
HTLV-1 Tax-Mediated Inhibition of FOXO3a Activity Is Critical for the Persistence of Terminally Differentiated CD4 T Cells
HTLV - infection contributes to the development of Adult T cell Leukemia (ATL) or the neurological disorder HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP). HTLV-1 principally targets CD4+ T lymphocytes and causes profound changes in activation, immune function and cell death. The molecular mechanisms involved in the persistence of infected CD4+ T cells following primary HTLV-1 infection remain unclear. We demonstrate here that the Tax oncoprotein inactivates the FOXO3a transcription factor to facilitate the long-term survival of a population of highly activated and terminally differentiated T cells that maintain the capacity to spread infectious viral particles. Mechanistically, expression of Tax oncoprotein in primary human CD4+ T cells resulted in the phosphorylation-dependent inactivation of FOXO3a, via the AKT kinase. Tax-mediated CD4+ T cell persistence was also reversed by chemical inhibition of the AKT pathway, and reproduced by the expression of a dominant negative version of FOXO3a itself or by silencing its transcriptionally active form using specific siRNA. Overall this study provides new mechanistic insights used by Tax to potentiate the long-term maintenance of CD4+ T lymphocytes following HTLV-1 infection and suggests that modulation of FOXO3a activity, using a range of inhibitors targeting the PI3K-AKT-FOXO3a pathway, may offer a valuable addition to current therapeutic approaches.
Published in the journal:
. PLoS Pathog 10(12): e32767. doi:10.1371/journal.ppat.1004575
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004575
Summary
HTLV - infection contributes to the development of Adult T cell Leukemia (ATL) or the neurological disorder HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP). HTLV-1 principally targets CD4+ T lymphocytes and causes profound changes in activation, immune function and cell death. The molecular mechanisms involved in the persistence of infected CD4+ T cells following primary HTLV-1 infection remain unclear. We demonstrate here that the Tax oncoprotein inactivates the FOXO3a transcription factor to facilitate the long-term survival of a population of highly activated and terminally differentiated T cells that maintain the capacity to spread infectious viral particles. Mechanistically, expression of Tax oncoprotein in primary human CD4+ T cells resulted in the phosphorylation-dependent inactivation of FOXO3a, via the AKT kinase. Tax-mediated CD4+ T cell persistence was also reversed by chemical inhibition of the AKT pathway, and reproduced by the expression of a dominant negative version of FOXO3a itself or by silencing its transcriptionally active form using specific siRNA. Overall this study provides new mechanistic insights used by Tax to potentiate the long-term maintenance of CD4+ T lymphocytes following HTLV-1 infection and suggests that modulation of FOXO3a activity, using a range of inhibitors targeting the PI3K-AKT-FOXO3a pathway, may offer a valuable addition to current therapeutic approaches.
Introduction
Infection with the human T cell leukemia virus type I (HTLV-1) affects more than 20 million people worldwide [1] and HTLV-1-associated diseases are a major cause of mortality and morbidity in endemic areas where infection rates range from 2 to 30%. Chronic infection with HTLV-1 can result in a number of severe pathologies, including the aggressive adult T cell leukemia (ATL) and the progressive neurological disorder termed myelopathy/tropical spastic paraperasis (HAM/TSP) [1]. The majority of HTLV-1-infected individuals remain asymptomatic carriers (AC) of the virus but a proportion of AC (1–5%) will develop ATL or HAM/TSP. CD4+ T cells are the main targets for viral infection [1], [2], although HTLV-1 can also infect cells of the myeloid lineage including dendritic cells and monocytes [3], [4].
HTLV-1-associated diseases are characterized by profound deregulation of CD4+ T cells in terms of activation, immune function and apoptosis [5], [6], all of which are facilitated by the pleiotropic functions of the viral oncoprotein Tax [7]–[10]. In addition to controlling viral gene expression and replication, Tax contributes to malignant transformation of CD4+ T cells by modulating host signalling pathways including NF-κB, PI3K-AKT, and JAK-STAT [7]–[10].
The chronic nature of retrovirus infection has been linked to the activity of the Forkhhead box (FOXO) transcription factor family, and particularly to FOXO3a, which can alter the activation, survival and proliferative capacity of CD4+ T cell compartment [11]–[15]. FOXO3a is constitutively expressed in most cell types including T lymphocytes, where it regulates apoptosis, tumorigenesis and inflammation [16]–[18], processes that are also deregulated in HTLV-1-associated diseases [5], [19], [20]. Specifically, FOXO3a stimulates expression of pro-apoptotic and anti-proliferative target genes such as BIM, FASL and p130 [21]. The FOXO family is subject to numerous post-translational modifications [17] and FOXO phosphorylation can serve either an inhibitory or an activating role in FOXO functions; phosphorylation by JNK activates FOXO3a function [22] while phosphorylation of specific residues (Ser 253 and Thr32) by the serine/threonine kinase AKT inactivates FOXO3a [23].
Previous studies demonstrated that FOXO3a activity contributes to the progressive depletion of central memory CD4+ T cells in HIV-1-infected patients [15]. Modulation of FOXO3a activity also occurs during de novo HIV-1 infection, where HIV Tat protein induces FOXO3a activity leading to HIV-specific apoptosis [24], [25].
In the present study, we demonstrate that expression of HTLV-1 Tax in primary human CD4+ T cells, either by productive HTLV-1 infection or lentiviral-mediated transduction results in the phosphorylation-dependent inactivation of FOXO3a via the upstream kinase AKT. FOXO3a inhibition resulted in long-term survival of terminally differentiated, Tax+CD27negCCR7neg CD4+ T cells that were capable of disseminating infectious HTLV-1. These results provide insight into the mechanisms used by HTLV-1 to increase the long-term maintenance of Tax+CD4+ T lymphocytes during the early stages of HTLV-1 pathogenesis.
Results
Productive HTLV-1 infection is associated with phosphorylation of FOXO3a and persistence of infectious CD4+CD27negCCR7neg T cells
Primary CD3/CD28 activated CD4+ T cells were infected with HTLV-1 in a dose dependent manner (Fig. 1A) using an in vitro trans-infection system in which CD4+ T cells were co-cultured with HTLV-1 shedding MT-2 cells [26]. Following multiple rounds of T cell receptor (TCR) triggering, HTLV-1 infected T cells [Tax+ cells; blue] persisted for 21–28 days without a significant reduction in cell number, (P<0.05) (Fig. 1B); in contrast, T cells that were not infected [Taxneg cells; black] displayed a reduction in cell number by 14 days post-infection (pi) (P<0.01). The half-life of gated TaxnegCD4+ T cells was 18.1 days pi, whereas a half-life calculation could not be determined for Tax+ T cells before 28 days.
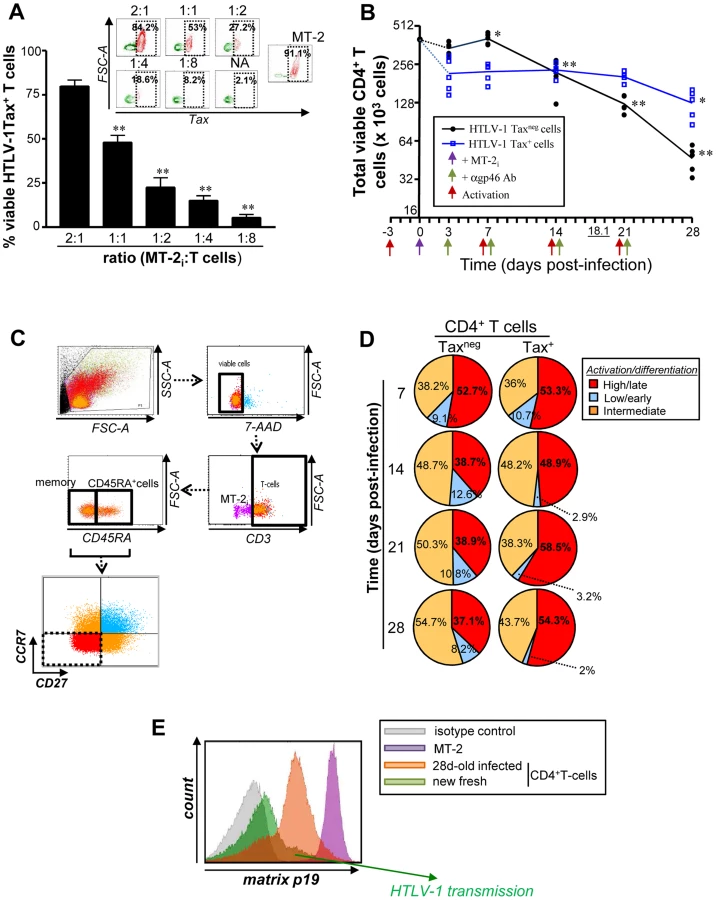
Using a combination of CD3, CD45RA, CCR7 and CD27 surface markers, we evaluated the generation and maintenance of terminally differentiated (CD3+CD45RA+/−CCR7negCD27neg) T cells during a 28-day cycle of HTLV-1 infection (Fig. 1C) [27], [28]. Repeated TCR triggering reduced the proportion of viable terminally differentiated CD27negCCR7neg effector cells among gated CD3+Taxneg T cells, whereas the proportion and cell number of Tax+ T lymphocytes increased, and maintained activation status (55.7±4.6% and 36.2±1.3% of CD3+CCR7negCD27negcells, respectively for Tax+ and Taxneg T cell population at 28 days; P<0.01) (Fig. 1D and S1A Fig.). In addition, terminally differentiated Tax+CD4+ T cells produced infectious HTLV-1, even after four weeks in culture, based on their capacity to transmit virus to freshly isolated autologous CD4+ T lymphocytes (Fig. 1E). Overall these results demonstrate that HTLV-1 infection promotes the in vitro maintenance of terminally differentiated, virus-producing CD4+ T cells (CD3+CCR7negCD27neg).
We hypothesized that the enhanced cellular survival observed in HTLV-1 infected CD4+ T cells may be associated with the deregulation of FOXO3a signalling, given its important role in regulating cell proliferation and apoptosis in other retroviral infections [14], [15], [24], [29]. We first investigated at 2 days pi the activation status of AKT, one of the upstream kinases responsible for phosphorylation of FOXO3a [23] (Fig. 2A). Based on phosphorylation of FOXO3a at Ser473 residue, as detected by PhosFlow and Western Blotting approaches [9], we concluded that the upstream kinase AKT was significantly activated in Tax-expressing cells (P = 0.0317 and P<0.001, respectively) (Fig. 2B–D). HTLV-1 infection led to an increase in phosphorylation of FOXO3a at residues S253 (P<0.01) and Thr32 residues (P<0.001) at 2 days pi (Fig. 2C, D), residues that inactivate FOXO3a function [23]. Consistent with this observation, productively infected cells displayed reduced expression of FOXO3a downstream target genes p130 and Bim [15], [23]. The phosphorylation status of IKKα/β, another upstream kinase of FOXO3a, was however unchanged (Fig. 2C, D). Overall these results demonstrate that productive HTLV-1 infection provides a survival and proliferative persistence advantage to infected CD4+ T cells, and is associated with AKT-mediated inactivation of FOXO3a transcriptional activity.

Tax expression inhibits FOXO3a activity via activation of AKT
Among the proteins encoded by HTLV-1, the Tax oncoprotein exerts its essential role in viral transcription, as well as in T cell transformation [30]–[32]. To determine whether Tax expression alone was sufficient to drive FOXO3a inactivation, HTLV-1 Tax was introduced into activated CD4+ T cells using lentiviral particle (LVP)-mediated transduction. Tax was detected using intracellular staining by flow cytometry (Fig. 3A) and a concentration of 80 ng LVP/106cells resulted in ∼40% Tax+ cells. LVPTax-transduced CD4+ T cells displayed higher expression levels of HTLV-1 Tax when compared to infected cells (S1B Fig.; fold increase ∼2.07; P = 0.0091). A BioMark transcriptional high throughput qPCR analysis of LVP-transduced T cells demonstrated that Tax expression modulated mRNA levels of several Tax-modulated genes [8], [31], including an increase in IL-2 and a decrease in type I IFN-associated genes. Tax expression led to higher mRNA expression levels of CXCR4, SOCS1 and myc proto-oncogene, as previously shown [7], [33]–[35]. This analysis also demonstrated the activated/differentiated status of Tax-transduced CD4+ T cells, based on the increased expression of CD40L, CTLA-4, IFNγ and IL7R mRNA (Fig. 3B). Interestingly, Tax expression not only inhibited p130 and Bim expression (Fig. 3B, C), but also down-regulated several other FOXO3a target genes; BCL6, p27, BIM, FASL, NOXA and PUMA were all downregulated at the mRNA levels at 24 and 48 h post-transduction (Fig. 3B, C). Tax transduction also induced AKT activation (P<0.001) [9], phosphorylation of FOXO3a (P<0.05) and inhibition of p130 (P<0.05) at the protein level, all of which were significantly reversed by the addition of an AKT inhibitor, AKT inhibitor IV (AKTi) (Fig. 4A, B). Conversely, treatment of transduced CD4+ T cells with 100 µg/mL IKK inhibitor II (Calbiochem) did not significantly alter the expression levels of phosphorylated FOXO3a (S2B–C Fig.). Tax expression in CD4+ T cells did not modulate FOXO3a stability, as we found no significant change in its expression in the presence or absence of Tax even after 6 days of transduction (S3A–B Fig.). Nonetheless Tax transduction resulted in increased nuclear localization of inactive pFOXO3a forms (Ser253 and Thr32 residues) (S3C–D Fig.) [13]. Overall this data demonstrate that Tax expression is sufficient to transcriptionally inactivate FOXO3a signaling, via upstream activation of AKT.

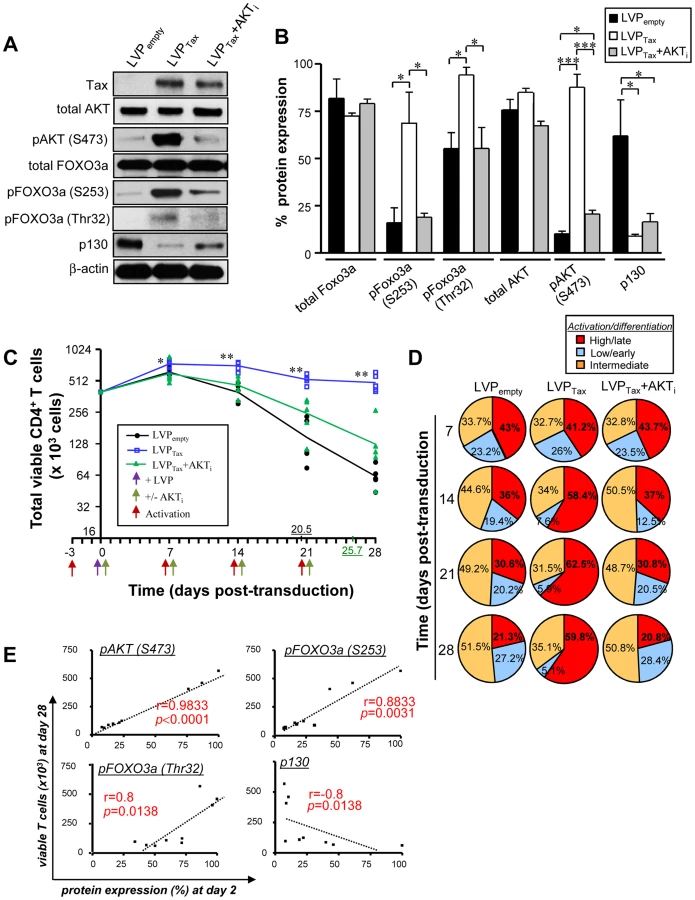
Tax-mediated FOXO3a inactivation is responsible for CD4+ T cell persistence
We further investigated whether Tax promoted the persistence of activated CD4+ T cells through AKT induction and subsequent FOXO3a inactivation. Tax transduction alone [blue] was sufficient to maintain T cell survival for 28 days, and maintenance of the differentiated T cell population was mediated through AKT signaling, since the addition of the AKTi reduced T cell viability to basal levels (Fig. 4C) (half-lives of transduced T cells with LVPempty [black] and LVPTax+AKTi [green] were 20.5 and 25.7 days, respectively). Tax expression was also associated with the terminally differentiated phenotype, similar to that of HTLV-1 productively infected-T cells (Fig. 4D). A majority of the Tax-transduced CD4+ T cells belonged to the CD3+CCR7negCD27negsubset at 28 days after Tax transduction (55.2±6.3% and 23.9±6.3% for LVPTax and LVPempty infected T cells, respectively; P<0.01). In addition, a strong correlation was established between the number of viable primary CD4+ T cells at 28 days post-transduction (Fig. 4C) and the inhibition of FOXO3a signaling observed as early as 2 days pi (Fig. 4A, B), and measured by phosphorylation of AKT (P<0.0001), pFOXO3a-S253 (P = 0.0031), pFOXO3a-Thr32 (P = 0.0138), and expression of p130 (P = 0.0138) (Fig. 4E). Altogether, these results demonstrate that long-term survival of activated CD4+ T lymphocytes is mediated by a Tax-dependent, AKT phosphorylation and inactivation FOXO3a transcriptional activity.
Specific inhibition of FOXO3a activity mimics Tax expression
Based on the above results, we rationalized that the T cell persistence observed during HTLV-1 infection or Tax transduction could be reproduced by introduction of a dominant negative form of FOXO3a, termed FOXO3a Nt [15], that encompasses the N-terminal DNA binding domain of FOXO3a (aa1–304) but lacks the C-terminal transactivation domain. FOXO3a Nt acts as a competitive DNA binding inhibitor of transcriptionally active FOXO3a [15], [36] and interferes with FOXO3a activation of pro-apoptotic and anti-proliferative target genes. As shown in Fig. 5A, lentiviral-mediated transduction of FOXO3a Nt prevented primary T cells from undergoing apoptosis (Fig. 5A) and thus mimicked Tax function. Expression of FOXO3a Nt also inhibited endogenous FOXO3a activity, as determined by reduced expression of p130 and Bim (Fig. 5B). FOXO3a Nt expression resulted in persistence of a highly activated, terminally differentiated CD4+ T cell population, similar to that observed with Tax expression (Fig. 5C, D). In addition, we found an increased percentage of terminally differentiated CD4+ T cells in the presence of specific FOXO3a siRNA at 14 days of culture (Fig. 5E, F and S4 Fig.). Finally, we sought to determine if Tax physically interacted with an inactivated FOXO3a; by co-immunoprecipitation from Tax-transduced primary T cells, we did not observe an interaction between Tax and FOXO3a, although the interaction between Tax and PI3K was detected (S5 Fig.) [9]. It is possible that Tax-PI3K association stimulates AKT activity and thus indirectly contributes to phosphorylation of FOXO3a by AKT. Collectively, these results demonstrate that Tax expression enhanced T cell longevity and activation, through the inhibition of FOXO3a transcriptional activity, mediated by AKT phosphorylation at S253 and Thr32 residues.

Discussion
HTLV-1 infection is associated with the expansion and leukemic transformation of CD4+ T lymphocytes, driven in large part by the chronic disruption of host signaling networks by the HTLV-1 Tax oncoprotein [8], [31], [37], [38]. In the present study, we demonstrate that HTLV-1 infection enhances the in vitro cellular persistence of activated CD4+ T cells, the expansion of terminally differentiated (CD3+CCR7negCD27neg) cells and the functional inactivation of the FOXO3a pathway (illustrated by the increased localization of inactive FOXO3a in the nucleus and the inhibition of several targets such as Bim and p130). Mechanistically, both de novo HTLV-1 infection and Tax transduction stimulated AKT activation and downstream phosphorylation of FOXO3a at residues S253 and Thr32 (Fig. 2 and 4A, B). Mechanistically, we did not observe an interaction between Tax and FOXO3a (S5 Fig.), although the interactions between Tax and PI3K was detected as previously reported [9]. It is possible that Tax-PI3K association stimulates AKT activity and thus indirectly contributes to phosphorylation of FOXO3a by AKT. Also, we cannot exclude the possibility that post-translational modifications other than phosphorylation (such as acetylation, methylation, ubiquitination) may impact FOXO3a activity [36], [39]. In addition, since mRNA and protein levels of Tax are generally barely detectable in ATL cells displaying constitutively active AKT [40], [41], it is possible that other Tax-independent mechanisms of FOXO3a inactivation may be used by HTLV-1. Nevertheless, Tax-transduced T cells displayed a global inhibition of FOXO3a activity, illustrated by reduced expression of many pro-apoptotic and anti-proliferative target genes such as BIM, FASL, NOXA, p27 and p130 at 24–48 h post-transduction (Fig. 3B, C). Overall this study provides new mechanistic insights by which Tax potentiates the long-term maintenance of CD4+ T lymphocytes following HTLV-1 infection.
FOXO3a activity is also targeted by another HTLV-1 accessory protein, HBZ, which was shown to inhibit FOXO3a by interfering with its localization and ability to bind DNA [13]. The mechanistically distinct, yet functionally redundant, inhibition of FOXO3a signaling may be explained by the distinct kinetics of expression of these two regulatory proteins. It has been reported that, in contrast to Tax, HBZ is transcribed at high levels in chronically infected patient samples [42]. Conversely, even though Tax mRNA expression is relatively moderate, it is at its highest during the early stages of infection, specifically within the first week [43]. Additionally, Tax controls FOXO4 activity through degradation by the proteasome during ATL development [11]. The inhibition of FOXO3a or FOXO4 activity by distinct HTLV-1 accessory mechanisms also highlights the importance of FOXO inactivation as a strategy to perpetuate HTLV-1 infected CD4+ T lymphocytes and to contribute in the ATL development.
Using a BioMark high throughput qPCR analysis (S1 Table), we demonstrated that Tax not only mediated CD4+ T cell persistence through the inactivation of the FOXO3a pathway, but also down regulated type I IFN responses (S6A Fig.), in part mediated by the negative regulator of the JAK-STAT1 pathway SOCS1 [7], [34]. Taken together with our findings, these data indicate an involvement of Tax oncoprotein in targeting FOXO3a to concomitantly modulate the cell survival, as well as the type I IFN antiviral responses in CD4+ T cells and thus facilitate HTLV-1 infection.
The identification of a pivotal role for FOXO3a in de novo HTLV-1 infection of CD4+ T cells in terms of cellular differentiation and persistence survival may have important consequences for retroviral pathogenesis. For instance, alterations in the microenvironment mediated by HIV infection significantly increase FOXO3a activity, with a major impact on T and B cell immunity and survival [14], [15], [44], [45]. Kino et al. reported that the HIV accessory protein Vpr inhibited the ability of insulin to induce FOXO3a phosphorylation via AKT, thus interfering with its exclusion from the nucleus [46]. The expression of HIV-1 regulatory molecule Tat in specific T cells and macrophages also induced FOXO3a-mediated apoptosis [24], [47]. FOXO3a activity also impacts the pathogenesis and the outcome of Abelson murine leukemia virus [29].
Several gene networks/pathways that were deregulated in Tax expressing CD4+ T cells were similarly disrupted in transcriptome analyses of PBMC from HTLV-1 infected individuals [34], [48], [49] (S5B Fig.). For instance, transcriptional analysis of differentially regulated pathways demonstrated that cytokines IL15, IL17R, IL7R were down regulated, and chemokine CXCR4 was up regulated in ATL patients; in contrast TNFRSF17 was down regulated while granzyme B and IL-2 were up regulated, in HAM/TSP and AC individuals (S5B Fig.). It is tempting to speculate that disruption of signalling mechanisms identified early after de novo HTLV-1 infection are also important in the development and maintenance of HTLV-1 associated pathologies and could be targeted for clinical treatment.
Due to poor prognosis of patients diagnosed with ATL, coupled with limited therapeutic options, novel immunological approaches including recombinant IL-7, IFN-α, and neutralizing anti-CD25 or anti-CXCR4 antibodies are currently being used to treat ATL and HAM/TSP patients [50]–[53]. The present study indicates that the PI3K-AKT-FOXO3a pathway may also represent a potential therapeutic target in ATL patients. Since AKT inhibitors are already in clinical development [54], they may offer a valuable addition to current therapeutic approaches.
Materials and Methods
Products
RPMI-1640 media, FBS and antibiotics were provided by Wisent Technologies (CA, USA). Unconjugated anti-Tax mAbs (clone LT4) was generously provided by Dr. Yuetsu Tanaka (Kitasato University, Kanagawa, Japan). MT-2 cell lines were obtained from the ATCC (VA, USA). All antibodies used for flow cytometry were purchased from BD Biosciences, except for the antibody to CD45RA-ECD, which was from Beckman Coulter. All primary antibodies used in Western Blots (anti-phospho forms of FOXO3a, anti-Bim, anti-ERK, anti-AKT, anti-PI3K p85, anti-IKK, and anti-phospho-IKK Abs) were purchased from Cell Signaling Technology Inc., whereas anti-p130 and anti-actin were purchased from Sigma Aldrich; anti-FOXO3a from Abcam. 7-Aminoactinomycin D (7-AAD) came from Invitrogen. Anti-Tax antibody (clone 1A3) was purchased from Santa Cruz Biotechnology.
Ethics statement
Leukaphereses from healthy donors were obtained from the Royal Victoria Hospital, Montreal (QC, Canada). Written informed consent approved by the Royal Victoria Hospital and the Jewish General Hospital review boards was provided to study participants. Research was conformed to ethical guidelines established by the ethics committee of the Royal Victoria Hospital, the Jewish General Hospital, and McGill University (# BMB-2001-028).
Purification and activation of CD4+ T cells
PBMCs were isolated using Ficoll-Hypaque gradient and CD4+ T cells were then purified using the untouched CD4 isolation kit (EasySep Human CD4+ T cell Enrichment Kit; StemCell Technologies, Vancouver, BC, Canada), allowing for more than 94% purification without any cell stimulation and apoptosis. CD4+ T cells were then activated 72 hours in RPMI complete in the presence of 1 µg/mL anti-CD28 (BD Biosciences) in 6 well plates pre-coated 24 hours earlier with 0.5 µg/mL anti-CD3 (clone: OKT-3, BioLegend; 2.106 cells/well).
HTLV-1 trans-infection
Cell-cell transmission of HTLV-1 was performed essentially as previously described [26]. 20.106 HTLV-1 produced cell line MT-2 were first irradiated at 15,000 rads and then mixed at various ratios (2∶1 to 1∶8) of irradiated MT-2 to activated CD4+ T cells. At several time points post-infection (pi), collected cells were treated with Cell Dissociation Solution non-enzymatic according to the Sigma manufacturer's protocol and finally filtered (70 µm), prior further analyses.
Flow cytometry
Surface staining
At day 2 pi, cells were first stained in calcium buffer with anti-CD3-PE Cy7 antibody (Ab) and Annexin-V-V450 in the presence or absence of anti-CD25-APC Cy7 or anti-HLA DR-Alexa700 Abs for 10 minutes at 4°C.
HTLV-1 Tax quantitation
Cells were washed twice and fixed at room temperature (RT) in BD FACS Lysing Buffer (Becton Dickinson) and incubated with anti-Tax-FITC and anti-p19-Alexa647 mAbs for 20 minutes at RT in 0.25% saponin. Anti-p19 IgG1 antibody was conjugated to Alexa647 dye using the Zenon mouse IgG1 labeling kit (number: Z25008; Life Technologies Inc., ON, USA) according to the manufacturer's protocol.
pAKT measurement
AKT phosphorylation was measured using BD Bioscience PhosFlow anti-pAKT (S473 residue) specific Ab, as previously described [23].
Western blotting and Tax co-immunoprecipitation (co-IP)
Protein lysates (2–10 µg) from highly purified CD4+ T cell subsets were subjected to Western blot analysis as previously described [4]. Densitometric quantifications of protein of interest (normalized to β-actin whose expression level was used as loading control) were calculated using ImageJ software.
Anti-Tax co-IP
MT-2 cells were collected to determine molecular interactions with HTLV-1 Tax. Briefly, cells were lysed using CHAPS buffer with protease inhibitors as previously described [4]. 500 µg of proteins were used for this assay. Monoclonal antibodies (clones 1A3 and LT4) were used to immunoprecipitate and immunoblot for Tax respectively. Proteins (20 µg) were also collected and referred to as the "input" fraction.
Generation of recombinant lentiviruses
The lentiviral vector pWPI (empty vector), packaging plasmid psPAX2 and envelope plasmid pMD2G were generously provided by VGTI-Florida, whereas pCLXSN-Tax vector was purchased from Addgene (ref: 44038; MA, USA). The FOXO3a N-terminal (Nt) fragment was cloned into pWPI, and as previously described [15], lentiviral particles were produced in 293T cells that harboured either pWPI or pCLXSN (empty controls), pWPI-FOXO3a Nt, or pCLXSN-Tax expression vector. Titers (ng/mL) of lentiviral constructs were assessed using HIV p24 ELISA (Zeptometrix Corporation, USA).
Long-term persistence assays
CD4+ T cells (4.104) were co-cultured in the presence of irradiated MT-2 cells (ratio = 1∶1) in complete RPMI. At 3 days pi, cells were washed to remove the maximum of dead MT-2i and cultured in the presence of 1 µg/mL anti-gp46 Abs to avoid any de novo infection. On days 7, 14 and 21 pi, cells were re-stimulated by adding fresh anti-CD3 and anti-CD28 Ab in the presence of anti-gp46 Abs (at 10 µg/mL; Abcam) to avoid any de novo infection. The efficiency of neutralizing anti-gp46 Ab was confirmed by the absence of p19 staining on primary cells that were treated since the onset of co-culture. At days 3, 7, 14, 21 and 28 pi, viable cultured cells were counted by trypan blue exclusion and stained with 7-AAD, anti-CD3-PE, anti-CD45RA-ECD, anti-CD27-APC H7, anti-CCR7-PE Cy7 and anti-Tax-Alexa 647 Ab for flow cytometry analyses.
FOXO3a silencing
A total of 107 activated CD4+ T cells were first electroporated in the presence of control or FOXO3a-specific siRNA (Invitrogen; 3 µg) using nucleofector II technology, according the manufacturer's protocol (Amaxa human T cell nucleofector kit). Transfected cells were then washed, and cultured alone for 14 days as described above in Long-term persistence assays.
Fluidigm BioMark assays
Total RNA was isolated from cells using RNeasy Kit (Qiagen, Valencia, USA) as per manufacturer's instructions. RNA was reverse transcribed using the SuperScript VILO cDNA synthesis kit according to manufacturer's instructions (Invitrogen, Carlsbad, USA). PCR primers were designed using Roche's Universal Probe Library Assay Design Center (www.universalprobelibrary.com) and ordered from the Integrated DNA Technology company (IDT, USA) (S1 Table). cDNA along with the entire pool of primers were pre-amplified for 14 cycles using TaqMan PreAmp Master Mix as per manufacturer's protocol (Applied Biosystems, Foster City, USA). cDNA were exonuclease treated to get rid of excess primers using Exonuclease I (E. coli) (New England Biolabs, Ipswich, USA). cDNA samples were prepared with 2× FastStart TaqMan Probe Master (Roche, Penzberg, Germany), GE sample loading buffer (Fluidigm, San Francisco, USA) and Taq Polymerase (Invitrogen, NY, USA). Assays were prepared with 2× assay loading reagent (Fluidigm, NY, USA), primers (IDT) and probes (Roche, Penzberg, Germany). Samples and assays were loaded in their appropriate inlets on a 48.48 BioMark chip. Chip was run on the Biomark HD System (Fluidigm, San Francisco USA) and enabled quantitative measurement of up to 48 different mRNAs in 48 samples under identical reaction conditions. Raw Ct values were calculated after 40 cycles by the real time PCR analysis software (Fluidigm, San Francisco, USA) and software designated failed reactions were discarded from analysis. All data are presented as a relative quantification with efficiency correction based on the relative expression of target gene versus the mean of (gapdh+actin+β2 microglobulin) as the invariant control. The N-fold differential expression of mRNA gene samples was expressed as 2ΔΔCt. The heatmap was produced with the R package pheatmap (http://CRAN.R-project.org/package=pheatmap) and gene expression is shown as gene-wise standardized expression (Z score).
Statistical analysis
Statistical analyses were performed as previously described [4]. ***, P<0.001; **, P<0.01 and *, P<0.05.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. CookLB, ElemansM, RowanAG, AsquithB (2013) HTLV-1: persistence and pathogenesis. Virology 435 : 131–140.
2. VerdonckK, GonzalezE, Van DoorenS, VandammeAM, VanhamG, et al. (2007) Human T-lymphotropic virus 1: recent knowledge about an ancient infection. Lancet Infect Dis 7 : 266–281.
3. JonesKS, Petrow-SadowskiC, HuangYK, BertoletteDC, RuscettiFW (2008) Cell-free HTLV-1 infects dendritic cells leading to transmission and transformation of CD4(+) T cells. Nat Med 14 : 429–436.
4. SzeA, BelgnaouiSM, OlagnierD, LinR, HiscottJ, et al. (2013) Host restriction factor SAMHD1 limits human T cell leukemia virus type 1 infection of monocytes via STING-mediated apoptosis. Cell Host Microbe 14 : 422–434.
5. RatnerL (2004) Adult T cell leukemia lymphoma. Front Biosci 9 : 2852–2859.
6. YasunagaJ, MatsuokaM (2007) Human T-cell leukemia virus type I induces adult T-cell leukemia: from clinical aspects to molecular mechanisms. Cancer Control 14 : 133–140.
7. CharoenthongtrakulS, ZhouQ, ShembadeN, HarhajNS, HarhajEW (2011) Human T cell leukemia virus type 1 Tax inhibits innate antiviral signaling via NF-kappaB-dependent induction of SOCS1. J Virol 85 : 6955–6962.
8. MatsuokaM, JeangKT (2007) Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat Rev Cancer 7 : 270–280.
9. PeloponeseJMJr, JeangKT (2006) Role for Akt/protein kinase B and activator protein-1 in cellular proliferation induced by the human T-cell leukemia virus type 1 tax oncoprotein. J Biol Chem 281 : 8927–8938.
10. ShembadeN, HarhajNS, YamamotoM, AkiraS, HarhajEW (2007) The human T-cell leukemia virus type 1 Tax oncoprotein requires the ubiquitin-conjugating enzyme Ubc13 for NF-kappaB activation. J Virol 81 : 13735–13742.
11. OteizaA, MechtiN (2011) The human T-cell leukemia virus type 1 oncoprotein tax controls forkhead box O4 activity through degradation by the proteasome. J Virol 85 : 6480–6491.
12. ShembadeN, HarhajEW (2011) Outfoxing FoxO transcription factors: HTLV-1 Tax oncoprotein inactivates FoxO4 via the ubiquitin-proteasome pathway. Future Virol 6 : 1165–1168.
13. Tanaka-NakanishiA, YasunagaJ, TakaiK, MatsuokaM (2014) HTLV-1 bZIP factor suppresses apoptosis by attenuating the function of FoxO3a and altering its localization. Cancer Res 74 : 188–200.
14. van GrevenyngheJ, CubasRA, DaFonsecaS, MetcalfT, TremblayCL, et al. (2012) Foxo3a: an integrator of immune dysfunction during HIV infection. Cytokine Growth Factor Rev 23 : 215–221.
15. van GrevenyngheJ, ProcopioFA, HeZ, ChomontN, RiouC, et al. (2008) Transcription factor FOXO3a controls the persistence of memory CD4(+) T cells during HIV infection. Nat Med 14 : 266–274.
16. LeeJC, EspeliM, AndersonCA, LintermanMA, PocockJM, et al. (2013) Human SNP links differential outcomes in inflammatory and infectious disease to a FOXO3-regulated pathway. Cell 155 : 57–69.
17. MonsalveM, OlmosY (2011) The complex biology of FOXO. Curr Drug Targets 12 : 1322–1350.
18. YangJY, HungMC (2011) Deciphering the role of forkhead transcription factors in cancer therapy. Curr Drug Targets 12 : 1284–1290.
19. FuziiHT, da Silva DiasGA, de BarrosRJ, FalcaoLF, QuaresmaJA (2014) Immunopathogenesis of HTLV-1-assoaciated myelopathy/tropical spastic paraparesis (HAM/TSP). Life Sci 104 : 9–14.
20. RomanelliMG, DianiE, BergamoE, CasoliC, CiminaleV, et al. (2013) Highlights on distinctive structural and functional properties of HTLV Tax proteins. Front Microbiol 4 : 271.
21. HuangH, TindallDJ (2007) Dynamic FoxO transcription factors. J Cell Sci 120 : 2479–2487.
22. van den BergMC, van GoghIJ, SmitsAM, van TriestM, DansenTB, et al. (2013) The small GTPase RALA controls c-Jun N-terminal kinase-mediated FOXO activation by regulation of a JIP1 scaffold complex. J Biol Chem 288 : 21729–21741.
23. RiouC, Yassine-DiabB, Van grevenyngheJ, SomogyiR, GrellerLD, et al. (2007) Convergence of TCR and cytokine signaling leads to FOXO3a phosphorylation and drives the survival of CD4+ central memory T cells. J Exp Med 204 : 79–91.
24. DabrowskaA, KimN, AldoviniA (2008) Tat-induced FOXO3a is a key mediator of apoptosis in HIV-1-infected human CD4+ T lymphocytes. J Immunol 181 : 8460–8477.
25. KimN, KukkonenS, GuptaS, AldoviniA (2010) Association of Tat with promoters of PTEN and PP2A subunits is key to transcriptional activation of apoptotic pathways in HIV-infected CD4+ T cells. PLoS Pathog 6: e1001103.
26. JonesKS, Petrow-SadowskiC, BertoletteDC, HuangY, RuscettiFW (2005) Heparan sulfate proteoglycans mediate attachment and entry of human T-cell leukemia virus type 1 virions into CD4+ T cells. J Virol 79 : 12692–12702.
27. FritschRD, ShenX, SimsGP, HathcockKS, HodesRJ, et al. (2005) Stepwise differentiation of CD4 memory T cells defined by expression of CCR7 and CD27. J Immunol 175 : 6489–6497.
28. MaCS, HodgkinPD, TangyeSG (2004) Automatic generation of lymphocyte heterogeneity: Division-dependent changes in the expression of CD27, CCR7 and CD45 by activated human naive CD4+ T cells are independently regulated. Immunol Cell Biol 82 : 67–74.
29. WilsonMK, McWhirterSM, AminRH, HuangD, SchlisselMS (2010) Abelson virus transformation prevents TRAIL expression by inhibiting FoxO3a and NF-kappaB. Mol Cells 29 : 333–341.
30. BellonM, BaydounHH, YaoY, NicotC (2010) HTLV-I Tax-dependent and -independent events associated with immortalization of human primary T lymphocytes. Blood 115 : 2441–2448.
31. BoxusM, TwizereJC, LegrosS, DewulfJF, KettmannR, et al. (2008) The HTLV-1 Tax interactome. Retrovirology 5 : 76.
32. SaitoK, SaitoM, TaniuraN, OkuwaT, OharaY (2010) Activation of the PI3K-Akt pathway by human T cell leukemia virus type 1 (HTLV-1) oncoprotein Tax increases Bcl3 expression, which is associated with enhanced growth of HTLV-1-infected T cells. Virology 403 : 173–180.
33. HiscottJ, NguyenTL, ArguelloM, NakhaeiP, PazS (2006) Manipulation of the nuclear factor-kappaB pathway and the innate immune response by viruses. Oncogene 25 : 6844–6867.
34. OliereS, HernandezE, LezinA, ArguelloM, DouvilleR, et al. (2010) HTLV-1 evades type I interferon antiviral signaling by inducing the suppressor of cytokine signaling 1 (SOCS1). PLoS Pathog 6: e1001177.
35. TwizereJC, SpringaelJY, BoxusM, BurnyA, DequiedtF, et al. (2007) Human T-cell leukemia virus type-1 Tax oncoprotein regulates G-protein signaling. Blood 109 : 1051–1060.
36. CharvetC, AlbertiI, LucianoF, JacquelA, BernardA, et al. (2003) Proteolytic regulation of Forkhead transcription factor FOXO3a by caspase-3-like proteases. Oncogene 22 : 4557–4568.
37. KashanchiF, BradyJN (2005) Transcriptional and post-transcriptional gene regulation of HTLV-1. Oncogene 24 : 5938–5951.
38. OliereS, DouvilleR, SzeA, BelgnaouiSM, HiscottJ (2011) Modulation of innate immune responses during human T-cell leukemia virus (HTLV-1) pathogenesis. Cytokine Growth Factor Rev 22 : 197–210.
39. CalnanDR, BrunetA (2008) The FoxO code. Oncogene 27 : 2276–2288.
40. HironakaN, MochidaK, MoriN, MaedaM, YamamotoN, et al. (2004) Tax-independent constitutive IkappaB kinase activation in adult T-cell leukemia cells. Neoplasia 6 : 266–278.
41. NakahataS, IchikawaT, ManeesaayP, SaitoY, NagaiK, et al. (2014) Loss of NDRG2 expression activates PI3K-AKT signalling via PTEN phosphorylation in ATLL and other cancers. Nat Commun 5 : 3393.
42. SaitoM, MatsuzakiT, SatouY, YasunagaJ, SaitoK, et al. (2009) In vivo expression of the HBZ gene of HTLV-1 correlates with proviral load, inflammatory markers and disease severity in HTLV-1 associated myelopathy/tropical spastic paraparesis (HAM/TSP). Retrovirology 6 : 19.
43. LiM, KesicM, YinH, YuL, GreenPL (2009) Kinetic analysis of human T-cell leukemia virus type 1 gene expression in cell culture and infected animals. J Virol 83 : 3788–3797.
44. van GrevenyngheJ, CubasRA, NotoA, DaFonsecaS, HeZ, et al. (2011) Loss of memory B cells during chronic HIV infection is driven by Foxo3a - and TRAIL-mediated apoptosis. J Clin Invest 121 : 3877–3888.
45. van GrevenyngheJ, HalwaniR, ChomontN, AncutaP, PeretzY, et al. (2008) Lymph node architecture collapse and consequent modulation of FOXO3a pathway on memory T - and B-cells during HIV infection. Semin Immunol 20 : 196–203.
46. KinoT, De MartinoMU, CharmandariE, IchijoT, OutasT, et al. (2005) HIV-1 accessory protein Vpr inhibits the effect of insulin on the Foxo subfamily of forkhead transcription factors by interfering with their binding to 14-3-3 proteins: potential clinical implications regarding the insulin resistance of HIV-1-infected patients. Diabetes 54 : 23–31.
47. CuiM, HuangY, ZhaoY, ZhengJ (2008) Transcription factor FOXO3a mediates apoptosis in HIV-1-infected macrophages. J Immunol 180 : 898–906.
48. GoncalvesDU, ProiettiFA, Barbosa-StancioliEF, MartinsML, RibasJG, et al. (2008) HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP) inflammatory network. Inflamm Allergy Drug Targets 7 : 98–107.
49. TattermuschS, SkinnerJA, ChaussabelD, BanchereauJ, BerryMP, et al. (2012) Systems biology approaches reveal a specific interferon-inducible signature in HTLV-1 associated myelopathy. PLoS Pathog 8: e1002480.
50. FukushimaN, NishiuraY, NakamuraT, KohnoS, EguchiK (2007) Blockade of IL-2 receptor suppresses HTLV-I and IFN-gamma expression in patients with HTLV-I-associated myelopathy/tropical spastic paraparesis. Intern Med 46 : 347–351.
51. MarcaisA, SuarezF, SibonD, BazarbachiA, HermineO (2012) Clinical trials of adult T-cell leukaemia/lymphoma treatment. Leuk Res Treatment 2012 : 932175.
52. MarcaisA, SuarezF, SibonD, FrenzelL, HermineO, et al. (2013) Therapeutic options for adult T-cell leukemia/lymphoma. Curr Oncol Rep 15 : 457–464.
53. WhiteY, YoshimitsuM, KozakoT, MatsushitaK, KoriyamaC, et al. (2013) Effects of exogenous interleukin-7 on CD8(+) T-cell survival and function in human T-cell lymphotropic virus type 1 infection. Leuk Lymphoma 54 : 2243–2250.
54. PalSK, ReckampK, YuH, FiglinRA (2010) Akt inhibitors in clinical development for the treatment of cancer. Expert Opin Investig Drugs 19 : 1355–1366.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 12
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Plasma Membrane-Located Purine Nucleotide Transport Proteins Are Key Components for Host Exploitation by Microsporidian Intracellular Parasites
- Rubella Virus: First Calcium-Requiring Viral Fusion Protein
- Emergence of MERS-CoV in the Middle East: Origins, Transmission, Treatment, and Perspectives
- Neutral Sphingomyelinase in Physiological and Measles Virus Induced T Cell Suppression
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy