
A Negative Feedback Modulator of Antigen Processing Evolved from a Frameshift in the Cowpox Virus Genome
Virus-infected or malignant transformed cells are eliminated by cytotoxic T lymphocytes, which recognize antigenic peptide epitopes in complex with major histocompatibility complex class I (MHC I) molecules at the cell surface. The majority of such peptides are derived from proteasomal degradation in the cytosol and are then translocated into the ER lumen in an energy-consuming reaction via the transporter associated with antigen processing (TAP), which delivers the peptides onto MHC I molecules as final acceptors. Viruses have evolved sophisticated strategies to escape this immune surveillance. Here we show that the cowpox viral protein CPXV012 inhibits the ER peptide translocation machinery by allosterically blocking ATP binding and hydrolysis by TAP. The short ER resident active domain of the viral protein evolved from a reading frame shift in the cowpox virus genome and exploits the ER-lumenal negative feedback peptide sensor of TAP. This CPXV012-induced conformational arrest of TAP is signaled by a unique communication across the ER membrane to the cytosolic motor domains of the peptide pump. Furthermore, this study provides the rare opportunity to decipher on a molecular level how nature plays hide and seek with a pathogen and its host.
Published in the journal:
. PLoS Pathog 10(12): e32767. doi:10.1371/journal.ppat.1004554
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004554
Summary
Virus-infected or malignant transformed cells are eliminated by cytotoxic T lymphocytes, which recognize antigenic peptide epitopes in complex with major histocompatibility complex class I (MHC I) molecules at the cell surface. The majority of such peptides are derived from proteasomal degradation in the cytosol and are then translocated into the ER lumen in an energy-consuming reaction via the transporter associated with antigen processing (TAP), which delivers the peptides onto MHC I molecules as final acceptors. Viruses have evolved sophisticated strategies to escape this immune surveillance. Here we show that the cowpox viral protein CPXV012 inhibits the ER peptide translocation machinery by allosterically blocking ATP binding and hydrolysis by TAP. The short ER resident active domain of the viral protein evolved from a reading frame shift in the cowpox virus genome and exploits the ER-lumenal negative feedback peptide sensor of TAP. This CPXV012-induced conformational arrest of TAP is signaled by a unique communication across the ER membrane to the cytosolic motor domains of the peptide pump. Furthermore, this study provides the rare opportunity to decipher on a molecular level how nature plays hide and seek with a pathogen and its host.
Introduction
Coexistence of pathogens and their hosts represents a masterpiece of evolution, which relies on a fine-tuned balance between pathogen replication and clearance of pathogens by the host immune system [1]. To escape immune surveillance, viruses have developed sophisticated strategies [2], [3]. For example, Herpes simplex viruses and varicella-zoster virus establish latency in trigeminal and dorsal root ganglia, which express only low levels of major histocompatibility complex class I (MHC I) molecules [2], [4], [5]. Exploitation refers e.g. to memory T-cells that circulate through the body and hence provide excellent vehicles for virus dissemination during primary simian varicella virus infection [6]. Sabotage is mediated e.g. by a C-type lectin-like gene product of cytomegalovirus that functions as a decoy ligand to subvert missing-self recognition by natural killer cells (NK), thereby circumventing the elimination of the virus infected cell [5], [7].
Recognizing virus-specific epitopes displayed on MHC I at the cell surface is the essential step in priming and execution of an adaptive immune response against infection. These antigenic peptide epitopes are derived from degradation of the cellular proteome, including virus or tumor associated gene products, via the ubiquitin-proteasomal pathway. The generated peptides are translocated into the ER lumen by the transporter associated with antigen processing (TAP) and subsequently loaded onto MHC I molecules [5], [8]. This heterodimeric ATP-binding cassette (ABC) transport complex is composed of two transmembrane domains (TMDs) and two cytosolic nucleotide-binding domains (NBDs), which couple the chemical energy of ATP binding and hydrolysis to the peptide translocation across the ER membrane [9]. TAP is the central component of the peptide-loading complex (PLC), composed of TAP1/2, tapasin (Tsn), ERp57, calreticulin, and MHC I. After guiding antigenic peptides to MHC I molecules [5], [10], peptide-loaded MHC I complexes dissociate from the PLC and traffic via the secretory pathway to the cell surface, where their antigenic cargo is inspected by cytotoxic T lymphocytes (CTLs). TAP can be dissected into the coreTAP complex, which has been shown to be essential and sufficient for peptide translocation, and extra N-terminal transmembrane domains (TMD0), which bind Tsn and are essential for the assembly of the PLC [11]–[14].
CPXV012 encoded by cowpox viruses (CPXV) has been identified to inhibit antigen processing, extending the types of viruses beyond the Herpesviridae family that interfere with the delivery of antigenic peptides into the ER lumen [15], [16]. CPXV012 encodes an ER-resident type II membrane protein [15] of 69 amino acids, harboring a signal anchor sequence and a short C-terminal region in the ER lumen. CPXV012 associates with the PLC and prevents peptide loading onto MHC I molecules by inhibiting peptide translocation into the ER lumen [15], [16]. Notably, CPXV012 prevents CD8+ T-cell effector responses by inhibiting peptide translocation into the ER [15], [16].
CPXV belongs to Orthopoxvirus (OPV) genus of the Poxviridae family that also includes clinically relevant pathogens such as the variola virus, which causes smallpox. In contrast to herpesviruses, poxviruses are acute viruses that use a “kiss-and-run” strategy of propagation among host cells. CPXV can infect and replicate in the cells of many different mammalian species, including humans. In Europe and parts of Asia, endemic CPXV is the most common cause of human OPV infections [17]. The increased number of CPXV infections indicates that the abandonment of the smallpox vaccination in 1977 may render the population more vulnerable to CPXV. This emerging zoonotic hazard for humans raises public health concerns. CPXV infections of healthy humans are generally self-limiting and cause only localized skin lesions [18]. However, severe CPXV infections with lethal outcome have been reported in immune compromised and eczematous patients, particularly children [19], [20].
Given the medical relevance, it is of particular interest to understand the molecular basis of how CPXV mediates immune evasion. Here we identified the coreTAP complex as the direct target for CPXV012, and revealed how antigen translocation is mechanistically blocked. We further provide an explanation on how this poxviral factor has evolved its evasion potential to escape surveillance by the adaptive immune system.
Results
CPXV012 inhibits MHC I antigen presentation by directly targeting TAP
We first examined whether CPXV012 variants affect the antigen presentation monitored by MHC I surface expression. As shown by flow cytometry, both Flag-tagged flagCPXV012 and C8-tagged C8CPXV012 caused a down-regulation of MHC I surface expression in HeLa cells (Fig. 1A and S1 Figure). The expression of other known immune evasins, such as ICP47 of herpes simplex virus type 1 (HSV-1) that binds to the cytosolic face of the TAP complex and blocks peptide binding [21]–[23], the human cytomegalovirus (HCMV) type I membrane glycoprotein US6 that inhibits ATP binding by TAP via its ER-lumenal domain [24], [25], and the tail-anchored Epstein-Barr virus (EBV) protein BNLF2a that prevents peptide and ATP binding to TAP [26]–[29] had an even stronger effect on MHC I processing (Fig. 1B and S1 Figure). Other than herpesviridae that are known to express highly potent TAP inhibitors, CPXV might rely on a synergistic cooperation between CPXV012 and CPXV203, which blocks MHC I trafficking [30], to efficiently down-regulate MHC I expression at the cell surface.

CPXV012 has been reported to associate with the murine PLC without disrupting its overall assembly [16]; however, the direct interaction partner within this macromolecular complex has not been identified so far. We therefore expressed flagCPXV012 or C8CPXV012 in insect cells, which provide the key advantage that any combination of PLC subunits can be expressed in a cellular background that lacks all components of the PLC [11]. As shown by coimmunoprecipitation using TAP1, TAP2, or flag specific antibodies, CPXV012 associated directly with the TAP complex (Fig. 1C). Similar results were obtained for C8-tagged C8CPXV012 (see below). The interaction with TAP is specific, because CPXV012 did not coprecipitate with TAPL (Fig. 1D), a lysosomal peptide translocation complex, which shares approx. 40% amino acid sequence identity with TAP1 or TAP2 [31]. Recent studies proposed that CPXV012 interacts with Tsn, thereby impairing Tsn-mediated peptide binding to MHC I [15], [16]. Notably, Tsn was coprecipitated with CPXV012 only in the presence of the TAP complex. These results suggest that CPXV012 does not directly interact with Tsn (Fig. 1E), but is bound to the PLC via its direct interaction with TAP (Fig. 1F). Moreover, the presence of CPXV012 does not block the TAP-Tsn interaction.
CPXV012 blocks antigen translocation by coreTAP after peptide binding
CPXV012 inhibits the TAP-mediated peptide translocation into the ER lumen in the absence of other components of the PLC (Fig. 2A). As reported previously, a more pronounced inhibitory effect was observed for the herpesviral inhibitors UL49.5 [16] and EBV-BNLF2a. In contrast to BNLF2a, which blocks both, peptide binding and translocation [27]–[29], [32], CPXV012 did not interfere with peptide binding to TAP (Fig. 2B). Upon peptide binding, rearrangements within the TMDs of TAP induce a conformational change of the NBDs, which can be monitored by cross-linking using the amine-specific homobifunctional reagent EGS [24], [33]. Hence, we examined the influence of CPXV012 on the peptide-induced conformational change by EGS cross-linking. In the presence of peptides, cross-linking of the TAP1/2 heterodimer was detected at ∼160 kDa (Fig. 2C). In the presence of EBV-BNLF2a, the peptide-induced cross-linking of the TAP heterodimer is abolished [29]. In contrast, CPXV012 had no effect on the peptide-induced conformational change monitored by cross-linking of TAP1 and TAP2.

As the coreTAP complex is essential and sufficient for peptide binding and translocation [11], we also examined whether CPXV012 can block this minimal functional unit of antigen translocation. Interestingly, CPXV012 inhibited peptide translocation via coreTAP in a similar way as the full TAP complex (S2A Figure). Consistent with the data obtained for full-length TAP, CPXV012 did not interfere with peptide binding to coreTAP (S2B Figure). As demonstrated by coimmunoprecipitation using TAP1 and TAP2 specific antibodies, CPXV012 binds specifically to coreTAP (Fig. 2D). Taken together, our data demonstrate that CPXV012 interferes with antigen processing by blocking the coreTAP complex at a state after peptide binding and a peptide-induced conformational change.
CPXV012 inhibits ATP binding by TAP1 and TAP2
To shed further light on the inhibition mechanism of CPXV012, we examined whether the TAP1/2•CPXV012 complex can bind ATP. TAP1/2 or TAP1/2•C8CPXV012 complexes were affinity purified and subsequently photo cross-linked with 8-azido-ATP[γ]biotin. Cross-linked proteins were analyzed by immunoblotting using anti-biotin extravidin-HRP. A single biotinylated band at 70-kDa was observed in the absence of C8CPXV012, consistent with azido-ATP photo labeling of TAP1 or TAP2 (Fig. 3A). The photo labeling of TAP was specific, because it was blocked by an access of unlabeled ATP. Strikingly, ATP cross-linking was inhibited at the TAP1/2•CPXV012 complex. These results demonstrate that CPXV012 inhibits ATP binding or the rate of nucleotide exchange by TAP.

Notably, the TAP complex harbors a canonical ATP-binding site II and a non-canonical ATP-binding site I. However, the functional role of these non-equivalent sites is not completely understood [9]. Hence, we investigated whether CPXV012 affects ATP binding to both sites. To distinguish between TAP1 (71 kDa) and TAP2 (72 kDa) in SDS-PAGE, the N-terminus of TAP2 was fused to tapasin (Tsn) via a flexible linker of 34 amino acids (TsnTAP2). It is worth mentioning that the TAP1/TsnTAP2 transport complex is fully functional in peptide translocation and MHC I peptide loading [14]. Moreover, expression of flagCPXV012 inhibited the peptide transport of TsnTAP2/TAP1 (S3 Figure). Subsequently, we examined the effect of CPXV012 on 8-azido-ATP photo cross-linking of the TAP1/TsnTAP2 complex. In the absence of CPXV012, TAP1 and TAP2 were specifically photo-labeled by 8-azido-ATP (Fig. 3B). In contrast, the TAP•flagCPXV012 complex showed a strongly reduced ATP photo labeling of TAP1 and TAP2. Taken together, these results suggest that CPXV012 inhibits ATP binding or the rate of nucleotide exchange of TAP1 and TAP2.
US6 and BNLF2a prevent the formation of TAP•CPXV012 complexes
We next investigated whether CPXV012 might recognize a similar TAP conformation as the immune evasin BNLF2a or US6, which target specific, yet different conformations of the antigen translocation complex [29]. After coexpression of TAP1, TAP2, flagCPXV012, and either US6myc (Fig. 4A and B) or BNLF2aC8 (Fig. 4A and C), coimmunoprecipitations were performed using antibodies against flagCPXV012 (anti-flag), US6myc (anti-myc), or BNLF2aC8 (anti-C8), respectively. Strikingly, expression of either US6 or BNLF2a prohibits binding of CPXV012 to TAP, while TAP1/2 was affinity purified with either US6myc or BNLF2aC8. Moreover, if flagCPXV012 was immunoprecipitated, almost no TAP was found to be associated with the cowpox viral factor, while TAP1/2•US6 or TAP1/2•BNLF2a complexes were detected. On the other hand, the formation of TAP1/2•CPXV012 complexes was not impaired in the absence of the viral TAP inhibitors (S4 Figure). Hence, US6 and BNLF2a prevent the formation of TAP1/2•CPXV012 complexes.
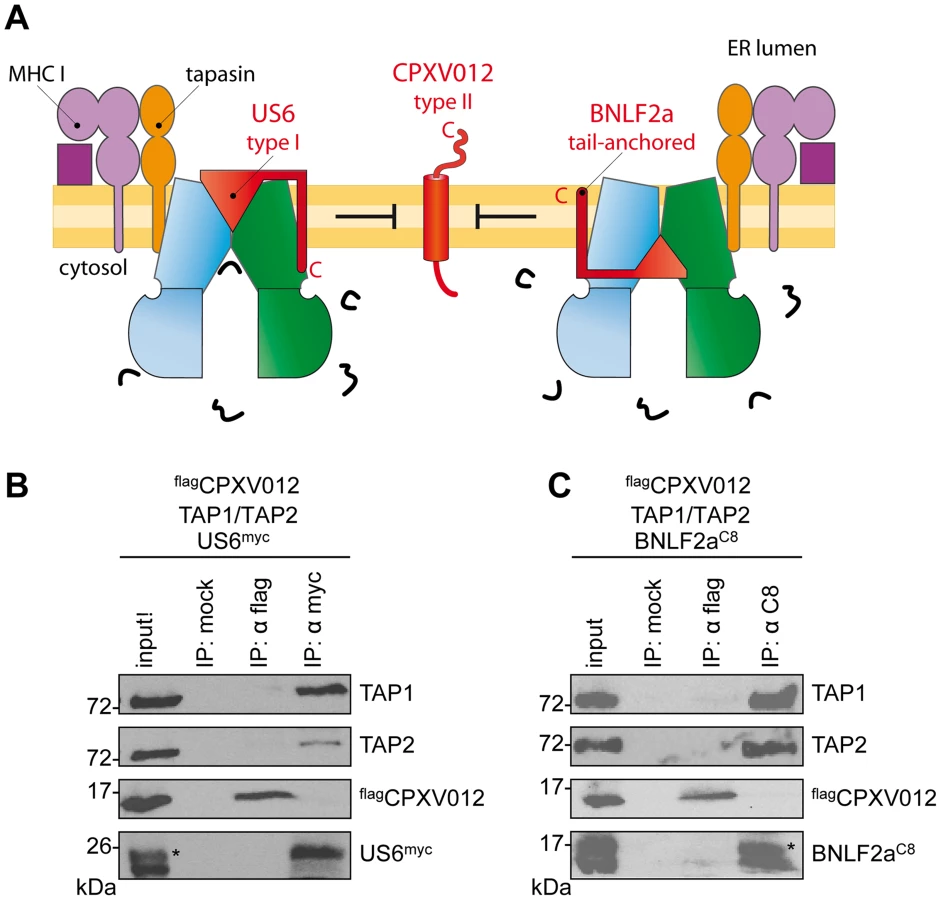
A genomic frameshift causes TAP inhibition
A phylogenetic comparison revealed a substantial genotypic and phenotypic diversity among the twelve CPXV strains sequenced so far [34]. CPXV012 used in this study is derived from the reference strain “Brighton Red” (BR, Supplemental Table 1). Notably, all CPXV012 orthologs share 45 N-terminal residues, which include the cytosolic region of residue 1–12 and the transmembrane helix (aa 13–35) of the type II membrane protein (Fig. 5A). In contrast, the C-terminal regions are divergent and the CPXX012 orthologs cluster in two main groups. The first group, represented by strain GRI90 or GER91, contains proteins harboring an ER-lumenal domain of 50 to 100 residues, which encodes a C-type lectin-like domain (CTLD). These proteins do not suppress MHC I antigen processing [15]. In contrast, the ER-lumenal domains of CPXV012 belonging to the “Brighton Red” subgroup are significantly shorter and lack similarity to any known domain.

Why are the C termini of both subgroups distinct to each other? Surprisingly, if analyzed on the DNA level, all CPXV012 ORFs turned out to be very similar (S5 Figure). The ORFs of CPXV012 are localized in the terminal genomic region, where many non-essential genes are clustered that encode for proteins affecting virulence or interaction with the host immune system [35], [36]. Unlike the central region of the Orthopoxvirus genome, which is conserved and encodes proteins for virus replication, the terminal genomic regions are diverse and are more likely prone to DNA rearrangements or mutations. Indeed, the “Brighton Red” CPXV012 ORF differs by a deletion of five A-T pairs, thus causing a frameshift. The alternative reading frame starts right after the TMD and ends with a stop codon after 25 residues. Since these last C-terminal residues are unique, we hypothesize that this region may be functionally important. Hence, we generated CPXV012 variants lacking five or six amino acids at their C or N terminus (C8CPXV012-CΔ5 or C8NΔ6-CPXV012, respectively). C8NΔ6-CPXV012 suppressed the MHC I surface expression of HeLa cells to the same extent as full-length C8CPXV012, whereas C8CPXV012-CΔ5 was inactive (Fig. 5B and C), even though almost equivalent amounts of each CPXV012 variant were expressed (Fig. 5D). This lack of function was not due to an impaired interaction with TAP, because C8CPXV012-CΔ5 can still interact with heterodimeric TAP1/2 complexes (Fig. 5E). Hence, the transmembrane region of CPXV012 is likely responsible for the interaction with TAP. Interestingly, the CPXV012 variant D10L, which contains a C-type lectin domain at its C-terminus and can therefore not inhibit TAP [15], interacts with the transporter (S6 Figure). This is due to the fact that both proteins share a high conserved N-terminal region including the transmembrane domain. In conclusion, the transmembrane domain mediates binding of the viral inhibitor to the TAP complex, whereas the last five C-terminal residues of CPXV012 are crucial for TAP inhibition.
CPXV012 exploits a trans-inhibition mechanism
Because this active C-terminal domain of CPXV012 is located within the ER lumen, these results further imply that CPXV012 inhibits ATP binding by TAP via an allosteric crosstalk across the ER membrane. Interestingly, the C terminus of CPXV012 exhibits all hallmarks of a high-affinity TAP substrate [37]: Two positive residues (Arg59 and Arg60), which correspond to positions one and two of a 11mer substrate peptide that ends with the hydrophobic Ile69 (S7 Figure). Strikingly, replacement of Arg59 and Arg60 by alanine (C8CPXV012RR59AA) diminished the ability of the viral inhibitor to suppress MHC I surface expression, because the peptide recognition motif of TAP within the C terminus of CPXV012 was abrogated (S7 Figure). Since ATP hydrolysis of TAP is tightly coupled to substrate binding and translocation [38], we assessed how the frameshifted C-terminal region of CPXV012 affects the peptide-stimulated ATPase activity of purified coreTAP complexes. Peptides representing the last 5, 10, 15, 20 or 25 residues of CPXV012 were synthesized with an acetylated N terminus (Fig. 6A). This modification is known to dramatically decrease the affinity for TAP avoiding that these CPXV012 fragments act as “classical” TAP substrates [37], [39]. The 25, 20, 15, and 10mer fragments significantly inhibited the peptide-stimulated ATP hydrolysis (Fig. 6B). The IC50 value for TAP inhibition by the 10mer CPXV012 fragment was 72±20 µM (Fig. 6C). This suggests that the C-terminal region of CPXV012 mimics a TAP substrate just before its release into the ER lumen, which induces the functional arrest of TAP by inhibiting ATP binding to the transport complex.

The direct proof of a novel trans-inhibition mechanism of CPXV012 was obtained by functional reconstitution of purified components into liposomes. After reconstitution, approximately half of the TAP complex face their NBD to the outside [40] and hence were exclusively energized by external ATP, thereby assuring uni-directional translocation of fluorescently labeled peptides into the lumen of the proteoliposomes [41]. Peptide transport was inhibited by competition with excess of unlabeled peptides as expected, but not by a 100-fold excess of the C-terminal CPXV012 fragment (Fig. 6D). This demonstrates that the active domain of CPXV012 is unable to block TAP from the cytosol. In contrast, encapsulation of the 10mer CPXV012 fragment into the proteoliposomes abolished peptide translocation by the TAP complex almost completely. Thus, the C-terminal fragment of CPXV012 blocks antigen translocation in trans. Since the tethered C-terminal peptide imitates a high-affinity peptide substrate, it is tempting to speculate that CPXV012 mechanistically exploits a novel trans-inhibition mechanism of the antigen processing machinery.
Discussion
Virus spread is accomplished through subversion of antiviral host immunity by an arsenal of factors that target key molecular steps necessary to mount an immune response [42]. In particular, gene products encoded by Herpesviridae are known to attack multiple steps in MHC I antigen presentation, including the generation of peptides by the ubiquitin-proteasome pathway, translocation of peptides into the ER by TAP, and the mistargeting of MHC I [3], [43]. The poxvirus gene product CPXV012 was classified as an inhibitor of antigen delivery to MHC I [15], [16]. However, the direct target, the inhibition mechanism, and the evolutionary origin remained illusive.
By expressing CPXV012 in mammalian and insect cells, we establish that CPXV012 directly inhibits the coreTAP complex. Notably, CPXV012 interferes with ATP binding to TAP1 and TAP2 and hence peptide induced ATP hydrolysis via an allosteric crosstalk across the ER membrane. This is mediated by the C-terminal region of CPXV012 located in the ER lumen, at the opposite membrane side of the NBDs (Fig. 6E). Similar to US6 encoded by HCMV, CPXV012 does not influence peptide binding, but inhibits ATP binding to the TAP complex. However, besides using a related strategy, our results demonstrate that the inhibition mechanism of CPXV012 is unique and distinct from US6: US6 inhibits ATP binding to TAP1 [24], whereas CPXV012 blocks ATP binding or nucleotide exchange of both TAP subunits. In addition, CPXV012 acts as a small peptide, whereas the active region of US6 consists of a folded ER-lumenal domain of 125 residues. In conclusion, CPXV012 and US6 prevent ATP binding to TAP via two distinct mechanisms, which presents a striking example of convergent evolution. Since BNLF2a and US6, which target distinct conformation states of TAP [29], prevent CPXV012 binding to TAP, it is tempting to speculate that CPXV012 transiently intercepts the translocation cycle at a state, which is not accessible if TAP is arrested by either US6 or BNLF2a. However, we cannot exclude entirely that binding of either US6 or BNLF2a to TAP mask directly the CPXV012 interaction site on TAP due to steric competition.
In this study, we provide several lines of evidence that the active domain of the immune evasin is located at the C-terminal tip of CPXV012, which is tethered to the ER membrane and inhibits the coreTAP complex. First, a C-terminally deletion CPXV012-CΔ5 is inactive in blocking MHC I antigen processing, whereas an N-terminal deletion is active to the same extent as full-length CPXV012. Second, the ten C-terminal residues of CPXV012 are sufficient to inhibit the peptide-stimulated ATPase activity of coreTAP. Notably, inhibition by the 10mer fragment is compartment-specific, because it only arrests TAP from the lumen, not from the cytosol. This active C-terminal segment of CPXV012, which emerged from a frameshift in the CPXV012 gene, exhibits all hallmarks of a high-affinity TAP substrate: positively charged or aromatic residues at position 1–3 and a hydrophobic residue at its C terminus [37]. Disturbing this consensus sequence by replacing the N-terminal positively charged residues (Arg59 and Arg60) by an alanine (C8CPXV012RR59AA) rendered the viral inhibitor inactive. If combined, these results are in line with a recently discovered negative feedback mechanism of TAP (trans-inhibition) [41]: substrate peptides are transported against the gradient into the lumen of TAP-containing proteoliposomes until the transporter is inhibited in trans by accumulated lumenal peptides.
In vivo, a significant proportion of TAP-translocated peptides does not associate with MHC I molecules, since they are more selective in peptide binding, if compared to TAP [44]. Accumulation of non-optimal MHC I substrates may compete with newly transported peptides for MHC I loading. Moreover, high concentrations of ER-lumenal peptides might even induce the unfolded protein response (UPR) and ER stress as a direct consequence of peptide accumulation, or indirectly as a result of competition between peptides and proteins for folding, assembly and retrograde translocation [44]. There is growing evidence that the UPR intersects with the functioning of cells of the immune system [45]. For instance, ER stress induces a decrease of MHC I surface expression [46] and a reduction in TAP1 protein levels, which is caused by the expression of microRNA-346 that directly targets human TAP1 mRNA [47]. In summary, it seems reasonable to assume that a trans-inhibition mechanism of TAP, as identified recently [41], would prevent detrimental accumulation of peptides in the ER lumen and hence ER stress. In addition, trans-inhibition might synergistically cooperate with pathways that export TAP-imported peptides from the ER lumen [48], [49]. Since the C terminus of CPXV012 resembles a high-affinity TAP peptide substrate that is sufficient to inhibit the transporter as soluble lumenal peptide, we propose that CPXV012 exploits this trans-inhibition mechanism of the TAP machinery. The active C-terminal fragment of CPXV012 is membrane-anchored and bound to TAP via its TMD, thereby mimicking a high ER lumenal peptide concentration. This high local concentration may trigger the ER-lumenal low-affinity [41] peptide binding site of TAP, causing the inhibition of the transport complex in trans (Fig. 6E).
CPXV is the suggested ancestor of other orthopoxviruses and contains the most complete set of immune modulators [35]. They target host pathways that regulate immediate immune responses, particularly the IFNs, the chemokines, the pro-inflammatory cytokines, the complement system, and mediators that orchestrate adaptive immunity [50]. With regard to this repertoire of mechanisms, two immune evasins of CPXV, namely CPXV012 and CPXV203, are known to inhibit synergistically the MHC I antigen presentation pathway: As shown here, CPXV012 diminishes the peptide supply in the ER by exploiting a novel trans-inhibition mechanism, whereas CPXV203 interferes with trafficking of MHC I from the ER to the plasma membrane by reprogramming KDEL-receptor recycling [15], [16], [51]–[53].
Down-regulation of MHC I antigen presentation by CPXV012 and CPXV203 does not inhibit the priming of CPXV-specific CD8+ T lymphocytes (CTLs) during acute infection, but blocks these CTLs at the effector response level by preventing recognition of infected cells in vivo [54]. Since direct priming should be also inhibited by CPXV-induced MHC I down-regulation, these results supported a model in which cross-presentation is used for CD8+ T cell priming during CPXV infection [54]. On the other hand, MHC I down-regulation would also leave infected cells vulnerable to be killed by NK cells. Usually, mature NK cells are tolerated toward normal cells by integrating stimulatory and inhibitory signals transmitted via receptor-ligand interactions [55]. According to the “missing-self” hypothesis [56], NK cells recognize and eliminate cells that fail to express MHC I molecules. Hence, it is not surprising that orthopoxviruses encode immune evasins that inhibit NK-cell activation. For example, CPXV expresses the orthopoxvirus MHC class I-like protein OMCP [57], a high-affinity antagonist of the activating NK-receptor NKG2D [58]. An opposite strategy is the expression of decoy ligands for the NK cell inhibitory receptor NKR-P1B, which plays a critical role in the MHC I independent “missing self” recognition by detecting the C-type lectin-related ligand Clr-b [59], [60]. Indeed, rat cytomegalovirus expresses a C-type lectin-like (RCTL) gene product that resembles Clr-b, which inhibits NK killing of infected cells via direct interaction with NKR-P1B [7]. Interestingly, in a subgroup of CPXV strains, CPXV012 codes for D10L that contains the C-type lectin domain at the C terminus (Fig. 5A). D10L is highly homologous to Clr-b, but it is beyond the scope of this manuscript to determine whether D10L indeed functions as a decoy ligand for NKR-P1B. Nevertheless, D10L binds to the TAP complex, since the N-terminal cytosolic and transmembrane region of D10L and CPXV012 are highly conserved. The physiological relevance of this interaction remains to be determined. More importantly, D10L lacks the C-terminal alternative reading frame, which mimics a TAP substrate consensus sequence, and is therefore unable to inhibit MHC I surface presentation [15]. In combination with the fact that the transmembrane region of all CPXV012 variants mediate the interaction with TAP, their differences at the C terminus explains on a molecular level how a gene product adapts “incidentally” a new function, thereby crossing the frontier of sabotage and stealth.
While this manuscript was undergoing final revision, a report by Luteijn et al. was published that also describes data relevant to the viral immune evasions strategy of CPXV012 [61]. Consistent with our findings, it was shown that CPXV012 inhibits ATP binding, but not peptide binding to TAP. Substitutions of long polyalanine stretches within the ER-luminal region indicate that residues 41–65 of the viral protein are involved in TAP inhibition [61]. However, these experiments could not distinguish whether the loss-of-function is either caused because these long alanine stretches replace functionally important amino acids within the CPXV012 functional domain, or induce secondary effects, which then render the protein inactive. Here, we developed an opposite experimental strategy based on maintaining CPXV012 function. We show that the isolated C-terminal 10mer CPXV012 fragment is sufficient to inhibit ATPase activity of purified TAP. Moreover, by reconstitution of the inhibition pathway of CPXV012 by purified components, we provide direct evidence that this fragment is only active if provided in trans, on the ER-lumenal side of TAP. Based on these results, we conclude that the stretch of ten residues at the C terminus of CPXV012 is the active domain of the viral protein, which is necessary and sufficient to inhibit TAP. Interestingly, the ER-lumenal domain of CPXV012 displays a strong affinity for phospholipids and might reside at the lipid-water interface of the ER membrane in close proximity to the TMDs of TAP [61]. Since it is currently completely unknown where and how peptides are released form TAP inside the ER lumen, it is plausible that the low-affinity ER-lumenal peptide sensor [41], which initiates trans-inhibition, is located at the membrane-TMD interface of the translocation machinery.
Several key findings are disclosed by the present study. First, we decipher the molecular basis of how CPXV012 inhibits antigen presentation via MHC I by direct inhibition of ATP binding to coreTAP. Second, we show that the inhibition mechanism is distinct of all other yet identified viral TAP inhibitors. Thus, CPXV012 is an excellent tool to study the structure and function of TAP as well as new antigen processing pathways. Third, we propose that CPXV012 blocks TAP by hijacking the newly identified negative feedback mechanism of the antigen translocation machinery. Therefore, our findings provide an important tool to uncover the yet uncharacterized negative feedback regulation of TAP. Finally, by elucidating the functional evolution of the viral immune evasive protein CPXV012, this study provides the rare opportunity to decipher on a molecular level how nature plays hide and seek with a pathogen and its host.
Materials and Methods
Cloning, constructs and recombinant viruses
CPXV012 (Gene ID: 1485887) with an N-terminal His10-flag tag (flagCPXV012) was codon optimized for expression in mammalian and insect cells and synthesized de novo (GenScript, Piscataway, NJ, USA). flagCPXV012 was cloned into pFastBac1 (Invitrogen, Carlsbad, CA, USA) vectors for the baculovirus production or was used as a template for PCR amplification. PCR reactions were performed under standard conditions using Phusion DNA polymerase (Finnzymes, Vantaa, Finland) and synthetic oligonucleotide primers (endonuclease cleavage sites are underlined). All constructs were verified by DNA sequencing. C8CPXV012 was generated using C8-CPVX012-BamHI-fo (GCGGATCC ATGCCGCGCGGCCCGGATCGCCCGGAAfGGCATTGAAGAATTCATCATGAGAGAGTCTATC), and CPXV012-EcoRI-rev (GCGAATTCTTAGATGATGCTATCCAGC) primers and was subcloned into pFASTBac1 (Invitrogen). Recombinant baculoviruses were generated using the Bac-to-Bac baculovirus expression system (Invitrogen).
For expression in human cells, PCR-generated products were cloned into pIRES2-EGFP (Clontech Laboratories Inc., Palo Alto, CA, USA) via the respective restriction sites upstream of the internal ribosome entry site (IRES) and enhanced GFP. C8CPXV012, flagCPXV012 (also containing an N-terminal His10-tag), and C8CPXV012-CΔ5 were amplified with the following primers: C8-CPXV012-NheI-fo (AATGCTAGCACCATGGCA CCGCGCGGCCCGGATCGCC), Flag-CPXV012-NheI-fo (AATGCTAGCACCATGGCACACCAT CATCACCATCATCACCAC), CPXV012-full-length-BamHI-rev (TTAGGATCCTCAGATGATG CTATCCAGCTTGTGGTAGTAC), and CPXV012-CΔ5-BamHI-rev (TTAGGATCCTCACTTGTG GTAGTACCTGCGGAACAG), respectively. C8NΔ6-CPXV012 was generated by three sequential PCR reactions using the overlapping forward primers CPXV012-NΔ6-PCR1-fo (CGGAAGGCATTGAAGAAAGCGGATCTATCTACCGTGTGATGATCG), CPXV012-PCR2-fo (CCGCGCGGCCCGGATCGCCCGGAAGGCATTGAAGAAAGCGGA), and C8-CPXV012-NheI-fo, and the reverse primer CPXV012-full-length-BamHI-rev. Residues Arg59 and Arg60 of CPXV012 were simultaneously replaced by two alanines (C8CPXV012RR59AA) via site-directed mutagenesis of the template C8CPXV012-pIRES2-EGFP using the primers CPXV012-R59AR60A-fo (GTCCCTGCTGTTCGCGGCATACTACCACAAGCTG) and CPXV012-R59A/R60A-rev (CAGCTTGTGGTAGTATGCCGCGAACAGCAGGGAC). D10L (Gene ID: 90660243) with an N-terminal C8 tag (C8D10L) was synthesized de novo and cloned into pIRES2-EGFP using NheI and BamHI restriction sites. BNLF2aC8-NST-pIRES2-EGFP has been described previously [29]. Human coreTAP1mVenus-His10 (TAP1 residues 227-808; Q03518) and coreTAP2mCerulean-StrepII (TAP2 residues 125-704; Q59H06) have been described previously [62].
Cell lines, infection and transfection
Spodoptera frugiperda (Sf9) insect cells were grown in Sf900II medium (Invitrogen) following standard procedures. Sf9 cells were infected with recombinant baculovirus encoding for flagCPXV012, C8CPXV012, US6myc [29], BNLF2aC8-NST [29], codon-optimized human TAP1/2 [63], and human wild-type TAP1His6/TAP2 [64], coreTAP1/2 [11] respectively. 48 h after infection, cells were harvested, and membranes were prepared as described [64]. HeLa and HEK293T cells (ATCC CRL-11268) were cultured in DMEM (PAA Laboratories, Cölbe, Germany) supplemented with 10% fetal calf serum (FCS; Biochrom AG, Berlin, Germany) at 37°C in a 5% CO2-humidified atmosphere. For flow cytometry, HeLa cells (ATCC CCL-2) were seeded in 6-well plates with a density of 2×105 cells/well the day before transfection. Transient transfection was conducted according to the Magnetofection protocol (Chemicell, Berlin, Germany) using 2 µg of plasmid DNA and 2 µl of PolyMAG (Chemicell) per well. For the tandem-affinity purification of TAP complexes, HEK293T cells were seeded in 150 mm dishes with a density of 2×106 cells/dish. On the next day, cells were transfected using 90 µg of polyethyleneimine (Sigma-Aldrich, Taufkirchen, Germany) and 30 µg of plasmid DNA according to standard procedures.
Antibodies and peptides
Monoclonal mouse anti-TAP1 (mAb 148.3) and anti-TAP2 (mAb 435.3) antibodies were used for immunoblotting and immunoprecipitation [64]. In certain experiments, polyclonal rabbit anti-TAP1 (1p2) and anti-TAP2 (2p4) antibodies were used for immunoprecipitation [65]. C8-tagged CPXV012 and BNLF2a were immunoprecipitated and detected by an anti-C8 antibody [66]. Flag-tagged CPXV012 was immunoprecipitated and detected by anti-flag antibodies purchased from Sigma-Aldrich (F1804). US6myc was immunoprecipitated and detected by a monoclonal anti-myc antibody (4A6) from Millipore (Schwalbach, Germany). The HC10 antibody, which recognizes human MHC I heavy chain [67], was kindly provided by Hidde L. Ploegh (MIT, Cambridge, MA). Peptide-loaded MHC I molecules were detected by phycoerythrin (PE) coupled anti-human HLA-ABC (W6/32). Mouse IgG2a isotype control antibodies were purchased from BioLegend (San Diego, CA, USA). Actin was detected by a mouse anti-actin antibody purchased from Sigma-Aldrich (A2228). Peptides were prepared by solid-phase synthesis using the Fmoc/tBu strategy. Peptides were purified by reverse-phase C18 HPLC, and their identity was confirmed by mass spectrometry. Peptides representing the last 25, 20, 15, 10, or 5 residues of CPXV012 were acetylated at their N termini.
Flow cytometry
Transiently transfected HeLa cells were harvested 48 h after transfection and washed once with cold FACS buffer (PBS/2% FCS). After blocking with 5% BSA in FACS buffer on ice, cells were washed twice and incubated with W6/32-PE or the PE-coupled isotype control antibody for 15 min on ice in the dark. Cells were again washed twice and FACS analysis was performed using an Attune Acoustic Focusing Cytometer (Applied Biosystems, Life Technologies, Carlsbad, CA, USA). Data were analyzed with FlowJo software (TreeStar, Ashland, OR, USA). For evaluation, a FSC/SSC gate was drawn around the main cell population and only GFP-positive cells were analyzed with respect to MHC I surface expression. To confirm protein expression, parallel samples of transfected HeLa cells were solubilized in RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific, Pierce, Rockford, IL, USA) and analyzed by SDS-PAGE (12%) and subsequent immunoblotting with anti-C8 antibodies.
Immunoprecipitation and immunoblotting
Membranes prepared from insect cells (1 mg of total protein) were resuspended in 0.4 ml lysis buffer L1 (20 mM Tris/HCl, pH 7.5, 150 mM NaCl, 5 mM MgCl2, 2% (w/v) digitonin (Carl Roth, Karlsruhe), 1.5 mM phenylmethylsulfonylfluoride, 3 mM benzamidine). After solubilization for 60 min on ice, non-solubilized proteins were removed by centrifugation at 100,000× g for 30 min at 4°C. The supernatant was incubated with M-280 sheep anti-mouse IgG Dynabeads (Dynal Biotech, Hamburg), which had been pre-loaded with antibodies. Beads were washed three times with 1 ml of washing buffer (20 mM Tris/HCl, pH 7.5, 150 mM NaCl, 2 mM EDTA, 0.2% (w/v) digitonin). Proteins were eluted in 1× SDS sample buffer (2% SDS, 50 mM Tris/HCl, pH 8.0, 200 mM DTT, 10% glycerol, 0.05% bromophenol blue) for 3 min at 65°C. Samples were denatured for 20 min at 65°C and separated by SDS-PAGE (12% or 6%). After electro transfer onto nitrocellulose membranes, proteins were detected with specific antibodies as indicated. Horseradish peroxidase (HP) conjugated secondary antibodies were detected with Lumi-Imager F1 (Roche). One representative blot out of three independent experiments is shown.
Peptide transport
Membranes prepared from insect cells (0.1 mg of total protein) were resuspended in 50 µl of AP buffer (5 mM MgCl2 in PBS, pH 7.4) in the presence of 3 mM ATP. The transport reaction was started by adding 1 µM of the peptide RRYQNSTC(F)L (C(F), fluorescein-labeled cysteine) for 3 min at 32°C and terminated with 1 ml ice-cold stop buffer (10 mM EDTA in PBS, pH 7.0). After centrifugation at 20,000× g for 8 min, the pellet was solubilized in 0.5 ml of lysis buffer L2 (50 mM Tris/HCl pH 7.5, 150 mM NaCl, 5 mM KCl, 1 mM CaCl2, 1 mM MnCl2, 1% Nonidet P-40) for 30 min at 4°C. Non-solubilized proteins were removed by centrifugation and the supernatant was incubated with 60 µl ConA Sepharose (50% w/v, Sigma-Aldrich) for 1 h at 4°C. After three washing steps with 0.5 ml lysis buffer each, ConA-bound peptides were specifically eluted with methyl-α-D-mannopyranoside (200 mM) and quantified by a fluorescence plate reader (λex/em 485/520 nm; Polarstar Galaxy, BMG Labtech, Offenburg, Germany). Background transport was determined in the presence of apyrase (1 U/sample). All measurements were performed in triplicate.
Peptide binding
Membranes prepared from insect cells (0.1 mg of total protein) were incubated with 0.5 µM of the peptide RRYC(F)KSTEL (C(F), fluorescein-labeled cysteine) in 50 µl of AP buffer for 15 min at 4°C. Free peptides were removed by washing the membranes twice with 1 ml of ice-cold AP buffer and subsequent centrifugation at 20,000× g for 8 min. Membranes were lysed with 0.3 ml AP buffer containing 1% SDS, and peptides were quantified by a fluorescence plate reader. All measurements were performed in triplicate. Background binding was determined in 100-fold excess of unlabeled high-affinity substrate peptide R9LQK (RRYQKSTEL).
Chemical cross-linking
Membranes prepared from insect cells (0.5 mg of total protein) were resuspended in 100 µl of ice-cold PBS buffer and incubated with the homobifunctional cross-linker ethylene glycol bis(succinimidyl succinate) (EGS, Thermo Scientific, Rockford, IL) at a final concentration of 0.5 mM. After incubation for 30 min at 4°C, the reaction was stopped by adding Tris/HCl buffer, pH 7.5 (50 mM final concentration). Membranes were collected by centrifugation (20,000× g for 8 min at 4°C) and analyzed by SDS-PAGE (6%) and immunoblotting using TAP1 or TAP2 specific antibodies. As specifically indicated, membranes were pre-incubated with 10 µM peptide R9LQK at 4°C for 1 h prior to cross-linking. One representative blot out of three independent experiments is shown.
8-Azido-ATP photo cross-linking
TAP1/2 complexes were purified from Sf9 membranes (600 µg of total protein) by immunoprecipitation using anti-TAP1 (mAb 148.3) or anti-C8 antibodies as described above. Dynabead immobilized complexes were pre-incubated in ATP binding buffer (20 mM HEPES, pH 7.4, 137 mM NaCl, 3 mM MgCl2) containing 15 µM of 8-azido-ATP[γ]biotin (Biolog Life Science Institute, Bremen, Germany) for 5 min on ice. Controls contained 15 µM 8-azido-ATP[γ]biotin and 5 mM unlabeled ATP. Photo cross-linking was initiated by UV irradiation (254 nm hand-held UV lamp) for 5 min on ice. After three washing steps in ATP-binding buffer, samples were denatured for 20 min at 65°C and separated by SDS-PAGE (12%). After electro transfer onto nitrocellulose membranes, proteins were detected with specific antibodies as indicated. Biotinylated proteins were visualized using an extravidin-HRP conjugate. Alternatively, membranes prepared from insect cells (600 µg of total protein) were resuspended in 150 µl ATP binding buffer and pre-incubated with 15 µM of 8-azido-ATP[γ]biotin in the presence and absence of an excess of ATP (5 mM) for 5 min on ice. After subsequent photo cross-linking for 5 min on ice, membranes were collected by sedimentation (20,000× g for 8 min), washed three times in ATP binding buffer, and then solubilized in 300 µl lysis buffer L1. Immunoprecipitations were performed using antibodies against either TAP1 (mAb 148.3) or the C8 tag as described above. Samples were analyzed by SDS-PAGE (12%) and subsequent immunoblotting with extravidin-HRP or the corresponding antibodies. One representative blot out of three independent experiments is shown.
Isolation of TAP complexes by tandem-affinity purification
Since overexpressed TAP1 or TAP2 subunits might form homodimers, a orthogonal purification strategy was applied that ensures the isolation of heterodimeric TAP1/2 complexes [62]. TAP1/2-expressing HEK293T cells were resuspended in ice-cold buffer A (50 mM Tris/HCl, pH 8.0, 250 mM NaCl, 10% glycerol, 0.05% (w/v) digitonin) supplemented with 1% (w/v) digitonin and incubated for 1 h in an overhead shaker. All steps were carried out at 4°C. Non-solubilized proteins were removed by centrifugation at 150,000× g for 1 h at 4°C. Proteins were bound to 100 µl streptavidin agarose beads (Pierce, Rockford, IL, USA) for 1 h. The beads were washed two times with 1 ml buffer A. Proteins were eluted with 1 ml buffer A supplemented with 2.5 mM biotin. The streptavidin agarose eluate was incubated for 1 h with IgG Dynabeads, which had been pre-loaded with anti-TAP1 antibodies (mAb 148.3). After three washing steps, the affinity-purified protein was eluted with 2× SDS buffer for 30 min at 37°C and analyzed by SDS-PAGE and immunoblotting.
Cell-free translation in rabbit reticulocyte lysate
To prepare coreTAP-containing insect ER microsomes, Sf9 cells were infected with recombinant baculovirus encoding for coreTAP1/2 [11]. 48 h after infection, cells were harvested, and membranes were prepared as described [68]. For the generation of mRNAs, C8CPXV012 or C8D10L was amplified directly from the corresponding pIRES2-EGFP plasmid using SP6-pIRES-fo (GATTTAGGTGACACTATAGAATACCACCGTCTATA TAAGCAGAGCTGGTTTAGTGAACC) and pIRES-GFP-rev (GTTTACGTCGCCGTCCAGC) primers. Purified PCR product was transcribed in vitro using SP6 RNA polymerase as before [29]. In vitro translation of purified mRNA (typically 50 µl reactions, 30°C, 40 min) was performed in the presence of rabbit reticulocyte lysate (Promega, Madison, WI, USA), Sf9 microsomes, and [35S]Met (0.4 µCi/µl). After translation, samples were analyzed by immunoprecipitation and phosphoimaging.
ATP hydrolysis activity of purified TAP
Peptide stimulated ATPase activity was determined by a colorimetric assay based on the complex formation of free inorganic phosphate and ammonium molybdate with malachite green [69]. CoreTAP1mVenus-His10 and coreTAP2mCerulean-StrepII were expressed in Pichia pastoris and solubilized in 2% (w/v) digitonin as described previously [40], [62]. CoreTAP1/2 heterodimers were purified by an orthogonal strategy using metal-affinity chromatography and subsequent streptactin-affinity chromatography as described [62]. For ATP hydrolysis measurements, 0.2 µM of purified coreTAP1/2 was incubated with 1 mM MgATP and the high-affinity substrate peptide R9LQK (1.0 µM) for 30 min at 37°C as described previously [63].
Peptide translocation into proteoliposomes
For reconstitution of purified coreTAP1/2 into liposomes, large unilamellar vesicles consisting of Escherichia coli polar lipid extract (Avanti Polar Lipids Inc., Alabaster, AL, USA) and 1,2-dioleoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids, Inc.) with a molar ratio of 7∶3 were prepared as previously [63]. Detergent-destabilized vesicles and purified coreTAP1/2 were mixed with a lipid to protein ratio of 20∶1 (w/w) and incubated for 30 min at 4°C. Detergent was removed by incubation with polystyrene beads as described previously [40]. Proteoliposomes were pelleted for 30 min at 80,000 rpm at 4°C. Protein aggregates, empty vesicles and proteoliposomes were separated by centrifugation on a continuous Ficoll density gradient [40]. After washing, proteoliposomes were resuspended to a final concentration of 5 mg/ml in reaction buffer (20 mM HEPES, 200 mM NaCl, 50 mM KCl, 5% glycerol, pH 7.3). For peptide transport, coreTAP1/2 (0.5 µg) containing proteoliposomes were pre-incubated on ice in 45 µl reaction buffer containing 1 µM fluorescence labeled peptide RRYC(F)KSTEL. Transport was started by adding 3 mM MgATP and performed for 10 min at 37°C. The reaction was stopped as described previously [63]. For encapsulation of peptides, TAP proteoliposomes in reaction buffer containing the active CPXV012 fragment were snap frozen in liquid nitrogen and thawed on ice. After three freeze and thaw cycles, proteoliposomes were washed twice to remove the remaining, not encapsulated peptide. Peptide transport assay was performed as described above.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. VossenMT, WesterhoutEM, Soderberg-NauclerC, WiertzEJ (2002) Viral immune evasion: a masterpiece of evolution. Immunogenetics 54 : 527–542.
2. BrodskyFM (1999) Stealth, sabotage and exploitation. Immunol Rev 168 : 5–11.
3. HansenTH, BouvierM (2009) MHC class I antigen presentation: learning from viral evasion strategies. Nat Rev Immunol 9 : 503–513.
4. AbendrothA, ArvinA (1999) Varicella-zoster virus immune evasion. Immunol Rev 168 : 143–156.
5. BlumJS, WearschPA, CresswellP (2013) Pathways of antigen processing. Annu Rev Immunol 31 : 443–473.
6. OuwendijkWJ, MahalingamR, de SwartRL, HaagmansBL, van AmerongenG, et al. (2013) T-Cell tropism of simian varicella virus during primary infection. PLoS Pathog 9: e1003368.
7. VoigtS, MesciA, EttingerJ, FineJH, ChenP, et al. (2007) Cytomegalovirus evasion of innate immunity by subversion of the NKR-P1B:Clr-b missing-self axis. Immunity 26 : 617–627.
8. NeefjesJJ, HämmerlingGJ, MomburgF (1993) Folding and assembly of major histocompatibility complex class I heterodimers in the endoplasmic reticulum of intact cells precedes the binding of peptide. J Exp Med 178 : 1971–1980.
9. ParcejD, TampéR (2010) ABC proteins in antigen translocation and viral inhibition. Nat Chem Biol 6 : 572–580.
10. HulpkeS, TampéR (2013) The MHC I loading complex: a multitasking machinery in adaptive immunity. Trends Biochem Sci 38 : 412–420.
11. KochJ, GuntrumR, HeintkeS, KyritsisC, TampéR (2004) Functional dissection of the transmembrane domains of the transporter associated with antigen processing (TAP). J Biol Chem 279 : 10142–10147.
12. ProckoE, RaghuramanG, WileyDC, RaghavanM, GaudetR (2005) Identification of domain boundaries within the N-termini of TAP1 and TAP2 and their importance in tapasin binding and tapasin-mediated increase in peptide loading of MHC class I. Immunol Cell Biol 83 : 475–482.
13. HulpkeS, TomiokaM, KremmerE, UedaK, AbeleR, et al. (2012) Direct evidence that the N-terminal extensions of the TAP complex act as autonomous interaction scaffolds for the assembly of the MHC I peptide-loading complex. Cell Mol Life Sci 69 : 3317–3327.
14. HulpkeS, BaldaufC, TampéR (2012) Molecular architecture of the MHC I peptide-loading complex: one tapasin molecule is essential and sufficient for antigen processing. FASEB J 26 : 5071–5080.
15. AlzhanovaD, EdwardsDM, HammarlundE, ScholzIG, HorstD, et al. (2009) Cowpox virus inhibits the transporter associated with antigen processing to evade T cell recognition. Cell Host Microbe 6 : 433–445.
16. ByunM, VerweijMC, PickupDJ, WiertzEJ, HansenTH, et al. (2009) Two mechanistically distinct immune evasion proteins of cowpox virus combine to avoid antiviral CD8 T cells. Cell Host Microbe 6 : 422–432.
17. VorouRM, PapavassiliouVG, PierroutsakosIN (2008) Cowpox virus infection: an emerging health threat. Curr Opin Infect Dis 21 : 153–156.
18. BourquainD, DabrowskiPW, NitscheA (2013) Comparison of host cell gene expression in cowpox, monkeypox or vaccinia virus-infected cells reveals virus-specific regulation of immune response genes. Virol J 10 : 61.
19. CzernyCP, Eis-HubingerAM, MayrA, SchneweisKE, PfeiffB (1991) Animal poxviruses transmitted from cat to man: current event with lethal end. Zentralbl Veterinarmed B 38 : 421–431.
20. PelkonenPM, TarvainenK, HynninenA, KallioER, HenttonenK, et al. (2003) Cowpox with severe generalized eruption, Finland. Emerg Infect Dis 9 : 1458–1461.
21. AhnK, MeyerTH, UebelS, SempéP, DjaballahH, et al. (1996) Molecular mechanism and species specificity of TAP inhibition by herpes simplex virus ICP47. EMBO J 15 : 3247–3255.
22. TomazinR, HillAB, JugovicP, YorkI, van EndertP, et al. (1996) Stable binding of the herpes simplex virus ICP47 protein to the peptide binding site of TAP. EMBO J 15 : 3256–3266.
23. AisenbreyC, SizunC, KochJ, HergetM, AbeleR, et al. (2006) Structure and dynamics of membrane-associated ICP47, a viral inhibitor of the MHC I antigen-processing machinery. J Biol Chem 281 : 30365–30372.
24. HewittEW, GuptaSS, LehnerPJ (2001) The human cytomegalovirus gene product US6 inhibits ATP binding by TAP. EMBO J 20 : 387–396.
25. KyritsisC, GorbulevS, HutschenreiterS, PawlitschkoK, AbeleR, et al. (2001) Molecular mechanism and structural aspects of transporter associated with antigen processing inhibition by the cytomegalovirus protein US6. J Biol Chem 276 : 48031–48039.
26. RessingME, KeatingSE, van LeeuwenD, Koppers-LalicD, PappworthIY, et al. (2005) Impaired transporter associated with antigen processing-dependent peptide transport during productive EBV infection. J Immunol 174 : 6829–6838.
27. HorstD, van LeeuwenD, CroftNP, GarstkaMA, HislopAD, et al. (2009) Specific targeting of the EBV lytic phase protein BNLF2a to the transporter associated with antigen processing results in impairment of HLA class I-restricted antigen presentation. J Immunol 182 : 2313–2324.
28. CroftNP, Shannon-LoweC, BellAI, HorstD, KremmerE, et al. (2009) Stage-specific inhibition of MHC class I presentation by the Epstein-Barr virus BNLF2a protein during virus lytic cycle. PLoS Pathog 5: e1000490.
29. WyciskAI, LinJ, LochS, HobohmK, FunkeJ, et al. (2011) Epstein-Barr viral BNLF2a protein hijacks the tail-anchored protein insertion machinery to block antigen processing by the transport complex TAP. J Biol Chem 286 : 41402–41412.
30. McCoyWHt, WangX, YokoyamaWM, HansenTH, FremontDH (2013) Cowpox virus employs a two-pronged strategy to outflank MHCI antigen presentation. Mol Immunol 55 : 156–158.
31. DemirelO, JanI, WoltersD, BlanzJ, SaftigP, et al. (2012) The lysosomal polypeptide transporter TAPL is stabilized by interaction with LAMP-1 and LAMP-2. J Cell Sci 125 : 4230–4240.
32. HorstD, FavaloroV, VilardiF, van LeeuwenHC, GarstkaMA, et al. (2011) EBV protein BNLF2a exploits host tail-anchored protein integration machinery to inhibit TAP. J Immunol 186 : 3594–3605.
33. LacailleVG, AndrolewiczMJ (1998) Herpes simplex virus inhibitor ICP47 destabilizes the transporter associated with antigen processing (TAP) heterodimer. J Biol Chem 273 : 17386–17390.
34. CarrollDS, EmersonGL, LiY, SammonsS, OlsonV, et al. (2011) Chasing Jenner's vaccine: revisiting cowpox virus classification. PLoS One 6: e23086.
35. ShchelkunovSN, SafronovPF, TotmeninAV, PetrovNA, RyazankinaOI, et al. (1998) The genomic sequence analysis of the left and right species-specific terminal region of a cowpox virus strain reveals unique sequences and a cluster of intact ORFs for immunomodulatory and host range proteins. Virology 243 : 432–460.
36. GubserC, HueS, KellamP, SmithGL (2004) Poxvirus genomes: a phylogenetic analysis. J Gen Virol 85 : 105–117.
37. UebelS, KraasW, KienleS, WiesmullerKH, JungG, et al. (1997) Recognition principle of the TAP transporter disclosed by combinatorial peptide libraries. Proc Natl Acad Sci U S A 94 : 8976–8981.
38. GorbulevS, AbeleR, TampéR (2001) Allosteric crosstalk between peptide-binding, transport, and ATP hydrolysis of the ABC transporter TAP. Proc Natl Acad Sci U S A 98 : 3732–3737.
39. SchumacherTN, KantesariaDV, HeemelsMT, Ashton-RickardtPG, ShepherdJC, et al. (1994) Peptide length and sequence specificity of the mouse TAP1/TAP2 translocator. J Exp Med 179 : 533–540.
40. SchölzC, ParcejD, EjsingCS, RobenekH, UrbatschIL, et al. (2011) Specific lipids modulate the transporter associated with antigen processing (TAP). J Biol Chem 286 : 13346–13356.
41. GrossmannN, VakkasogluAS, HulpkeS, AbeleR, GaudetR, et al. (2014) Mechanistic determinants of the directionality and energetics of active export by a heterodimeric ABC transporter. Nat Commun 5 : 5419.
42. RevillezaMJ, WangR, MansJ, HongM, NatarajanK, et al. (2011) How the virus outsmarts the host: function and structure of cytomegalovirus MHC-I-like molecules in the evasion of natural killer cell surveillance. J Biomed Biotechnol 2011 : 724607.
43. RessingME, LuteijnRD, HorstD, WiertzEJ (2013) Viral interference with antigen presentation: trapping TAP. Mol Immunol 55 : 139–142.
44. LehnerPJ, HewittEW, RömischK (2000) Antigen presentation: peptides and proteins scramble for the exit. Curr Biol 10: R839–842.
45. ToddDJ, LeeAH, GlimcherLH (2008) The endoplasmic reticulum stress response in immunity and autoimmunity. Nat Rev Immunol 8 : 663–674.
46. UlianichL, TerrazzanoG, AnnunziatellaM, RuggieroG, BeguinotF, et al. (2011) ER stress impairs MHC Class I surface expression and increases susceptibility of thyroid cells to NK-mediated cytotoxicity. Biochim Biophys Acta 1812 : 431–438.
47. BartoszewskiR, BrewerJW, RabA, CrossmanDK, BartoszewskaS, et al. (2011) The unfolded protein response (UPR)-activated transcription factor X-box-binding protein 1 (XBP1) induces microRNA-346 expression that targets the human antigen peptide transporter 1 (TAP1) mRNA and governs immune regulatory genes. J Biol Chem 286 : 41862–41870.
48. ShepherdJC, SchumacherTN, Ashton-RickardtPG, ImaedaS, PloeghHL, et al. (1993) TAP1-dependent peptide translocation in vitro is ATP dependent and peptide selective. Cell 74 : 577–584.
49. KoopmannJO, AlbringJ, HuterE, BulbucN, SpeeP, et al. (2000) Export of antigenic peptides from the endoplasmic reticulum intersects with retrograde protein translocation through the Sec61p channel. Immunity 13 : 117–127.
50. SeetBT, JohnstonJB, BrunettiCR, BarrettJW, EverettH, et al. (2003) Poxviruses and immune evasion. Annu Rev Immunol 21 : 377–423.
51. ByunM, WangX, PakM, HansenTH, YokoyamaWM (2007) Cowpox virus exploits the endoplasmic reticulum retention pathway to inhibit MHC class I transport to the cell surface. Cell Host Microbe 2 : 306–315.
52. DasguptaA, HammarlundE, SlifkaMK, FruhK (2007) Cowpox virus evades CTL recognition and inhibits the intracellular transport of MHC class I molecules. J Immunol 178 : 1654–1661.
53. McCoyWHt, WangX, YokoyamaWM, HansenTH, FremontDH (2012) Structural mechanism of ER retrieval of MHC class I by cowpox. PLoS Biol 10: e1001432.
54. GaineyMD, RivenbarkJG, ChoH, YangL, YokoyamaWM (2012) Viral MHC class I inhibition evades CD8+ T-cell effector responses in vivo but not CD8+ T-cell priming. Proc Natl Acad Sci U S A 109: E3260–3267.
55. CampbellKS, HasegawaJ (2013) Natural killer cell biology: an update and future directions. J Allergy Clin Immunol 132 : 536–544.
56. LjunggrenHG, KarreK (1990) In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today 11 : 237–244.
57. CampbellJA, TrossmanDS, YokoyamaWM, CarayannopoulosLN (2007) Zoonotic orthopoxviruses encode a high-affinity antagonist of NKG2D. J Exp Med 204 : 1311–1317.
58. RauletDH (2003) Roles of the NKG2D immunoreceptor and its ligands. Nat Rev Immunol 3 : 781–790.
59. IizukaK, NaidenkoOV, PlougastelBF, FremontDH, YokoyamaWM (2003) Genetically linked C-type lectin-related ligands for the NKRP1 family of natural killer cell receptors. Nat Immunol 4 : 801–807.
60. CarlyleJR, JamiesonAM, GasserS, ClinganCS, AraseH, et al. (2004) Missing self-recognition of Ocil/Clr-b by inhibitory NKR-P1 natural killer cell receptors. Proc Natl Acad Sci U S A 101 : 3527–3532.
61. LuteijnRD, HoelenH, KruseE, van LeeuwenWF, GrootensJ, et al. (2014) Cowpox virus protein CPXV012 eludes CTLs by blocking ATP binding to TAP. J Immunol 193 : 1578–1589.
62. ParcejD, GuntrumR, SchmidtS, HinzA, TampéR (2013) Multicolour fluorescence-detection size-exclusion chromatography for structural genomics of membrane multiprotein complexes. PLoS One 8: e67112.
63. HergetM, KreissigN, KolbeC, SchölzC, TampéR, et al. (2009) Purification and reconstitution of the antigen transport complex TAP: a prerequisite for determination of peptide stoichiometry and ATP hydrolysis. J Biol Chem 284 : 33740–33749.
64. MeyerTH, van EndertPM, UebelS, EhringB, TampéR (1994) Functional expression and purification of the ABC transporter complex associated with antigen processing (TAP) in insect cells. FEBS Lett 351 : 443–447.
65. NijenhuisM, HämmerlingGJ (1996) Multiple regions of the transporter associated with antigen processing (TAP) contribute to its peptide binding site. J Immunol 157 : 5467–5477.
66. AbaciogluYH, FoutsTR, LamanJD, ClaassenE, PincusSH, et al. (1994) Epitope mapping and topology of baculovirus-expressed HIV-1 gp160 determined with a panel of murine monoclonal antibodies. AIDS Res Hum Retroviruses 10 : 371–381.
67. StamNJ, SpitsH, PloeghHL (1986) Monoclonal antibodies raised against denatured HLA-B locus heavy chains permit biochemical characterization of certain HLA-C locus products. J Immunol 137 : 2299–2306.
68. SaksenaS, ShaoY, BraunagelSC, SummersMD, JohnsonAE (2004) Cotranslational integration and initial sorting at the endoplasmic reticulum translocon of proteins destined for the inner nuclear membrane. Proc Natl Acad Sci U S A 101 : 12537–12542.
69. HenkelRD, VandeBergJL, WalshRA (1988) A microassay for ATPase. Anal Biochem 169 : 312–318.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 12
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Plasma Membrane-Located Purine Nucleotide Transport Proteins Are Key Components for Host Exploitation by Microsporidian Intracellular Parasites
- Rubella Virus: First Calcium-Requiring Viral Fusion Protein
- Emergence of MERS-CoV in the Middle East: Origins, Transmission, Treatment, and Perspectives
- Neutral Sphingomyelinase in Physiological and Measles Virus Induced T Cell Suppression
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy