
Unique Features of HIV-1 Spread through T Cell Virological Synapses
article has not abstract
Published in the journal:
. PLoS Pathog 10(12): e32767. doi:10.1371/journal.ppat.1004513
Category:
Pearls
doi:
https://doi.org/10.1371/journal.ppat.1004513
Summary
article has not abstract
The spread of viral infections can be initiated by the release of cell-free virus particles that infect at a distance or via cell-associated virus, which can promote the direct transmission of viruses between adjacent cells. In the case of human immunodeficiency virus type 1 (HIV-1), cell–cell contact has been found to enhance infection through specialized structures called virological synapses (VS). Cell–cell interactions between virus scavenging dendritic cells and T cells or between infected and uninfected T cells are two major cell interactions that enhance HIV infection. Here we review the features of VS formed between infected and uninfected T cells and focus on how these differ from infection by cell-free virus. While virus particle production is a shared characteristic of both cell-free and cell–cell HIV transmission, cell–cell infection displays several unique features that contribute to the enhanced efficiency of this mode of transmission. Five distinguishing features of HIV spread through T cell virological synapses are discussed below.
Cell–Cell Adhesion Activates Virus Assembly at the VS
The T cell VS was initially characterized as an actin-dependent polarization of viral proteins Gag and Env to the site of cell–cell contact on infected donor cells and the recruitment of viral receptor CD4 to the site on uninfected target cells [1]. During cell–cell transmission of HIV-1, an infected T cell first forms a stable adhesive junction with an uninfected CD4+ T cell, which serves as a focal point for de novo viral assembly and transfer [2]. VS formation can be described as a two-part process that begins with adhesion triggered by Env-CD4 interactions and is stabilized by interactions between cellular adhesion molecules (i.e., LFA-1 and ICAM-1,3) [3]. When presented on an artificial membrane in the presence of ICAM-1, HIV Env is sufficient to trigger the arrest of CD4+ T cell migration and initiate VS formation (Fig. 1A) [4].
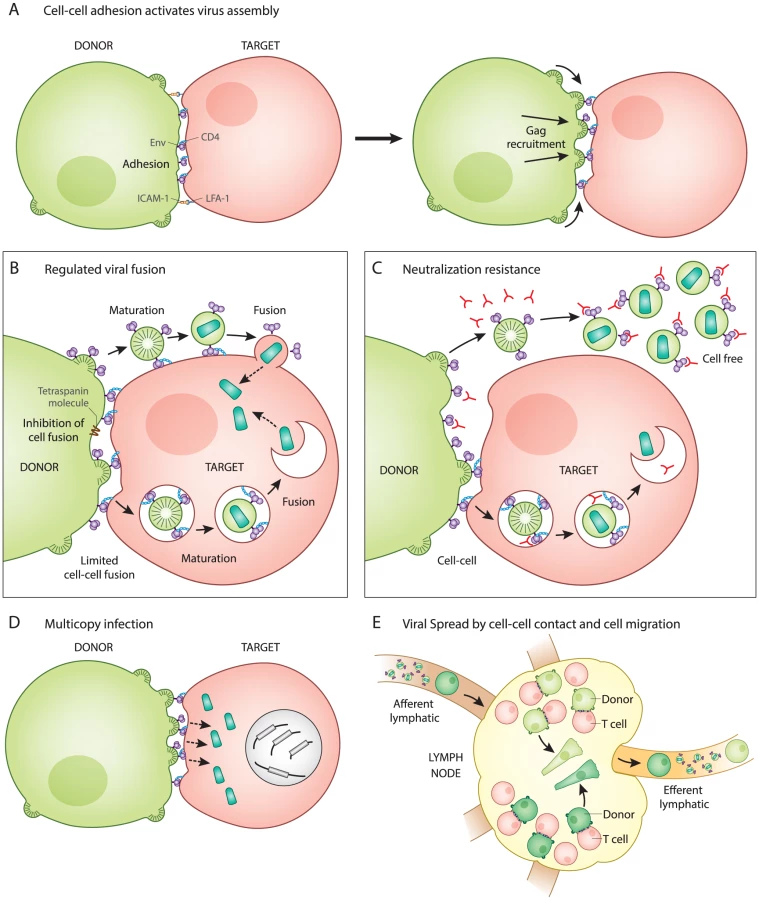
After cell–cell adhesion, the signaling of the VS partly resembles immune signaling through immunological synapses (IS). The tyrosine kinase Zap70 that is involved in IS signaling also promotes the recruitment of Gag to the site of contact between cells in the VS, but without the involvement of the T cell receptor [5]. In addition, the budding of HIV at VS and release of secretory microvesicles at IS both appear to exploit similar machinery for the directional secretion and budding of particles at their respective synapses [6], [7]. When VS were imaged by live confocal microscopy, fluorescently tagged Gag proteins rapidly mobilize and concentrate at the site of cell–cell contact [2]. Time-lapse imaging showed that cell adhesion occurred before Gag was recruited to sites of cell–cell contact, indicating that Env functions first as a cell adhesion molecule even before it associates with the newly forming virus particles [2]. The mechanism by which Env engagement triggers the recruitment of Gag to the VS remains an important question.
Regulation of Env-Mediated Membrane Fusion
While virus particle production is required for both cell-free and cell–cell infection [8], these two modes of transmission differ with regard to how and when Env-mediated membrane fusion is activated. Entry of cell-free HIV-1 occurs when the viral Env protein interacts with CD4 on the surface of uninfected cells, which directly triggers viral membrane fusion. In contrast, during cell–cell HIV-1 infection, the initial interaction between Env and CD4 occurs at the cell–cell junction and does not immediately activate Env-mediated fusion (Fig. 1B). The direct fusion of synapsed cells is thought be inhibited by the interactions of Env with the immature lattice of Gag at the VS [9]. Additionally, membrane proteins such as tetraspanins and actin-membrane organizing proteins such as ezrin, which are both concentrated at the VS, also inhibit cell–cell fusion [10], [11]. The absence of significant levels of syncytia during VS formation [12] implies that Env-mediated fusion is regulated during this process.
Given that fusion is regulated during synapse formation, it is important to consider how it is activated following the transfer of virus particles into target cells. In studies of cell-free virions, the uncleaved Gag lattice within immature particles can inhibit viral fusion by interacting with the Env cytoplasmic tail [13], [14]. During VS infection, viral assembly is tightly coordinated with transfer. Electron microscopy studies have captured nascent viral budding forms engaged with the target cell, both in vitro and in vivo in humanized mice [2], [15]. Live imaging studies examining viral transfer across the VS revealed a rapid, CD4-dependent, translocation of particles into a protected, trypsin-resistant, virus-containing compartment [2]. This process of internalization has been characterized by some as having several features of clathrin-dependent endocytosis [16], [17], while others have concluded the pathway is non-endocytic [1]. During VS transmission, we have proposed that the activation of fusion is not triggered by CD4, which is engaged early during VS, but, rather, by particle formation and subsequent maturation, which was observed as occurring within an intracellular compartment [18]. Through this mechanism, viral membrane fusion may be prevented from occurring prematurely, prior to the formation of mature virus particles. The pathway that the virus takes into the cell and how the fusogenicity of Env is regulated warrants further study and may play a role in understanding how cell–cell infection promotes immune evasion (discussed further below).
Neutralization Resistance
Recent studies have generally found that HIV-1 transmission through VS is more resistant to neutralizing antibodies as compared to infections with cell-free virus [19], [20]. Because cell-surface HIV-1 Env must engage CD4 on the target cell to initiate the VS, it is likely that Env is available for binding by neutralizing antibodies (Fig. 1C). We suggest that it is unlikely that Env is hidden in a privileged synaptic space if it must bind to CD4 at the cell surface. An important feature of the neutralization resistance of the VS is that the magnitude of the resistance depends upon the epitope on Env that is targeted [19]–[21]. This may imply that the antibody epitopes exposed during cell–cell transmission are conformationally restricted as compared to virus particles. Interestingly, the deletion of the cytoplasmic domain of Env enhanced the neutralization of cell–cell infection, while having very modest impact on cell-free virus [20]. Because this domain of the viral glycoprotein modulates the fusogenicity of Env in response to virus particle maturation [13], [14], it may be that cell-surface Env assumes unique conformational states that allow it to resist neutralization. To fully understand the resistance of cell–cell transmission to neutralization, it will be important to define the differences in Env conformation on the cell surface versus the virion surface.
Multicopy Infection and Selective Drug Resistance
During VS transmission, many virus particles can be observed to transfer simultaneously across single synapses (Fig. 1D) [2]. Experimentally, cell-to-cell infection results in a greater copy number of successful viral integration events than predicted by a Poisson distribution [22], [23]. Studies on HIV-infected patient splenocytes have found multiple proviral copies of HIV-1 (3–4) per infected human splenocyte when examined by in situ hybridization [24]. In vitro studies report an enhanced resistance of cell-to-cell transmission to certain classes of antiretroviral drugs, which is attributed to the high multiplicity of infection mediated by VS [25], [26]. This relative resistance was overcome with highly-active combination therapies [25]. Multicopy infection has also been observed in single cell PCR studies [27], leading us to consider how the presence of coinfected cells may impact viral fitness. When mutant viral sequences are co-inherited with wild-type sequences, then many defective genomes may contribute to the gene pool without themselves being replication-competent. This provides a physical basis for selection of HIV-1 on the level of quasispecies rather than individual clonal sequences [22]. Cell-to-cell transmission may therefore contribute to the fitness of viral quasispecies by enhancing the diversity of actively transmitted sequences.
HIV-1 Spread by Cell Migration and Cell−Cell Interaction
In contrast to virus dissemination by cell-free particles, dissemination by cell-associated mechanisms is potentially dependent upon the migration, compartmentalization, and cellular interactions of infected cells. While it remains difficult to directly measure the relative roles of cell-free and cell-associated virus in viral dissemination in vivo, a number of studies indicate that cellular migration through tissues is important for HIV spread in vivo.
Within lymphoid organs such as the spleen, HIV-1 replication has been described as spatially segregated where viruses with distinct sequences segregated between neighboring follicles [24]. This spatial segregation of viral sequences may indicate that spread within a tissue compartment is predominantly local and dependent on cell proximity (Fig. 1E). A revealing study of acute HIV-1 transmission in humanized mouse models reported that T cell migration is critical to promote systemic spread of HIV-1 [28]. In this study, a local infection could be inhibited from spreading to distal sites by blocking lymphocyte egress from the draining lymph node using an inhibitor of the sphingosine monophosphate receptor (S1PR) [28]. Future studies are needed to determine the extent to which lymphocyte migration and VS contribute to the systemic spread of HIV-1.
Overview
While the basic steps of the virus life cycle are conserved between cell-free and VS-mediated infection, the latter exhibits a unique temporal and spatial organization of virus production that is integrated with cell–cell signaling and coordinated with cell migration within the body. Given the intimate connections of the VS with the biology of T helper cells, a deeper understanding of the VS will be required to gain a full appreciation of how HIV-1 spreads and causes disease.
Zdroje
1. JollyC, KashefiK, HollinsheadM, SattentauQJ (2004) HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J Exp Med 199 : 283–293.
2. HubnerW, McNerneyGP, ChenP, DaleBM, GordonRE, et al. (2009) Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science 323 : 1743–1747.
3. JollyC, MitarI, SattentauQJ (2007) Adhesion molecule interactions facilitate human immunodeficiency virus type 1-induced virological synapse formation between T cells. J Virol 81 : 13916–13921.
4. Vasiliver-ShamisG, TuenM, WuTW, StarrT, CameronTO, et al. (2008) Human immunodeficiency virus type 1 envelope gp120 induces a stop signal and virological synapse formation in noninfected CD4+ T cells. J Virol 82 : 9445–9457.
5. Sol-FoulonN, SourisseauM, PorrotF, ThoulouzeMI, TrouilletC, et al. (2007) ZAP-70 kinase regulates HIV cell-to-cell spread and virological synapse formation. EMBO J 26 : 516–526.
6. ChoudhuriK, LlodraJ, RothEW, TsaiJ, GordoS, et al. (2014) Polarized release of T-cell-receptor-enriched microvesicles at the immunological synapse. Nature 507 : 118–123.
7. JollyC, WelschS, MichorS, SattentauQJ (2011) The regulated secretory pathway in CD4(+) T cells contributes to human immunodeficiency virus type-1 cell-to-cell spread at the virological synapse. PLoS Pathog 7: e1002226.
8. MonelB, BeaumontE, VendrameD, SchwartzO, BrandD, et al. (2012) HIV cell-to-cell transmission requires the production of infectious virus particles and does not proceed through env-mediated fusion pores. J Virol 86 : 3924–3933.
9. RoyNH, ChanJ, LambeleM, ThaliM (2013) Clustering and mobility of HIV-1 Env at viral assembly sites predict its propensity to induce cell-cell fusion. J Virol 87 : 7516–7525.
10. WengJ, KrementsovDN, KhuranaS, RoyNH, ThaliM (2009) Formation of syncytia is repressed by tetraspanins in human immunodeficiency virus type 1-producing cells. J Virol 83 : 7467–7474.
11. RoyNH, LambeleM, ChanJ, SymeonidesM, ThaliM (2014) Ezrin Is a Component of the HIV-1 Virological Presynapse and Contributes to the Inhibition of Cell-Cell Fusion. J Virol 88 : 7645–7658.
12. SourisseauM, Sol-FoulonN, PorrotF, BlanchetF, SchwartzO (2007) Inefficient human immunodeficiency virus replication in mobile lymphocytes. J Virol 81 : 1000–1012.
13. MurakamiT, AblanS, FreedEO, TanakaY (2004) Regulation of human immunodeficiency virus type 1 Env-mediated membrane fusion by viral protease activity. J Virol 78 : 1026–1031.
14. WymaDJ, JiangJ, ShiJ, ZhouJ, LinebergerJE, et al. (2004) Coupling of human immunodeficiency virus type 1 fusion to virion maturation: a novel role of the gp41 cytoplasmic tail. J Virol 78 : 3429–3435.
15. LadinskyMS, KiefferC, OlsonG, DeruazM, VrbanacV, et al. (2014) Electron tomography of HIV-1 infection in gut-associated lymphoid tissue. PLoS Pathog 10: e1003899.
16. BoschB, GrigorovB, SenserrichJ, ClotetB, DarlixJL, et al. (2008) A clathrin-dynamin-dependent endocytic pathway for the uptake of HIV-1 by direct T cell-T cell transmission. Antiviral Res 80 : 185–193.
17. SloanRD, KuhlBD, MespledeT, MunchJ, DonahueDA, et al. (2013) Productive entry of HIV-1 during cell-to-cell transmission via dynamin-dependent endocytosis. J Virol 87 : 8110–8123.
18. DaleBM, McNerneyGP, ThompsonDL, HubnerW, de Los ReyesK, et al. (2011) Cell-to-cell transfer of HIV-1 via virological synapses leads to endosomal virion maturation that activates viral membrane fusion. Cell Host Microbe 10 : 551–562.
19. AbelaIA, BerlingerL, SchanzM, ReynellL, GunthardHF, et al. (2012) Cell-cell transmission enables HIV-1 to evade inhibition by potent CD4bs directed antibodies. PLoS Pathog 8: e1002634.
20. DurhamND, YewdallAW, ChenP, LeeR, ZonyC, et al. (2012) Neutralization Resistance of Virological Synapse-Mediated HIV-1 Infection Is Regulated by the gp41 Cytoplasmic Tail. J Virol 86 : 7484–7495.
21. MalbecM, PorrotF, RuaR, HorwitzJ, KleinF, et al. (2013) Broadly neutralizing antibodies that inhibit HIV-1 cell to cell transmission. J Exp Med 210 : 2813–2821.
22. Del PortilloA, TripodiJ, NajfeldV, WodarzD, LevyDN, et al. (2011) Multiploid inheritance of HIV-1 during cell-to-cell infection. J Virol 85 : 7169–7176.
23. RussellRA, MartinN, MitarI, JonesE, SattentauQJ (2013) Multiple proviral integration events after virological synapse-mediated HIV-1 spread. Virology 443 : 143–149.
24. JungA, MaierR, VartanianJP, BocharovG, JungV, et al. (2002) Multiply infected spleen cells in HIV patients. Nature 418 : 144.
25. AgostoLM, ZhongP, MunroJ, MothesW (2014) Highly active antiretroviral therapies are effective against HIV-1 cell-to-cell transmission. PLoS Pathog 10: e1003982.
26. SigalA, KimJT, BalazsAB, DekelE, MayoA, et al. (2011) Cell-to-cell spread of HIV permits ongoing replication despite antiretroviral therapy. Nature 477 : 95–98.
27. JosefssonL, KingMS, MakitaloB, BrannstromJ, ShaoW, et al. (2011) Majority of CD4+ T cells from peripheral blood of HIV-1-infected individuals contain only one HIV DNA molecule. Proc Natl Acad Sci U S A 108 : 11199–11204.
28. MurookaTT, DeruazM, MarangoniF, VrbanacVD, SeungE, et al. (2012) HIV-infected T cells are migratory vehicles for viral dissemination. Nature 490 : 283–287.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 12
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Plasma Membrane-Located Purine Nucleotide Transport Proteins Are Key Components for Host Exploitation by Microsporidian Intracellular Parasites
- Rubella Virus: First Calcium-Requiring Viral Fusion Protein
- Emergence of MERS-CoV in the Middle East: Origins, Transmission, Treatment, and Perspectives
- Neutral Sphingomyelinase in Physiological and Measles Virus Induced T Cell Suppression
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy