAL4 flies (Bloomington) at room temperature. On the day of injection, the progeny were heat-shocked at 37°C for 1 hour and shocked every 2 days throughout the experiment. Adults of the stated genotypes were challenged with WNV-KUN or VSV as previously described [22]. Groups of at least 20 flies were challenged for mortality studies. For viral titers, groups of 15 flies per experimental treatment were crushed and processed at 6 days post-infection for plaque assays on BHK cells [31].
Quantitative PCR
Total RNA was isolated from infected cells using Trizol (Invitrogen). For northern blots, RNA species were analyzed as previously described [105]. For RT-qPCR, cDNA was generated using random hexamers to prime reverse transcription reactions using MMLV reverse transcriptase. cDNA samples were treated with 100 U DNase I (Qiagen) according to manufacturer's protocol. Quantitative PCR (qPCR) was performed with the cDNA using Power SYBR Green PCR Master Mix (Applied Biosystems) and primers targeting VSV and WNV (VSV For-CGGAGGATTGACGACTAATGC, Rev-ACCATCCGAGCCATTCGA: WNV For-ACATCAAACGTGGTTGTTCCGCTG, Rev-TTGAGGCTAGAGCCAAGCATAGCA) in accordance with manufacturer's protocol. qPCR conditions were as follows; initial 94°C for 5 min, then 30 cycles of 94°C for 30 sec, 55°C for 30 sec, and 72°C for 30 sec. Relative viral copy numbers were generated by normalizing to cells treated with control dsRNA.
Mammalian RNAi
For siRNA treatments, mammalian cells were reverse transfected with 20 nM siRNA (Ambion: Negative Control #1 (AM4611), GFP (AM4626), XPO1 (s14937), RUVBL1 (s16370)), KAT5 (s20630, s20631) using HiPerfect according to the manufacturers protocol (Qiagen). 60 hours post-transfection, for immunofluorescence, cells were replated in a 96-well format and infected with either VSV or WNV-KUN virus 12 hours later. For FACS the cells were infected with WNV for 24 hours and stained for NS1 or TIP60 [82] or infected and processed for northern blot or immunoblot at 12 hr p.i. for VSV and 20 hr p.i. for WNV-KUN.
For shRNA treatments, lentiviruses (pLK0.1) encoding shRNA targeting RUVBL1 (clone 1: GCTGGAGATGTGATTTACATT; clone 2: GCTGGCAAAGATCAATGGCAA; clone 3: GCCACAGAGTTTGACCTTGAA; clone 4: GCAAGATATTCTGTCTATGAT) or a control (luciferase) were obtained from RNAi Core facility at Washington University School of Medicine. Lentivirus particles were generated after co-transfection of HEK-293T cells with packaging plasmids. Supernatants were collected at 48 hours later and added to neuron cultures. Three days after transduction, neurons were infected with WNV (New York 1999 strain) at an MOI of 0.1. One day later, supernatants were harvested and titered for virus infection by focus-forming assay [106].
Drug treatments
One day prior to infection U2OS or DL1 cells were seeded in 96-well plates at 20,000 or 70,000 cells per well respectively. Leptomycin B (SIGMA) was added at 50 ng/ml to mammalian cells cells 2 hr prior to infection with WNV-KUN or VSV. Dichloroacetic Acid (SIGMA) or Hexokinase II inhibitor II (Calbiochem) were added to U2OS or DL1 cells respectively, 30 minutes prior to infection. Cells were fixed 20 (WNV-KUN) or 12 (VSV) hr post infection, stained and imaged as previously described.
Supporting Information
Zdroje
1. NashD, MostashariF, FineA, MillerJ, O'LearyD, et al. (2001) The outbreak of West Nile virus infection in the New York City area in 1999. N Engl J Med 344 : 1807–1814.
2. CampbellGL, MarfinAA, LanciottiRS, GublerDJ (2002) West Nile virus. Lancet Infect Dis 2 : 519–529.
3. RoehrB (2012) Texas records worst outbreak of West Nile virus on record. BMJ 345: e6019.
4. RoehrB (2012) US hit by massive West Nile virus outbreak centred around Texas. BMJ 345: e5633.
5. HallRA, BroomAK, SmithDW, MackenzieJS (2002) The ecology and epidemiology of Kunjin virus. Curr Top Microbiol Immunol 267 : 253–269.
6. Gubler D, Kuno G, Markoff L (2007) Flaviviruses. In: D.M K, Howley PM, editors. Virology. Philadelphia: Lippincott Williams & Wilkins.
7. GublerDJ (2002) The global emergence/resurgence of arboviral diseases as public health problems. Arch Med Res 33 : 330–342.
8. BhattS, GethingPW, BradyOJ, MessinaJP, FarlowAW, et al. (2013) The global distribution and burden of dengue. Nature 496 : 504–507.
9. HeinzFX, StiasnyK (2012) Flaviviruses and flavivirus vaccines. Vaccine 30 : 4301–4306.
10. WenglerG, WenglerG, GrossHJ (1978) Studies on virus-specific nucleic acids synthesized in vertebrate and mosquito cells infected with flaviviruses. Virology 89 : 423–437.
11. BressanelliS, StiasnyK, AllisonSL, SturaEA, DuquerroyS, et al. (2004) Structure of a flavivirus envelope glycoprotein in its low-pH-induced membrane fusion conformation. EMBO J 23 : 728–738.
12. Sanchez-San MartinC, LiuCY, KielianM (2009) Dealing with low pH: entry and exit of alphaviruses and flaviviruses. Trends Microbiol 17 : 514–521.
13. ChambersTJ, HahnCS, GallerR, RiceCM (1990) Flavivirus genome organization, expression, and replication. Annu Rev Microbiol 44 : 649–688.
14. WestawayEG, MackenzieJM, KhromykhAA (2003) Kunjin RNA replication and applications of Kunjin replicons. Adv Virus Res 59 : 99–140.
15. Martin-AcebesMA, BlazquezAB, Jimenez de OyaN, Escribano-RomeroE, SaizJC (2011) West Nile virus replication requires fatty acid synthesis but is independent on phosphatidylinositol-4-phosphate lipids. PLoS One 6: e24970.
16. HeatonNS, RandallG (2010) Dengue virus-induced autophagy regulates lipid metabolism. Cell host & microbe 8 : 422–432.
17. WelschS, MillerS, Romero-BreyI, MerzA, BleckCK, et al. (2009) Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell host & microbe 5 : 365–375.
18. MukhopadhyayS, KuhnRJ, RossmannMG (2005) A structural perspective of the flavivirus life cycle. Nat Rev Microbiol 3 : 13–22.
19. ZambonRA, NandakumarM, VakhariaVN, WuLP (2005) The Toll pathway is important for an antiviral response in Drosophila. Proc Natl Acad Sci U S A 102 : 7257–7262.
20. XiZ, RamirezJL, DimopoulosG (2008) The Aedes aegypti toll pathway controls dengue virus infection. PLoS Pathog 4: e1000098.
21. SabinLR, ZhouR, GruberJJ, LukinovaN, BambinaS, et al. (2009) Ars2 regulates both miRNA - and siRNA - dependent silencing and suppresses RNA virus infection in Drosophila. Cell 138 : 340–351.
22. CherryS, PerrimonN (2004) Entry is a rate-limiting step for viral infection in a Drosophila melanogaster model of pathogenesis. Nat Immunol 5 : 81–87.
23. HaoL, SakuraiA, WatanabeT, SorensenE, NidomCA, et al. (2008) Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 454 : 890–893.
24. SessionsOM, BarrowsNJ, Souza-NetoJA, RobinsonTJ, HersheyCL, et al. (2009) Discovery of insect and human dengue virus host factors. Nature 458 : 1047–1050.
25. RosePP, HannaSL, SpiridigliozziA, WannissornN, BeitingDP, et al. (2011) Natural resistance-associated macrophage protein is a cellular receptor for sindbis virus in both insect and Mammalian hosts. Cell Host Microbe 10 : 97–104.
26. PhilipsJA, RubinEJ, PerrimonN (2005) Drosophila RNAi screen reveals CD36 family member required for mycobacterial infection. Science 309 : 1251–1253.
27. AgaisseH, BurrackLS, PhilipsJA, RubinEJ, PerrimonN, et al. (2005) Genome-wide RNAi screen for host factors required for intracellular bacterial infection. Science 309 : 1248–1251.
28. DorerMS, KirtonD, BaderJS, IsbergRR (2006) RNA interference analysis of Legionella in Drosophila cells: exploitation of early secretory apparatus dynamics. PLoS Pathog 2: e34.
29. DerreI, PypaertM, Dautry-VarsatA, AgaisseH (2007) RNAi screen in Drosophila cells reveals the involvement of the Tom complex in Chlamydia infection. PLoS Pathog 3 : 1446–1458.
30. CherryS, DoukasT, ArmknechtS, WhelanS, WangH, et al. (2005) Genome-wide RNAi screen reveals a specific sensitivity of IRES-containing RNA viruses to host translation inhibition. Genes Dev 19 : 445–452.
31. XuJ, GrantG, SabinLR, Gordesky-GoldB, YasunagaA, et al. (2012) Transcriptional pausing controls a rapid antiviral innate immune response in Drosophila. Cell host & microbe 12 : 531–543.
32. KrishnanMN, NgA, SukumaranB, GilfoyFD, UchilPD, et al. (2008) RNA interference screen for human genes associated with West Nile virus infection. Nature 455 : 242–245.
33. EbelGD, DupuisAP2nd, NgoK, NicholasD, KauffmanE, et al. (2001) Partial genetic characterization of West Nile virus strains, New York State, 2000. Emerg Infect Dis 7 : 650–653.
34. NawaM, TakasakiT, YamadaK, KuraneI, AkatsukaT (2003) Interference in Japanese encephalitis virus infection of Vero cells by a cationic amphiphilic drug, chlorpromazine. J Gen Virol 84 : 1737–1741.
35. ChuJJ, LeongPW, NgML (2006) Analysis of the endocytic pathway mediating the infectious entry of mosquito-borne flavivirus West Nile into Aedes albopictus mosquito (C6/36) cells. Virology 349 : 463–475.
36. AndersonJF, RahalJJ (2002) Efficacy of interferon alpha-2b and ribavirin against West Nile virus in vitro. Emerg Infect Dis 8 : 107–108.
37. ChungKM, NybakkenGE, ThompsonBS, EngleMJ, MarriA, et al. (2006) Antibodies against West Nile Virus nonstructural protein NS1 prevent lethal infection through Fc gamma receptor-dependent and -independent mechanisms. J Virol 80 : 1340–1351.
38. KempC, MuellerS, GotoA, BarbierV, ParoS, et al. (2013) Broad RNA interference-mediated antiviral immunity and virus-specific inducible responses in Drosophila. Journal of immunology 190 : 650–658.
39. SabinLR, HannaSL, CherryS (2010) Innate antiviral immunity in Drosophila. Curr Opin Immunol 22 : 4–9.
40. ShellyS, LukinovaN, BambinaS, BermanA, CherryS (2009) Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 30 : 588–598.
41. FiloneCM, HannaSL, CainoMC, BambinaS, DomsRW, et al. (2010) Rift valley Fever virus infection of human cells and insect hosts is promoted by protein kinase C epsilon. PLoS One 5: e15483.
42. ChotkowskiHL, CiotaAT, JiaY, Puig-BasagoitiF, KramerLD, et al. (2008) West Nile virus infection of Drosophila melanogaster induces a protective RNAi response. Virology 377 : 197–206.
43. JhaS, DuttaA (2009) RVB1/RVB2: running rings around molecular biology. Mol Cell 34 : 521–533.
44. GrigolettoA, LestienneP, RosenbaumJ (2011) The multifaceted proteins Reptin and Pontin as major players in cancer. Biochim Biophys Acta 1815 : 147–157.
45. LeeJS, KimY, BhinJ, ShinHJ, NamHJ, et al. (2011) Hypoxia-induced methylation of a pontin chromatin remodeling factor. Proc Natl Acad Sci U S A 108 : 13510–13515.
46. DiopSB, BertauxK, VasanthiD, SarkeshikA, GoirandB, et al. (2008) Reptin and Pontin function antagonistically with PcG and TrxG complexes to mediate Hox gene control. EMBO Rep 9 : 260–266.
47. McKeeganKS, DebieuxCM, WatkinsNJ (2009) Evidence that the AAA+ proteins TIP48 and TIP49 bridge interactions between 15.5K and the related NOP56 and NOP58 proteins during box C/D snoRNP biogenesis. Mol Cell Biol 29 : 4971–4981.
48. ChoiJ, HeoK, AnW (2009) Cooperative action of TIP48 and TIP49 in H2A.Z exchange catalyzed by acetylation of nucleosomal H2A. Nucleic Acids Res 37 : 5993–6007.
49. IzumiN, YamashitaA, IwamatsuA, KurataR, NakamuraH, et al. (2010) AAA+ proteins RUVBL1 and RUVBL2 coordinate PIKK activity and function in nonsense-mediated mRNA decay. Sci Signal 3: ra27.
50. ScherretJH, MackenzieJS, HallRA, DeubelV, GouldEA (2002) Phylogeny and molecular epidemiology of West Nile and Kunjin viruses. Curr Top Microbiol Immunol 267 : 373–390.
51. DeddoucheS, MattN, BuddA, MuellerS, KempC, et al. (2008) The DExD/H-box helicase Dicer-2 mediates the induction of antiviral activity in drosophila. Nat Immunol 9 : 1425–1432.
52. DostertC, JouanguyE, IrvingP, TroxlerL, Galiana-ArnouxD, et al. (2005) The Jak-STAT signaling pathway is required but not sufficient for the antiviral response of drosophila. Nat Immunol 6 : 946–953.
53. Souza-NetoJA, SimS, DimopoulosG (2009) An evolutionary conserved function of the JAK-STAT pathway in anti-dengue defense. Proc Natl Acad Sci U S A 106 : 17841–17846.
54. BellostaP, HulfT, Balla DiopS, UsseglioF, PradelJ, et al. (2005) Myc interacts genetically with Tip48/Reptin and Tip49/Pontin to control growth and proliferation during Drosophila development. Proc Natl Acad Sci U S A 102 : 11799–11804.
55. JonssonZO, JhaS, WohlschlegelJA, DuttaA (2004) Rvb1p/Rvb2p recruit Arp5p and assemble a functional Ino80 chromatin remodeling complex. Mol Cell 16 : 465–477.
56. DoyonY, SelleckW, LaneWS, TanS, CoteJ (2004) Structural and functional conservation of the NuA4 histone acetyltransferase complex from yeast to humans. Mol Cell Biol 24 : 1884–1896.
57. WanzelM, HeroldS, EilersM (2003) Transcriptional repression by Myc. Trends Cell Biol 13 : 146–150.
58. BauerA, ChauvetS, HuberO, UsseglioF, RothbacherU, et al. (2000) Pontin52 and reptin52 function as antagonistic regulators of beta-catenin signalling activity. EMBO J 19 : 6121–6130.
59. WeaverSC, ReisenWK (2010) Present and future arboviral threats. Antiviral Res 85 : 328–345.
60. KurkelaS, RattiO, HuhtamoE, UzcateguiNY, NuortiJP, et al. (2008) Sindbis virus infection in resident birds, migratory birds, and humans, Finland. Emerg Infect Dis 14 : 41–47.
61. LetchworthGJ, RodriguezLL, Del cbarreraJ (1999) Vesicular stomatitis. Vet J 157 : 239–260.
62. MoutaillerS, KridaG, SchaffnerF, VazeilleM, FaillouxAB (2008) Potential vectors of Rift Valley fever virus in the Mediterranean region. Vector Borne Zoonotic Dis 8 : 749–753.
63. NeneV, WortmanJR, LawsonD, HaasB, KodiraC, et al. (2007) Genome sequence of Aedes aegypti, a major arbovirus vector. Science 316 : 1718–1723.
64. BarlettaAB, SilvaMC, SorgineMH (2012) Validation of Aedes aegypti Aag-2 cells as a model for insect immune studies. Parasit Vectors 5 : 148.
65. TurnerJG, SullivanDM (2008) CRM1-mediated nuclear export of proteins and drug resistance in cancer. Curr Med Chem 15 : 2648–2655.
66. ZempI, KutayU (2007) Nuclear export and cytoplasmic maturation of ribosomal subunits. FEBS Lett 581 : 2783–2793.
67. CollierS, ChanHY, TodaT, McKimmieC, JohnsonG, et al. (2000) The Drosophila embargoed gene is required for larval progression and encodes the functional homolog of schizosaccharomyces Crm1. Genetics 155 : 1799–1807.
68. AvadhanulaV, WeasnerBP, HardyGG, KumarJP, HardyRW (2009) A novel system for the launch of alphavirus RNA synthesis reveals a role for the Imd pathway in arthropod antiviral response. PLoS Pathog 5: e1000582.
69. CostaA, JanE, SarnowP, SchneiderD (2009) The Imd pathway is involved in antiviral immune responses in Drosophila. PLoS One 4: e7436.
70. WolffB, SanglierJJ, WangY (1997) Leptomycin B is an inhibitor of nuclear export: inhibition of nucleo-cytoplasmic translocation of the human immunodeficiency virus type 1 (HIV-1) Rev protein and Rev-dependent mRNA. Chem Biol 4 : 139–147.
71. HeroldA, TeixeiraL, IzaurraldeE (2003) Genome-wide analysis of nuclear mRNA export pathways in Drosophila. EMBO J 22 : 2472–2483.
72. ScharteM, HanX, UchiyamaT, TawadrousZ, DeludeRL, et al. (2006) LPS increases hepatic HIF-1alpha protein and expression of the HIF-1-dependent gene aldolase A in rats. J Surg Res 135 : 262–267.
73. PelicanoH, MartinDS, XuRH, HuangP (2006) Glycolysis inhibition for anticancer treatment. Oncogene 25 : 4633–4646.
74. Ansarah-SobrinhoC, NelsonS, JostCA, WhiteheadSS, PiersonTC (2008) Temperature-dependent production of pseudoinfectious dengue reporter virus particles by complementation. Virology 381 : 67–74.
75. PiersonTC, XuQ, NelsonS, OliphantT, NybakkenGE, et al. (2007) The stoichiometry of antibody-mediated neutralization and enhancement of West Nile virus infection. Cell host & microbe 1 : 135–145.
76. LucasM, MashimoT, FrenkielMP, Simon-ChazottesD, MontagutelliX, et al. (2003) Infection of mouse neurones by West Nile virus is modulated by the interferon-inducible 2′-5′ oligoadenylate synthetase 1b protein. Immunol Cell Biol 81 : 230–236.
77. BrienJD, DaffisS, LazearHM, ChoH, SutharMS, et al. (2011) Interferon regulatory factor-1 (IRF-1) shapes both innate and CD8(+) T cell immune responses against West Nile virus infection. PLoS Pathog 7: e1002230.
78. JiangD, WeidnerJM, QingM, PanXB, GuoH, et al. (2010) Identification of five interferon-induced cellular proteins that inhibit west nile virus and dengue virus infections. J Virol 84 : 8332–8341.
79. DiamondMS, GaleMJr (2012) Cell-intrinsic innate immune control of West Nile virus infection. Trends Immunol 33 : 522–530.
80. SchogginsJW, WilsonSJ, PanisM, MurphyMY, JonesCT, et al. (2011) A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472 : 481–485.
81. LiQ, BrassAL, NgA, HuZ, XavierRJ, et al. (2009) A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proceedings of the National Academy of Sciences of the United States of America 106 : 16410–16415.
82. LiJ, DingSC, ChoH, ChungBC, GaleMJr, et al. (2013) A short hairpin RNA screen of interferon-stimulated genes identifies a novel negative regulator of the cellular antiviral response. mBio 4: e00385–00313.
83. HargreavesDC, HorngT, MedzhitovR (2009) Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138 : 129–145.
84. Ramirez-CarrozziVR, BraasD, BhattDM, ChengCS, HongC, et al. (2009) A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 138 : 114–128.
85. BhattDM, Pandya-JonesA, TongAJ, BarozziI, LissnerMM, et al. (2012) Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell 150 : 279–290.
86. KakugawaS, ShimojimaM, NeumannG, GotoH, KawaokaY (2009) RuvB-like protein 2 is a suppressor of influenza A virus polymerases. Journal of virology 83 : 6429–6434.
87. GuptaA, JhaS, EngelDA, OrnellesDA, DuttaA (2012) Tip60 degradation by adenovirus relieves transcriptional repression of viral transcriptional activator EIA. Oncogene
88. KamineJ, ElangovanB, SubramanianT, ColemanD, ChinnaduraiG (1996) Identification of a cellular protein that specifically interacts with the essential cysteine region of the HIV-1 Tat transactivator. Virology 216 : 357–366.
89. LiR, ZhuJ, XieZ, LiaoG, LiuJ, et al. (2011) Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell host & microbe 10 : 390–400.
90. SmithJA, WhiteEA, SowaME, PowellML, OttingerM, et al. (2010) Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proceedings of the National Academy of Sciences of the United States of America 107 : 3752–3757.
91. PetosaC, SchoehnG, AskjaerP, BauerU, MoulinM, et al. (2004) Architecture of CRM1/Exportin1 suggests how cooperativity is achieved during formation of a nuclear export complex. Mol Cell 16 : 761–775.
92. CullenBR (2003) Nuclear RNA export. J Cell Sci 116 : 587–597.
93. HuttenS, KehlenbachRH (2007) CRM1-mediated nuclear export: to the pore and beyond. Trends Cell Biol 17 : 193–201.
94. FariaPA, ChakrabortyP, LevayA, BarberGN, EzelleHJ, et al. (2005) VSV disrupts the Rae1/mrnp41 mRNA nuclear export pathway. Mol Cell 17 : 93–102.
95. von KobbeC, van DeursenJM, RodriguesJP, SitterlinD, BachiA, et al. (2000) Vesicular stomatitis virus matrix protein inhibits host cell gene expression by targeting the nucleoporin Nup98. Mol Cell 6 : 1243–1252.
96. HerLS, LundE, DahlbergJE (1997) Inhibition of Ran guanosine triphosphatase-dependent nuclear transport by the matrix protein of vesicular stomatitis virus. Science 276 : 1845–1848.
97. GustinKE, SarnowP (2001) Effects of poliovirus infection on nucleo-cytoplasmic trafficking and nuclear pore complex composition. EMBO J 20 : 240–249.
98. RicourC, DelhayeS, HatoSV, OlenyikTD, MichelB, et al. (2009) Inhibition of mRNA export and dimerization of interferon regulatory factor 3 by Theiler's virus leader protein. J Gen Virol 90 : 177–186.
99. KimuraT, HashimotoI, NagaseT, FujisawaJ (2004) CRM1-dependent, but not ARE-mediated, nuclear export of IFN-alpha1 mRNA. J Cell Sci 117 : 2259–2270.
100. AtashevaS, FishA, FornerodM, FrolovaEI (2010) Venezuelan equine Encephalitis virus capsid protein forms a tetrameric complex with CRM1 and importin alpha/beta that obstructs nuclear pore complex function. Journal of virology 84 : 4158–4171.
101. ReidSP, LeungLW, HartmanAL, MartinezO, ShawML, et al. (2006) Ebola virus VP24 binds karyopherin alpha1 and blocks STAT1 nuclear accumulation. Journal of virology 80 : 5156–5167.
102. MoserTS, SchiefferD, CherryS (2012) AMP-activated kinase restricts Rift Valley fever virus infection by inhibiting fatty acid synthesis. PLoS Pathog 8: e1002661.
103. SzretterKJ, DaffisS, PatelJ, SutharMS, KleinRS, et al. (2010) The innate immune adaptor molecule MyD88 restricts West Nile virus replication and spread in neurons of the central nervous system. J Virol 84 : 12125–12138.
104. HannaSL, PiersonTC, SanchezMD, AhmedAA, MurtadhaMM, et al. (2005) N-linked glycosylation of west nile virus envelope proteins influences particle assembly and infectivity. J Virol 79 : 13262–13274.
105. NakamotoM, MoyRH, XuJ, BambinaS, YasunagaA, et al. (2012) Virus recognition by Toll-7 activates antiviral autophagy in Drosophila. Immunity 36 : 658–667.
106. FuchsA, PintoAK, SchwaebleWJ, DiamondMS (2011) The lectin pathway of complement activation contributes to protection from West Nile virus infection. Virology 412 : 101–109.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 2
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Suppression of RNA Silencing by a Plant DNA Virus Satellite Requires a Host Calmodulin-Like Protein to Repress Expression
- Reversible Silencing of Cytomegalovirus Genomes by Type I Interferon Governs Virus Latency
- Identification of Host-Targeted Small Molecules That Restrict Intracellular Growth
- Implication of PMLIV in Both Intrinsic and Innate Immunity
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy