
A Compositional Look at the Human Gastrointestinal Microbiome and Immune Activation Parameters in HIV Infected Subjects
HIV progression is characterized by immune activation and microbial translocation. One factor that may be contributing to HIV progression could be a dysbiotic microbiome. We therefore hypothesized that the GI mucosal microbiome is altered in HIV patients and this alteration correlates with immune activation in HIV. 121 specimens were collected from 21 HIV positive and 22 control human subjects during colonoscopy. The composition of the lower gastrointestinal tract mucosal and luminal bacterial microbiome was characterized using 16S rDNA pyrosequencing and was correlated to clinical parameters as well as immune activation and circulating bacterial products in HIV patients on ART. The composition of the HIV microbiome was significantly different than that of controls; it was less diverse in the right colon and terminal ileum, and was characterized by loss of bacterial taxa that are typically considered commensals. In HIV samples, there was a gain of some pathogenic bacterial taxa. This is the first report characterizing the terminal ileal and colonic mucosal microbiome in HIV patients with next generation sequencing. Limitations include use of HIV-infected subjects on HAART therapy.
Published in the journal:
. PLoS Pathog 10(2): e32767. doi:10.1371/journal.ppat.1003829
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003829
Summary
HIV progression is characterized by immune activation and microbial translocation. One factor that may be contributing to HIV progression could be a dysbiotic microbiome. We therefore hypothesized that the GI mucosal microbiome is altered in HIV patients and this alteration correlates with immune activation in HIV. 121 specimens were collected from 21 HIV positive and 22 control human subjects during colonoscopy. The composition of the lower gastrointestinal tract mucosal and luminal bacterial microbiome was characterized using 16S rDNA pyrosequencing and was correlated to clinical parameters as well as immune activation and circulating bacterial products in HIV patients on ART. The composition of the HIV microbiome was significantly different than that of controls; it was less diverse in the right colon and terminal ileum, and was characterized by loss of bacterial taxa that are typically considered commensals. In HIV samples, there was a gain of some pathogenic bacterial taxa. This is the first report characterizing the terminal ileal and colonic mucosal microbiome in HIV patients with next generation sequencing. Limitations include use of HIV-infected subjects on HAART therapy.
Introduction
Human immunodeficiency virus (HIV) infection is a chronic illness characterized by progressive CD4+ T cell loss. With the advent of highly active anti-retroviral therapy (HAART), HIV infection is controlled, resulting in reduced death from opportunistic infections. However, despite successful viral suppression, many HIV patients have persistent inflammation/immune activation resulting in the development of non-HIV comorbidities including cardiovascular disease, osteoporosis, neurocognitive decline, cancer, as well as increased mortality [1], [2].
The mechanism of persistent inflammation and immune activation in HIV patients whose viral loads are successfully suppressed by HAART is not fully understood. However, several studies suggest that immune activation could be a consequence of gut-triggered systemic inflammation and microbial translocation [3]: This is not surprising because one of the earliest and principal sites of inflammation/immune activation and resultant CD4+ T cell infection by HIV is the gastrointestinal (GI) tract [4]. In fact, the GI tract houses the largest mucosal immune system in the body, and is a major interface between immune cells and the environment, with its substantial surface area. It also contains a significant amount of microbial mass, collectively referred to as the gastrointestinal microbiome, the majority of which is bacterial and has an estimated metabolic activity comparable to a human liver.
Little is known about the human GI tract microbiome in disease states including HIV, despite the fact that current evidence points toward a potential central role for the GI tract microbiome in HIV progression. Studies in HIV-infected humans show that bacterial products are increased in the circulation of patients with HIV [5], and this can occur even at early stages of HIV infection, before peripheral CD4+ T cell depletion reaches levels that can lead to clinically apparent disease and opportunistic infections [6]. Furthermore, polymicrobial bacterial DNA is detected in the circulation even in patients with suppressed viral loads [7]. The source of this increase is thought to be gastrointestinal tract permeability, which in turn is influenced by the composition and the function of the gastrointestinal microbiome, and the microbial - immune cell interactions across the gastrointestinal epithelium.
Direct evidence of alterations in the composition of the gastrointestinal tract microbiome has also been found for some bacterial taxa: Current data indicate increases in commensals that can be pathogenic such as Pseudomonas in the feces of subjects with HIV infection; and decreases in mostly beneficial commensals such as Lactobacilli and Bifidobacteria [6].Furthermore, prebiotics have been tested in HIV in an effort to alter the microbiome to have a more favorable composition, resulting in reduced CD4+ T cell activation and improvement in NK cell activity [8].
On the contrary to the human observations, studies on animal models have not supported the presence of an alteration in the bacterial microbiome [3], [9]:In a simian model of HIV with colitis, the composition of the GI tract microbiome was altered with increases in Campylobacter due to the colitis resulting from the presence of SIV, however, no specific differences were found attributable to SIV infection itself [10]. Similarly, in a metagenomic study of the fecal virome in SIV-infected macaques, no major differences were noted when the bacterial sequences were examined [9]. Possible reasons for this discrepancy in findings in the SIV model and human HIV could be related to the SIV model itself, or to the study of fecal samples to date. It is theoretically plausible that mucosal samples could provide higher relevance to immune activation given the close proximity of the microbiota to the GI tract epithelium and immune cells at the mucosal surface. On the other hand, human studies in HIV inherently carry major limitations by nature of the subjects themselves being on antiretroviral therapy or having additional lifestyle factors that cannot be tightly controlled for. Nevertheless, HIV-infected human studies are expected to provide significant information as to the mechanisms at play in the clinical setting, and are needed to confirm or refute the findings observed in SIV-infected macaques.
To date, a detailed and comprehensive look at the GI tract microbiome, especially the ileal and colonic mucosal microbiome in relation to the fecal microbiome has not been reported in HIV -infected humans Major advances in next generation sequencing technologies now allow for a comprehensive and rapid evaluation of the GI tract microbiome in humans at a fraction of the effort and cost, compared to the weeks and months required for such a sequencing effort less than a decade ago. The advent of these technologies has brought about rapid characterization of the bacterial microbiome in the GI tract by sequencing of bacterial genes, such as the 16SrDNA that are used in the taxonomical classification of bacteria. Therefore, these technologies represent an opportunity to characterize the HIV associated GI tract microbiome.
We aimed to study the composition of the gastrointestinal tract mucosal and luminal bacterial microbiome using 16S rDNA sequencing and to correlate the microbiome to clinical parameters as well as immune activation and circulating bacterial products in HIV-infected patients. We hypothesized that the GI mucosal microbiome is altered in HIV-infected patients and such an alteration correlates with immune activation.
Results
Description of the data
We obtained samples from the terminal ileum (TI), the right colon (RC), the left colon (LC) during colonoscopy, as well as fecal samples from the colonic lumen (F). The sample sites are shown in Table 1. There were no statistically significant differences in terms of age, gender, and race among the HIV and control subjects (Table S1). The mean CD4 count was 425+/−259 among the HIV subjects (Table S2). The viral load was <75 for 17 out of 21 patients; the remaining four subjects had a mean viral load of 1571+/−2059 cps/ml (Table S2). In 18 out of 21 HIV-infected subjects, colonoscopy was performed for colon cancer screening; and the remaining three subjects had constipation (n = 1), minor rectal bleeding (n = 2), and intermittent loose stools (n = 1). 13/21 HIV subjects were MSM; four had confirmed heterosexual acquisition of HIV; and in 4 subjects, the mode of HIV acquisition was not known.

We obtained 1,079,589 raw sequences, and 322,061,108 raw bases with an average of 8849 sequences/sample at an average length of 298 bps/sequence in two separate runs on a 454 instrument, as described in the methods. After quality–filtering (also as described in the methods), 455,452 total sequences and an average of 3733 sequences per sample were available, that were denoised, >250 bp long, de-multiplexed, reverse-primer-truncated and chimera-filtered for the rest of the analysis. The sequences were rarified to the minimum number of high quality sequences in all samples and normalized by total count for the alpha and beta diversity analyses conducted below.
HIV infection is associated with a decrease in microbial diversity in the terminal ileum and the colon
Sample richness was assessed by operational taxonomic unit (OTU) counts in each individual sample: The number of observed OTUs in the HIV samples was less than that of the control samples (Figure 1a). When looked at by sample type, all sample types had less OTUs in the HIV group compared to the healthy controls. The differences between the HIV and controls groups were especially heightened for samples from the ileum and the right colon which showed numerically higher reductions in sample richness (Figure 2a–d).


Within sample bacterial diversity was also assessed by both the chao1 and the Phylogenetic Diversity (PD) Whole Tree metric. Both metrics demonstrated reduced overall diversity among the samples from HIV-infected subjects compared to controls (Figures 1b and 1c). The differences were also seen for each individual sample type, however samples from the mucosal surface of HIV-infected subjects were more divergent from those of controls, compared to the differences noted between fecal samples from the HIV and the control groups (Figures 2e–h, and 2i–l). Collectively, this data points towards a less diverse bacterial population in HIV throughout the TI and the colon as well as the luminal compartment, although the differences in diversity were most noticeable on the mucosal surfaces, and especially the right colon.
The GI tract bacterial microbiome composition is significantly altered in HIV-infected subjects
In order to examine the global differences in bacterial composition between the HIV-infected subjects and the controls, we calculated distances between each sample using the Bray-Curtis similarity and unweighted Unifrac. We chose these two different measures for the following reason: The Bray-Curtis similarity is based on shared OTU counts between samples [11], whereas the Unifrac involves placing samples on a phylogenetic tree and calculating and summing branch lengths unique to a given sample on the tree [12]. Consequently, Bray-Curtis similarity gives equal importance to differences in every taxa, whereas in Unifrac, phylogenetically related taxa cause less divergent Unifrac values compared to distant or unrelated taxa which cause larger differences.
- a
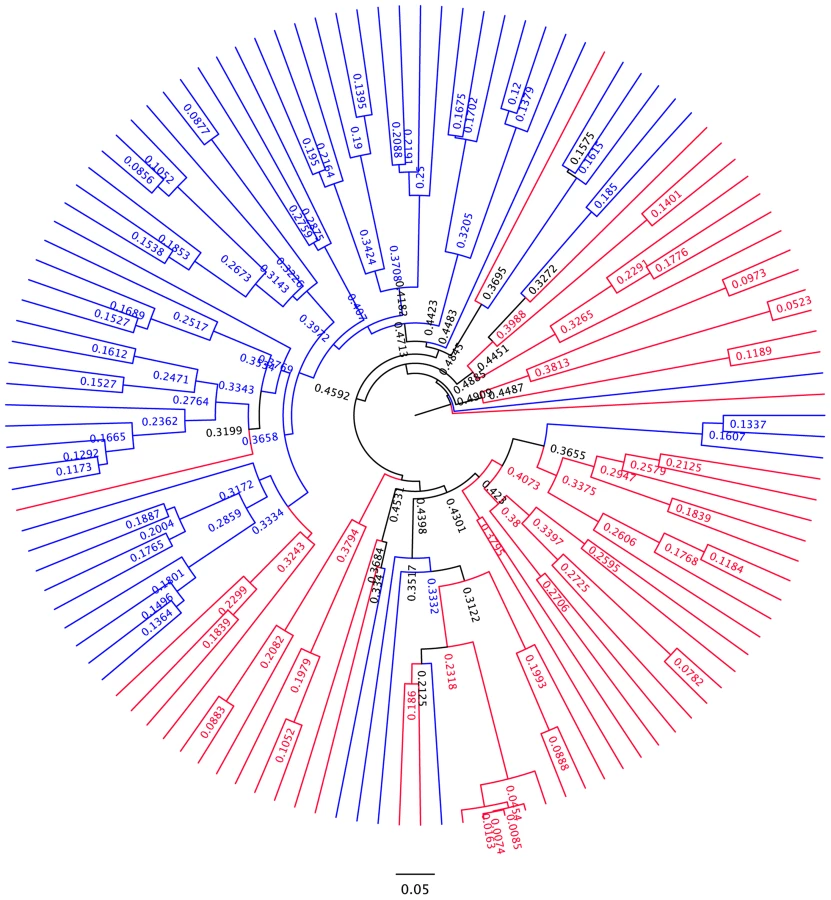
Differences in overall community composition in HIV-infected subjects vs. controls. To uncover differences in bacterial community composition between the samples, we performed nonmetric multidimensional scaling (NMDS) of all the samples using the count-based Bray-Curtis similarity at the OTU level. NMDS is a powerful ordination method that can uncover nonlinear relationships between samples and is widely used in ecology. As shown in the results in Figure 3a, there was differential grouping of the control samples to the left of the graph, meanwhile samples from HIV-infected subjects were more dispersed than the control samples and were to the right. Hierarchical clustering of the samples with the unweighted pair group method with arithmetic mean (UPGMA) using the Bray-Curtis similarity additionally confirmed that samples from HIV –infected subjects clustered differently than those from controls (Figure 4). In order to test whether the visual differences seen with NMDS ordination and the clustering observed by UPGMA are significant, we compared global bacterial composition in the HIV and control groups at the genus level using perMANOVA with the Bray-Curtis similarity in a one-fixed-factor and one-level nested mixed random effects model (i.e. replicates within sample sites within disease presence (i.e. HIV vs. Controls)). We noted that the visual differences between HIV and controls were statistically significant (Table 2).


- b
Differences in overall community composition in HIV-infected subjects vs. controls by site of sampling. We then compared samples from HIV-infected subjects and controls at each site (TI, RC, LC, F) using Unifrac based PCO ordination: Separation of samples was seen for each sample type, however the differential dispersion of samples was graphically more apparent for the TI and RC samples, with a few cases overlapping in the LC and F samples (Figure 3c–f). In order to assure that the findings we have observed are not an artifact of the sequence rarification and to get an estimate of the robustness of the results to our sequencing effort, we also performed jackknifed estimates of the Unifrac PCO analysis and the results obtained did not alter the conclusions (Figure S1, S2, S3, S4, S5).
- c
Comparison of bacterial OTUs observed in HIV-infected subjects and controls with network analysis. In order to evaluate bacterial OTUs associated with HIV and controls, we constructed a bipartite network of the bacterial OTUs and the samples, in which OTU-nodes are connected via edges to sample nodes in which their sequences are found. We have chosen the network analysis approach because it easily allows for visualization of thousands of OTUs in limited number of samples and can reveal significant associations. In the network, edge weights are determined by the number of sequences in an OTU; and clustering of the OTUs and samples are determined by a stochastic spring embedded algorithm as implemented in Cytoscape. The sample connections within the network are analyzed statistically with a G test of independence: Sample nodes from HIV-infected subjects and controls are tested to see whether they are more connected to a particular group than expected by chance. The network diagram is shown in Figure S6: In the center of the graph, sample nodes which are colored by HIV (in red) and controls (in blue) reveal significant clustering of the samples to either the right or the left of the graph, respectively for these groups. Some OTU nodes (shown in white) are only connected to HIV samples (in pink lines) while an equally numerous OTU nodes are only connected to the control samples (in lavender lines). OTU nodes that are connected to single samples lie in the periphery of the network, while those OTU nodes that are connected to multiple samples lie in the center of the network. The network statistics are shown in Table S3, and suggest an overall network with little interconnectedness overall, largely owing to the fact that the OTU nodes are only connected through the sample nodes. However, network statistics also indicate when the network is divided into two subnetworks (i.e. HIV network and control network), there are differences: The HIV node subnetwork has less average neighbors, more heterogeneity and less density compared to both the overall and the control node subnetwork. The HIV nodes are also farther apart from each other than the control nodes. These findings are compatible with the heterogeneity and disarray of the microbiome in HIV.
HIV-infection is associated with increases in potentially pathogenic taxa in the GI tract microbiome
The mean and standard deviations of different bacterial taxa at each taxonomic level in the HIV and control samples are given in Tables S4, S5, S6, S7. Stacked histogram for all samples at a phylogenetic resolution at the family level is given in Figure 5. The histogram shows clear visual differences between the abundance of bacterial taxa in then control samples to the right (that have a lot of brown coloring) and HIV samples to the left (that have an increased abundance of Gamma Proteobacteria shown as blue coloring)

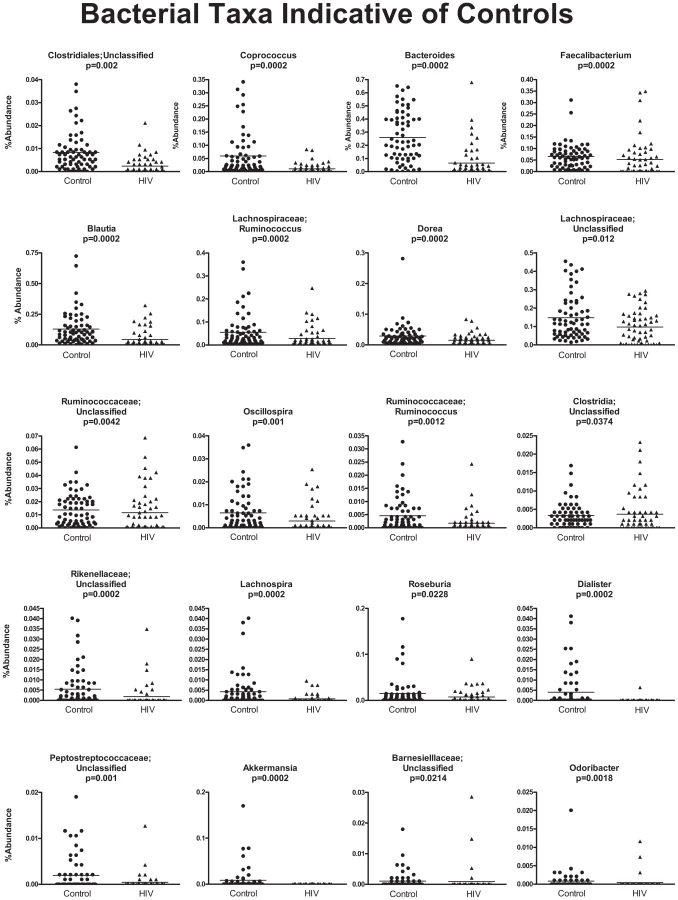
In order to discover which bacterial groups are driving the differences between the HIV and the control samples, we performed an indicator species analysis at the genus level for the HIV group vs. the control cases, controlled by type of sample. Bacterial taxa that have an indicator value >15 and are significantly different (p<0.05) are shown in Figures 6–8. The magnitude of the indicator values for each taxa in HIV and controls is shown in Figure 6. HIV was associated with a significant increase in Brachyspira, Campylobacter, Catenibacterium, Escherichia and other unclassified Enterobacteriaceae, unclassified Fusobacteriaceae, Mogibacterium, Prevotella, and Ralstonia (Figure 7, p<0.05 all). On the other hand, the healthy control group had more Akkermansia, Bacteroides, Blautia, Coprococcus, Dialister, Dorea, Faecalibacterium, Lachnospira, Roseburia, Ruminococcus, Odoribacter, Oscillospira genera, as well as unclassified bacteria from the following families, Barnesiellaceae, Lachnospiraceae, Peptostreptococcaceae, Rikenellaceae, and Ruminococcaceae (Figure 8, p<0.05 all).


Relationship between bacterial taxa in HIV-infected subject samples with serum soluble markers of immune activation
Soluble markers of immune activation (serum cytokines and microbial translocation markers) were measured from all subjects except five (3 subjects in the HIV-infected and 2 subjects in the control group) in whom samples were not available. We performed a canonical correspondence analysis (CCA) to look for an association between bacterial composition and serum interleukin-6 (IL-6), tumor necrosis factor alpha (TNF), lipoteichoic acid (LTA) and soluble CD14 (sCD14). CCA correlations between the cytokines (IL-6 and TNF) and microbial translocation products (LTA and sCD14) are given in Table S8. The presence of IL-6 and LTA were associated with a continuum of bacterial composition shifts from the healthy state towards HIV (shown along the first axis of the CCA) (Figure 9). The impacts of each of the cytokine and microbial translocation markers on the CCA are shown in Figures 9b–c. Increasing IL-6 was associated with controls whereas increasing LTA was associated with HIV. However, these cytokine levels failed to explain majority of the global composition changes between HIV and controls (spread across the first CCA axis), considering that the amount of cumulative variation explained was 5% of the total variance in the data. Bacterial composition-cytokine correlations were also not significant using a Monte-Carlo randomization test suggesting a weak association between these cytokines and microbial translocation products and global bacterial composition in the cases.

In situations when a handful of bacterial taxa may be driving the synthesis of these cytokines, it is certainly plausible that the individual bacterial taxa rather than the total microbial composition may be associated with cytokine levels or microbial translocation products. We tested for this by looking at Spearman's correlations between the individual bacterial taxa that were identified in the indicator species analysis and the cytokines and microbial translocation products in the HIV group. Significant potential associations with bacterial taxa in the HIV group are given in Table 3. Notably, some of these associations did not appear linear, suggesting a complex interrelationship with bacterial composition and cytokine secretion and microbial translocation products (data not shown).

Relationship of the microbiome in HIV with CD4 count and viral load and other clinical characteristics
- a
Clinical characteristics. We examined the possibility of any antibiotic exposures in the HIV patients or controls affecting our results and conclusions: There were four subjects who had antibiotic exposures in the HIV group within 1 month of sample collection. In total, 2/22 of the control subjects and 8/21 of the HIV subjects had a history of antibiotic use anytime within the past 1 year. The two control subjects had not used any antibiotics within the last 3 months, but were exposed to antibiotics within 12 months. Amongst the HIV subjects, one was on TMP-SMZ prophylaxis and another was prescribed this although did not report himself as taking it. In the other 6 HIV-positive subjects, the last antibiotic use in the subjects were three weeks prior to sample collection (n = 2), 4 months prior to sample collection (n = 1), 6 months prior to sample collection (n = 1), 7 months prior to sample collection (n = 1), and 12 months prior to sample collection (n = 1). When samples from subjects who have any antibiotic exposures were examined against the rest of the samples in terms of diversity indices, we noted no differences for samples collected from subjects with an antibiotic exposure within the past 1 month versus those with no antibiotic exposures (Figure S7). Similarly, we noted no difference between the samples collected from subjects with an antibiotic exposure within the past 3 months versus those with no antibiotic exposures (Figure S7). However, there appeared to be some difference between samples collected from subjects with an antibiotic exposure within the past 1 year versus those with no antibiotic exposures (Figure S7).
- b
CD4+ T - cell counts. Diversity indices did not show a difference between those HIV infected subjects with high and low CD4 counts at cutoffs of 200 and 500 in all subjects as well as those subjects without any antibiotic exposures (Figures S11 and S12). In PCO analysis using Unifrac distances and in NMDS analysis using Bray-Curtis distances, samples belonging to subjects whose CD4+ T cell counts were <200 were not appearing clustered together, and were not separated from samples belonging to subjects whose CD4+ T cell counts were >200. (Figure S13). No apparent separation was seen for samples coming from subjects whose CD4+ T cell counts were <500, versus those with CD4+ T cell counts >500 (Figure S13). There were no significant correlations between CD4+ T cell count and individual bacterial taxa.
- c
Viral load. In our dataset, seventeen out of 21 HIV - infected subjects had undetectable viral loads, so it was not possible to perform a correlation between the microbiome and viral load. In alpha diversity or ordination analyses, we did not note a significant difference between samples from viremic subjects versus non-viremic subjects (Figures S14 and S15).
Discussion
This study represents a first global look at the lower GI tract microbiome in HIV-infected subjects using sequencing technologies. Our data shows that the lower intestinal mucosal bacterial populations in HIV-infected subjects are less diverse, definitely distinct from non-HIV controls, and composed more frequently of bacterial populations that are potentially pathogenic. Such disarray in the lower gut microbiome during HIV-infection goes along with findings of increased gut permeability in HIV and may be one of the contributing etiologic factors to HIV progression, warranting further investigation. Notably, the microbiome in HIV-infected subjects is so different that the cases can be distinctly separated from healthy controls based on global microbiome composition. In many other pyrosequencing experiments in human subjects, in unconstrained ordination analyses, the variability of the GI tract microbiome itself between subjects or samples, usually outweighs the effects seen as a result of disease. Such a large magnitude of disarray in the GI tract microbiome in HIV signals the importance of the GI tract microbiome in HIV infection, and could be both a consequence of HIV infection itself and/or a contributor to disease progression.
When the individual genera associated with HIV-infected subjects and controls are examined, an important finding is the loss of a significant set of genera associated with controls in the HIV cases, typical of a dysbiotic microbiome. In many instances these are genera that are thought to be beneficial such as Bacteroides, which was negatively associated with IL-6 in our HIV cases. The network analysis also suggests that HIV is characterized by the loss of OTUs, and a change across all samples. In fact, while the HIV microbiome is a less diverse microbiome, the samples in the HIV group are farther apart from each other in our network analysis suggesting that the gain of bacteria among HIV samples is rather numerous and can vary across persons infected with HIV. Furthermore, nearly all of the bacterial taxa we have found to be associated with HIV in our study have also shown to be potentially pathogenic organisms in many other disease states, though some are rarely pathogenic. When we look at these one by one, the following can be said about the genera associated with HIV: Escherichia and other genera from Enterobacteriaceae, Campylobacter, various genera from Fusobacteriaceae are common pathogens of UTI, bacteremia, gastrointestinal infections, and periodontitis and necrotizing infections/abscesses. Catenibacterium has previously been associated with presence of uremia in subjects with end stage renal disease [13]. Mogibacterium has been found in the sputum of people with tuberculosis [14] and has also been associated with periodontitis [15], [16] and endodontic infections [17]. Ralstonia are plant pathogens but have been emerging in case reports of community acquired pneumonia [18], [19] as well as becoming increasingly identified as infectious agents in cystic fibrosis patients [20],immunocompromised individuals with hematological malignancies [21], [22], and in nosocomial infections [23]. Brachyspira is the cause of intestinal spirochetosis in both animals and humans; and have previously been described as a cause of chronic diarrhea in HIV subjects [24]. While Prevotella are commonly found in healthy individuals in the GI tract, they could be pathogenic at other sites across the body such as the oral mucosa or the vagina. Prevotella are associated with anaerobic infections of the respiratory tract such as aspiration pneumonia, lung abscess, chronic otitis media and sinusitis; abscesses round the mouth; urinary tract infections; brain abscesses; osteomyelitis ; periodontal disease; in addition to their well-documented presence in bacterial vaginosis.
Previous studies on the vaginal microbiome in HIV also have suggested a significant change at this site [25]–[29], with an increased diversity in the vaginal environment in HIV [30]. In contrast, our results suggest a decreased diversity in the GI tract microbiome of HIV-infected subjects. This may be due to the inherent nature of the bacterial communities at these different mucosal sites: There is low diversity typically seen in the vaginal environment with high specialization for certain functions, such as regulation of vaginal pH, which shows correlation with microbiome composition [31]. In contrast, high diversity is typically seen in the GI tract in the healthy state [31], with reductions in diversity seen in most disease states investigated to date across multiple studies (e.g. inflammatory bowel disease and colon cancer) [32]–[34]. One of the bacterial taxa associated with bacterial vaginosis in the vaginal environment in women with and without HIV has been Prevotella [30], which was also identified to be increased in the GI tract specimens from HIV cases in our study. This co-occurrence of Prevotella in the HIV GI tract specimens and in bacterial vaginosis may suggest that the gut could be one source of colonization of the vagina with Prevotella in bacterial vaginosis, which in turn is an important risk factor for heterosexual HIV transmission. Therefore, we suspect that the microbiome changes in HIV across the body could be wide-spread and could potentially be related to each other. Further studies that concomitantly examine the GI tract and the vaginal microbiome are needed to increase our understanding of the role of the GI tract microbiome in altering the vaginal environment in HIV and thereby as a risk factor for HIV transmission.
On the contrary to the human observations in our study, two separate studies on SIV-infected macaques have not shown an alteration in gut bacterial composition [3], [9]. This significant discrepancy between our findings in HIV-infected humans and those in SIV-infected macaques could relate to a number of factors: There could be intrinsic differences between SIV as model of disease versus HIV ; the presence of colitis in some SIV-infected macaques may have affected or surpassed the magnitude of the change in microbiome composition by SIV itself making these changes difficult to detect; and/or the discrepancy could be explained by the study of fecal samples in the case of SIV-infected macaques in the two previously published studies, versus the study of colonic tissue biopsies in the case of HIV-infected humans in our study. Alternatively, it is also possible that the major driver of bacterial compositional alterations in HIV-infected humans could be disease related factors such as the presence of HAART or other treatments that may have been administered to humans over the course of disease. Alternatively, lifestyle factors (that are tightly controlled in animal models) rather than the presence of HIV infection could have also resulted in changes in the bacterial microbiome in HIV-infected humans. Future studies should carefully characterize such potential confounders in humans such as diet, treatment duration, as well as antibiotic exposures in a longitudinal manner.
Compared to other studies that have profiled individual bacterial orders with PCR in the GI tract in HIV in humans, our study is confirmatory: Previous studies have shown increases in Enterobacteriales and Bacteroidales orders in HIV [6], [35] corresponding to the rises in Escherichia and other genera from Enterobacteriaceae and rises in Prevotella (at the expense of other Bacteroidales such as Bacteroides) in our study, respectively. We have also identified a number of other genera that have not been associated with HIV before. During the revision of this manuscript, an additional study examining the bacterial microbiome with a microarray technique in rectal biopsies of 25 HIV-infected humans has been published [36]. Findings in this study are largely parallel to ours: There is an increase in pathogenic Proteobacteria and a decrease of the Bacteroidia members in the dysbiotic mucosal-adherent bacterial community in the rectum. Similar to our study, there was not a strong association between peripheral blood CD4+ T cell counts and microbiome composition. Our study adds to these published findings in several ways: The dysbiosis in HIV occurs in multiple locations throughout the lower GI tract. We also note a reduction in the diversity of the microbiome which was most evident on the mucosal surface (especially the right colon) and less so in fecal samples. We note a disarray and heterogeneity in microbiome composition in the HIV group resulting in less connectedness of the microbiome in our network analysis, which in turn may affect the ability of the microbiome to suppress or constrain pathologic organisms in the gut and increase the susceptibility to further perturbations. Hence, our findings in conjunction with other studies suggest that there may be a potentially pathogenic but typically commensal microbiome pattern in HIV that could be modulated for therapeutic gains. Future larger scale studies are needed to confirm HIV specific microbiome patterns in the GI tract.
One possible mechanism by which the GI tract microbiome could modulate HIV progression and related disease states, could be through immune alterations resulting in the release of inflammatory cytokines that occur as a result of gut barrier dysfunction, which has been well described in HIV [7], [37]–[40]. Our study found associations with inflammatory cytokines in the blood and the microbiome. However, it appears that the effect of the serum cytokines and microbial translocation products tested is a weak one and may perhaps require larger numbers of subjects to study. Perhaps circulating cytokines or other immunological correlates of HIV infection other than the ones examined in this study (such as the recently identified increased kynurenine production [36]) or cytokine/chemokine levels in the gut mucosa, or additional mechanisms that result in soluble factors or direct cell to cell contact between bacteria and the intestinal mucosal surface have a larger magnitude of association with the potentially pathogenic GI tract microbiome in HIV, and now require further study.
The strength of our study is the use of state of the art pyrosequencing technologies to evaluate the lower intestinal microbiome; the use of mucosal samples throughout the lower GI tract for pyrosequencing in addition to fecal samples; and multiple approaches to data analysis to gain insights into the data. We believe mucosal biopsy samples may give a better understanding of the interactions between the microbiome and disease states due to the close proximity of epithelial cells with bacteria. While the microbiome in stool samples is thought to grossly mirror the microbiome on the mucosal surface in the healthy state, not all members of the fecal community are members of the mucosal community; and fecal microbiome composition is also subject to change with transient members that may come and go, as part of food/other ingested materials [41], [42]. The fecal microbiome is also generally more diverse than the mucosal microbiome. This increased level of microbial diversity could make subtle changes in the disease setting much harder to detect or less apparent in fecal samples. This may also be one reason to note a greater difference in the mucosal samples versus the fecal samples in our study. Our study is limited by its cross-sectional design and it is hypothesis generating; but future longitudinal studies are needed to infer cause-effect relationships between HIV transmission, spread and progression and the GI tract microbiome. Another limitation of our study is the fact that the patients in our study were on HAART. It is currently unknown whether the therapy itself could lead to changes in the GI tract microbiome. In the future, bacterial composition in the lower GI tract will also have to be explored in patients who are not on any medications and the changes seen in this study will have to be confirmed in untreated HIV populations.
Strategies modulating the GI tract microbiome have been underway as part of the efforts to devise therapeutic strategies directed against harmful immune activation in HIV. One such strategy could be dietary manipulation or manipulation of the microbiome with prebiotics and/or probiotics [8], [43]–[45]. Our findings add to the body of literature on this topic and also suggest that there may be a therapeutic potential for such interventions. Future studies exploring the diets of HIV patients in conjunction with disease stage and the GI tract microbiome are needed to explore the role of dietary manipulation, prebiotics and probiotics as part of HIV treatment.
In conclusion, there are changes in the lower GI tract microbiome in HIV characterized by loss of various commensal bacterial genera and gain of potentially pathogenic ones, and this finding now paves the way to future studies that examine further relationships between the GI microbiome and HIV transmission as well as progression.
Materials and Methods
Subjects and samples
Colonic biopsy samples and fecal samples were obtained from the tissue bank at Rush University Medical Center, Division of Gastroenterology, Hepatology and Nutrition, from 21 subjects with HIV and 22 control subjects, after obtaining study approval from the Rush IRB. All subjects gave written and verbal informed consent prior to tissue collection under the tissue bank IRB at Rush University and were recruited from the same geographic area and one single outpatient endoscopy lab. The utilized tissue bank obtained samples from the terminal ileum, right colon, left colon and luminal fecal samples at the time of the subject's colonoscopy with a standard 2.2 mm biopsy forceps or a luken's trap, respectively. All samples were placed in a cryovial and flash frozen in liquid nitrogen in the endoscopy room and stored in −80 C freezers. Samples were analyzed anonymously under a research protocol which was also approved by the Rush University Medical Center IRB. The characteristics of the human subjects and the collected tissues are given in Tables 1, S1 and S2. All subjects received a polyethylene glycol (PEG) based bowel prep within 24 hours of colonoscopy, except one subject who received a sodium phosphate based bowel prep. There were no differences in samples from subjects exposed to various PEG based preps in a PCO analysis using Unifrac (Figure S16).
Sequencing
DNA was extracted using a commercially available kit, FastDNA Spin Kit for Soil, (MP Biomedicals, Solon, OH 44139 USA), using the manufacturer's recommended protocol. The adequacy of the amount of extracted DNA from samples was verified with fluorometric quantitation (Qubit, Life Technologies, Grand Island, NY 14072) and samples with inadequate amounts of template DNA were not sequenced. 5′GAGTTTGATCNTGGCTCAG3′ forward primer and 5′GNTTTACNGCGGCKGCTG3′ reverse primers were used to pyrosequence the 16S rDNA on a 454 GS FLX platform, with barcoding, using titanium kits [46].
Sequence processing and quality assessment
Custom C# and python scripts as well as python scripts in the Quantitative Insights Into Microbial Ecology (QIIME) software pipeline (VirtualBox Versions 1.5 and 1.6) were used to process the sequencing files [47]–[50]. Two sequence runs were performed with about equal distribution of HIV and control cases in both runs. The sequence outputs were filtered for low quality sequences (defined as any sequences that are <200 bps or >1000 bps, sequences with any nucleotide mismatches to either the barcode or primer, sequences with homopolymer runs >6, sequences with an average quality score of <25, sequences with ambigious bases >6) and were truncated at the reverse primer. Sequences were denoised using USEARCH [51], and chimera checked with UCHIME [52] and Chimera Slayer [53]. Operational taxonomic units (OTUs) were picked using uclust [51]at 97% similarity, and representative sequences were generated. Sequences were aligned with PyNAST [54] and taxonomy assignment was performed in Qiime 1.6VB against the Qiime 1.6 version of Greengenes database [55], [56] using the RDP classifier [57] at a 80% bootstrap value threshold. An approximately-maximum-likelihood phylogenetic tree was created using FastTree v2.1.3 [58]. Differential dispersion of the samples by runs for batch effects were also checked usingPCO using Unifrac distances. While run related batch effects were seen, these failed to explain the group differences observed between HIV and controls (Figure S17).
Lab measurements
Serum specimens were evaluated in duplicate batches for circulating levels of IL-6, and TNF-α using the Human High Sensitivity Cytokine/Chemokine kit (EMD Millipore, Billerica, MA) according to manufacturer recommended protocols. All kits were read using a Luminex 100 IS System (Luminex Corp, Austin, TX) by the Rush Proteomics and Biomarkers Core Facility with biomarker concentrations calculated using a 5-parametric curve fit using xPonent 3.2 software (Luminex Corp). Median %CV and assay recovery values all fell within acceptable limits specified by EMD-Millipore. The serum levels of soluble CD14 (sCD14) and Lipoteichoic acid (LTA) was determined by ELISA.
Statistics
Qiime 1.5 was used calculate Bray-Curtis similarity and Unifrac distances based on OTUs, and NMDS and PCO coordinates, Unifrac tests; UPGMA clustering; and calculate the bipartite network attributes. UPGMA dendogram and phylogenetic trees were visualized in FigTree v1.3.1. Cytoscape v2.8.1 was used to visualize the network; and calculate network and node statistics. PerMANOVA, Indicator species analysis (ISA) [59], CCA and were performed in PC-ORD. For PerMANOVA analyses, we used the Bray-Curtis similarity at the genus level on rarified and log transformed data. In order to adjust for unequal sample sizes in groups, we randomly subsampled the groups 1000 times, stratified by sample site (I,RC,LC, and F), and performed a randomization test of significance of pseudo F values, with 4999 randomizations for each model, and collated the results from the 1000 subsamplings. For ISA analyses, we used rarified and log transformed data and the analysis was blocked for sample type. For CCA analyses, in order to adjust for unequal sample sizes in groups, we subsampled 8 cases from each site for each group randomly. Axis scores were standardized and centered to unit variance. Scores for graphing samples were linear combinations of genera. We performed a randomization test of the eigenvalues of the canonical axes with 1000 randomizations. We also performed a randomization test with 1000 randomizations for as species-environment correlations for which the null hypothesis was that there is no structure in the bacterial genera and therefore no relationship between the bacterial genera and the cytokine matrices. SPSS (Version 17.0.0, Chicago, IL, USA) was used to analyze clinical metadata and to correlate bacterial taxa (which were found to be significant in the indicator species analysis) to lab measurements in the HIV group. In SPSS, T - tests or ANOVA were used to analyze differences for parametric data satisfying test assumptions; Kruskal - Wallis or Mann-Whitney tests or median tests were used to analyze nonparametric data; Chi-Square or t–tests were used to detect differences in proportions between groups, as appropriate. In SPSS, bacterial taxa count data was log transformed and most taxa were observed to be non-normally distributed. Spearman's correlations were calculated. Correlations that were based mostly on zeros or few sequences were not reported as significant. GraphPadPrism was used to generate scatterplots of bacterial taxa.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. SandlerNG, WandH, RoqueA, LawM, NasonMC, et al. (2011) Plasma levels of soluble CD14 independently predict mortality in HIV infection. J Infect Dis 203 : 780–790.
2. OstrowskiSR, PiironenT, Hoyer-HansenG, GerstoftJ, PedersenBK, et al. (2005) High plasma levels of intact and cleaved soluble urokinase receptor reflect immune activation and are independent predictors of mortality in HIV-1-infected patients. J Acquir Immune Defic Syndr 39 : 23–31.
3. KlattNR, FunderburgNT, BrenchleyJM (2013) Microbial translocation, immune activation, and HIV disease. Trends Microbiol 21 : 6–13.
4. BrenchleyJM, SchackerTW, RuffLE, PriceDA, TaylorJH, et al. (2004) CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. J Exp Med 200 : 749–759.
5. BrenchleyJM, PriceDA, SchackerTW, AsherTE, SilvestriG, et al. (2006) Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 12 : 1365–1371.
6. GoriA, TincatiC, RizzardiniG, TortiC, QuirinoT, et al. (2008) Early impairment of gut function and gut flora supporting a role for alteration of gastrointestinal mucosa in human immunodeficiency virus pathogenesis. J Clin Microbiol 46 : 757–758.
7. MerliniE, BaiF, BellistriGM, TincatiC, d'Arminio MonforteA, et al. (2011) Evidence for polymicrobic flora translocating in peripheral blood of HIV-infected patients with poor immune response to antiretroviral therapy. PLoS One 6: e18580.
8. GoriA, RizzardiniG, Van't LandB, AmorKB, van SchaikJ, et al. (2011) Specific prebiotics modulate gut microbiota and immune activation in HAART-naive HIV-infected adults: results of the “COPA” pilot randomized trial. Mucosal Immunol 4 : 554–563.
9. HandleySA, ThackrayLB, ZhaoG, PrestiR, MillerAD, et al. (2012) Pathogenic simian immunodeficiency virus infection is associated with expansion of the enteric virome. Cell 151 : 253–266.
10. McKennaP, HoffmannC, MinkahN, AyePP, LacknerA, et al. (2008) The macaque gut microbiome in health, lentiviral infection, and chronic enterocolitis. PLoS Pathog 4: e20.
11. BrayJR, CurtisCT (1957) An ordination of upland forest communities of southern Wisconsin. Ecological Monographs 27 : 325–349.
12. LozuponeC, KnightR (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71 : 8228–8235.
13. VaziriND, WongJ, PahlM, PicenoYM, YuanJ, et al. (2013) Chronic kidney disease alters intestinal microbial flora. Kidney Int 83 : 308–315.
14. CheungMK, LamWY, FungWY, LawPT, AuCH, et al. (2013) Sputum Microbiota in Tuberculosis as Revealed by 16S rRNA Pyrosequencing. PLoS One 8: e54574.
15. ColomboAP, BochesSK, CottonSL, GoodsonJM, KentR, et al. (2009) Comparisons of subgingival microbial profiles of refractory periodontitis, severe periodontitis, and periodontal health using the human oral microbe identification microarray. J Periodontol 80 : 1421–1432.
16. SatoT, YamakiK, IshidaN, HashimotoK, TakeuchiY, et al. (2012) Cultivable anaerobic microbiota of infected root canals. Int J Dent 2012 : 609689.
17. RolphHJ, LennonA, RiggioMP, SaundersWP, MacKenzieD, et al. (2001) Molecular identification of microorganisms from endodontic infections. J Clin Microbiol 39 : 3282–3289.
18. RammaertB, BorandL, GoyetS, TeV, HemS, et al. (2010) Ralstonia pickettii community-acquired pneumonia in Cambodia. Int J Tuberc Lung Dis 14 : 1653–1654.
19. PanW, ZhaoZ, DongM (2011) Lobar pneumonia caused by Ralstonia pickettii in a sixty-five-year-old Han Chinese man: a case report. J Med Case Rep 5 : 377.
20. DaviesJC, RubinBK (2007) Emerging and unusual gram-negative infections in cystic fibrosis. Semin Respir Crit Care Med 28 : 312–321.
21. BarbutF, KosmannMJ, LalandeV, NeymeD, CoppoP, et al. (2006) Outbreak of Ralstonia pickettii pseudobacteremia among patients with hematological malignancies. Infect Control Hosp Epidemiol 27 : 642–644.
22. XuJ, MooreJE, MillarBC, AlexanderHD, McClurgR, et al. (2004) Improved laboratory diagnosis of bacterial and fungal infections in patients with hematological malignancies using PCR and ribosomal RNA sequence analysis. Leuk Lymphoma 45 : 1637–1641.
23. RyanMP, PembrokeJT, AdleyCC (2006) Ralstonia pickettii: a persistent gram-negative nosocomial infectious organism. J Hosp Infect 62 : 278–284.
24. EnaJ, Simon-AylonA, PasquauF (2009) Intestinal spirochetosis as a cause of chronic diarrhoea in patients with HIV infection: case report and review of the literature. Int J STD AIDS 20 : 803–805.
25. WattsDH (2012) Mother to child transmission of HIV–another complication of bacterial vaginosis? J Acquir Immune Defic Syndr 60 : 221–224.
26. FrankDN, ManigartO, LeroyV, MedaN, ValeaD, et al. (2012) Altered vaginal microbiota are associated with perinatal mother-to-child transmission of HIV in African women from Burkina Faso. J Acquir Immune Defic Syndr 60 : 299–306.
27. SaxenaD, LiY, YangL, PeiZ, PolesM, et al. (2012) Human microbiome and HIV/AIDS. Curr HIV/AIDS Rep 9 : 44–51.
28. SpearGT, GilbertD, LandayAL, ZariffardR, FrenchAL, et al. (2011) Pyrosequencing of the genital microbiotas of HIV-seropositive and -seronegative women reveals Lactobacillus iners as the predominant Lactobacillus Species. Appl Environ Microbiol 77 : 378–381.
29. HummelenR, FernandesAD, MacklaimJM, DicksonRJ, ChangaluchaJ, et al. (2010) Deep sequencing of the vaginal microbiota of women with HIV. PLoS One 5: e12078.
30. SpearGT, SikaroodiM, ZariffardMR, LandayAL, FrenchAL, et al. (2008) Comparison of the diversity of the vaginal microbiota in HIV-infected and HIV-uninfected women with or without bacterial vaginosis. J Infect Dis 198 : 1131–1140.
31. Structure, function and diversity of the healthy human microbiome. Nature 486 : 207–214.
32. SanapareddyN, LeggeRM, JovovB, McCoyA, BurcalL, et al. (2012) Increased rectal microbial richness is associated with the presence of colorectal adenomas in humans. ISME J 6 : 1858–1868.
33. HansenR, RussellRK, ReiffC, LouisP, McIntoshF, et al. (2012) Microbiota of de-novo pediatric IBD: increased Faecalibacterium prausnitzii and reduced bacterial diversity in Crohn's but not in ulcerative colitis. Am J Gastroenterol 107 : 1913–1922.
34. ManichanhC, Rigottier-GoisL, BonnaudE, GlouxK, PelletierE, et al. (2006) Reduced diversity of faecal microbiota in Crohn's disease revealed by a metagenomic approach. Gut 55 : 205–211.
35. EllisCL, MaZM, MannSK, LiCS, WuJ, et al. (2011) Molecular characterization of stool microbiota in HIV-infected subjects by panbacterial and order-level 16S ribosomal DNA (rDNA) quantification and correlations with immune activation. J Acquir Immune Defic Syndr 57 : 363–370.
36. Vujkovic-CvijinI, DunhamRM, IwaiS, MaherMC, AlbrightRG, et al. (2013) Dysbiosis of the Gut Microbiota Is Associated with HIV Disease Progression and Tryptophan Catabolism. Sci Transl Med 5 : 193ra191.
37. SankaranS, GeorgeMD, ReayE, GuadalupeM, FlammJ, et al. (2008) Rapid onset of intestinal epithelial barrier dysfunction in primary human immunodeficiency virus infection is driven by an imbalance between immune response and mucosal repair and regeneration. J Virol 82 : 538–545.
38. EppleHJ, SchneiderT, TroegerH, KunkelD, AllersK, et al. (2009) Impairment of the intestinal barrier is evident in untreated but absent in suppressively treated HIV-infected patients. Gut 58 : 220–227.
39. SmithAJ, SchackerTW, ReillyCS, HaaseAT (2010) A role for syndecan-1 and claudin-2 in microbial translocation during HIV-1 infection. J Acquir Immune Defic Syndr 55 : 306–315.
40. CostiniukCT, AngelJB (2012) Human immunodeficiency virus and the gastrointestinal immune system: does highly active antiretroviral therapy restore gut immunity? Mucosal Immunol 5 : 596–604.
41. AhmedS, MacfarlaneGT, FiteA, McBainAJ, GilbertP, et al. (2007) Mucosa-associated bacterial diversity in relation to human terminal ileum and colonic biopsy samples. Appl Environ Microbiol 73 : 7435–7442.
42. NavaGM, StappenbeckTS (2011) Diversity of the autochthonous colonic microbiota. Gut Microbes 2 : 99–104.
43. WilsonNL, MoneyhamLD, AlexandrovAW (2013) A Systematic Review of Probiotics as a Potential Intervention to Restore Gut Health in HIV Infection. J Assoc Nurses AIDS Care 24 : 98–111.
44. SchunterM, ChuH, HayesTL, McConnellD, CrawfordSS, et al. (2012) Randomized pilot trial of a synbiotic dietary supplement in chronic HIV-1 infection. BMC Complement Altern Med 12 : 84.
45. Gonzalez-HernandezLA, Jave-SuarezLF, Fafutis-MorrisM, Montes-SalcedoKE, Valle-GutierrezLG, et al. (2012) Synbiotic therapy decreases microbial translocation and inflammation and improves immunological status in HIV-infected patients: a double-blind randomized controlled pilot trial. Nutr J 11 : 90.
46. SmithDM, SnowDE, ReesE, ZischkauAM, HansonJD, et al. (2010) Evaluation of the bacterial diversity of pressure ulcers using bTEFAP pyrosequencing. BMC Med Genomics 3 : 41.
47. WolcottRD, GontcharovaV, SunY, DowdSE (2009) Evaluation of the bacterial diversity among and within individual venous leg ulcers using bacterial tag-encoded FLX and titanium amplicon pyrosequencing and metagenomic approaches. BMC Microbiol 9 : 226.
48. SenR, IshakHD, EstradaD, DowdSE, HongE, et al. (2009) Generalized antifungal activity and 454-screening of Pseudonocardia and Amycolatopsis bacteria in nests of fungus-growing ants. Proc Natl Acad Sci U S A 106 : 17805–17810.
49. IshakHD, PlowesR, SenR, KellnerK, MeyerE, et al. (2011) Bacterial diversity in Solenopsis invicta and Solenopsis geminata ant colonies characterized by 16S amplicon 454 pyrosequencing. Microb Ecol 61 : 821–831.
50. CaporasoJG, KuczynskiJ, StombaughJ, BittingerK, BushmanFD, et al. (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7 : 335–336.
51. EdgarRC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26 : 2460–2461.
52. EdgarRC, HaasBJ, ClementeJC, QuinceC, KnightR (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27 : 2194–2200.
53. HaasBJ, GeversD, EarlAM, FeldgardenM, WardDV, et al. (2011) Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 21 : 494–504.
54. CaporasoJG, BittingerK, BushmanFD, DeSantisTZ, AndersenGL, et al. (2010) PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26 : 266–267.
55. McDonaldD, PriceMN, GoodrichJ, NawrockiEP, DeSantisTZ, et al. (2012) An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6 : 610–618.
56. WernerJJ, KorenO, HugenholtzP, DeSantisTZ, WaltersWA, et al. (2012) Impact of training sets on classification of high-throughput bacterial 16s rRNA gene surveys. ISME J 6 : 94–103.
57. WangQ, GarrityGM, TiedjeJM, ColeJR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73 : 5261–5267.
58. PriceMN, DehalPS, ArkinAP (2010) FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5: e9490.
59. DufreneM, LegendreP (1997) Species assemblages and indicator species: the need for a flexible asymmetrical approach. Ecological Monographs 67 : 345–366.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 2
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Suppression of RNA Silencing by a Plant DNA Virus Satellite Requires a Host Calmodulin-Like Protein to Repress Expression
- Reversible Silencing of Cytomegalovirus Genomes by Type I Interferon Governs Virus Latency
- Identification of Host-Targeted Small Molecules That Restrict Intracellular Growth
- Implication of PMLIV in Both Intrinsic and Innate Immunity
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy