
SV40 Utilizes ATM Kinase Activity to Prevent Non-homologous End Joining of Broken Viral DNA Replication Products
Viruses from both Polyomaviridae and Papillomaviridae families share several characteristics. These include common modes of DNA replication and an accumulation of DNA damage signaling and repair proteins at replicating viral DNA. Several DNA repair proteins, with unknown functions during viral DNA replication, associate with the viral replication centers of the polyomavirus simian virus 40 (SV40). In this study we examined the mechanisms that regulate and recruit DNA repair machinery to replicating viral DNA during permissive SV40 infection. We found that the virus deploys DNA repair to broken viral DNA using cellular DNA damage signaling pathways. Our results shed light on why both Polyomaviridae and Papillomaviridae DNA replication elicits DNA damage signaling and repair. As no effective treatments currently exist for the Polyomaviridae family, our data identify pathways that might be therapeutically targeted to inhibit productive viral replication. Additionally, we categorize distinct functions for DNA repair and damage signaling pathways during viral replication. The results provide insights into how viruses exploit cellular processes to overwhelm the cell and propagate.
Published in the journal:
. PLoS Pathog 10(12): e32767. doi:10.1371/journal.ppat.1004536
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004536
Summary
Viruses from both Polyomaviridae and Papillomaviridae families share several characteristics. These include common modes of DNA replication and an accumulation of DNA damage signaling and repair proteins at replicating viral DNA. Several DNA repair proteins, with unknown functions during viral DNA replication, associate with the viral replication centers of the polyomavirus simian virus 40 (SV40). In this study we examined the mechanisms that regulate and recruit DNA repair machinery to replicating viral DNA during permissive SV40 infection. We found that the virus deploys DNA repair to broken viral DNA using cellular DNA damage signaling pathways. Our results shed light on why both Polyomaviridae and Papillomaviridae DNA replication elicits DNA damage signaling and repair. As no effective treatments currently exist for the Polyomaviridae family, our data identify pathways that might be therapeutically targeted to inhibit productive viral replication. Additionally, we categorize distinct functions for DNA repair and damage signaling pathways during viral replication. The results provide insights into how viruses exploit cellular processes to overwhelm the cell and propagate.
Introduction
A diverse set of protein functions is required to ensure the timely, accurate duplication of the genome. In addition to the components of the replication machinery itself [1], [2], accurate replication requires the S phase checkpoint kinase, ataxia telangiectasia-mutated and rad3-related (ATR). ATR and its related kinases, ataxia telangiectasia-mutated (ATM) and DNA-protein kinase catalytic subunit (DNA-PKcs), are members of the PI3K-related protein kinase (PIKK) family that regulate DNA damage signaling in response to various endogenous and exogenous stresses [3]. ATR kinase function is primarily activated by DNA replication stress through the capacity of the ATR/ATRIP complex to sense stretches of replication protein A (RPA)-bound single-stranded DNA [4]. ATM and DNA-PKcs function to promote DNA repair and are primarily activated in response to double strand breaks (DSB) [3]. To identify DSBs, ATM and DNA-PKcs rely on MRE11-RAD50-NBS1 (MRN) and KU70/80 (KU), respectively [5]. DNA-PKcs promotes non-homologous end joining (NHEJ) [6]. On the other hand, either ATM - or ATR-dependent phosphorylation events are accompanied by activation and recruitment of numerous factors that influence DNA repair and mediate arrest of both the cell cycle and DNA replication [3].
Several DNA repair proteins are required for the successful completion of cellular DNA replication, particularly those of the homology-directed repair (HDR) pathway. HDR is initiated by MRN recognition of DSB termini [7]. The S phase specific interaction of MRN with CtIP, a processivity factor for the MRE11 nuclease [8], [9], enables the initiation of 5′ to 3′ end resection to create a short 3′ tail. The recessed 5′ end can be subsequently digested by the more processive nucleases EXO1 and the BLM/DNA2 complex [10] to generate a lengthy 3′ tail that can be bound by the RAD51 recombinase to catalyze strand invasion and displacement loop formation [11], [12]. HDR is commonly activated following replication stress or by agents that elicit DSBs in S and G2 phases [13]. Inactivation of HDR factors causes slowed DNA synthesis [14], instability of nascent DNA strands [15], anaphase bridges owing to un-replicated DNA entering mitosis [16], [17], [18], and increased genome breakage in both the presence and absence of replication stress [19], [20], [21]. Several of these characteristics are reminiscent of the defects observed in Seckel syndrome cells, which harbor hypomorphic mutations in the ATR or CtIP genes, providing a possible link between HDR and DNA damage signaling [22], [23], [24], [25]. Thus, both ATR kinase signaling and HDR functions are required during S phase to promote genome stability.
Similar to studies of cellular DNA replication, recent evidence suggests that polyomaviruses and papillomaviruses might utilize a more extensive set of DNA replication factors than previously anticipated [26], [27], [28], [29], [30]. Infection of cells by polyomaviruses and papillomaviruses is accompanied by intense ATM and ATR activation [26], [28], [31], [32], [33]. However, viral DNA replication continues despite DNA damage checkpoint signaling (for review see [34]). ATM and ATR kinase activities facilitate the repair of replication-associated breaks on viral chromatin during SV40 DNA replication and failure to repair these breaks results in the accumulation of concatemers of the SV40 genome through rolling circle replication [30]. On the other hand, DNA-PKcs kinase activity is not required for normal viral replication [30]. SV40 has been used to identify and characterize numerous cellular DNA replication factors that together with the viral origin recognition complex/replicative DNA helicase, large T antigen (Tag), can replicate the viral genome [1], [34]. Thus, DNA damage signaling and repair proteins used by SV40 DNA replication might represent a set of factors employed during normal cellular DNA replication to prevent and/or repair replication-associated DSBs.
Here we show that, in addition to rolling circle formation, inhibition of ATM or ATR causes the accumulation of broken, non-replicating linear viral DNA. We examined which repair proteins co-localize with sites of viral replication during unperturbed infection to define further the cellular DNA repair factors that associate with replicating SV40 chromatin and contribute to viral replication. We find that HDR activities are strongly recruited to Tag foci, but NEHJ factors are not. The selective recruitment of HDR factors and exclusion of NHEJ proteins requires ATM but not ATR kinase activity. Although ATM and ATR contribute to cell cycle arrest in infected cells, the activity of ATM in regulating DSB repair choice is independent of its cell cycle checkpoint function. Our results suggest that while ATM and ATR kinase inhibition results in the accumulation of multiple forms of broken viral DNA, DNA-PKcs function at replicating viral DNA is limited to instances when ATM kinase activity is inhibited. Our results lend further insight into how SV40 manipulates the DNA damage response to promote its replication inside infected cells.
Results
ATM and ATR cooperate to prevent the accumulation of DNA DSBs during SV40 DNA replication
We previously observed that inhibition of either ATM or ATR caused increased accumulation of both broken unidirectional replication forks and head to tail repeats of the viral genome known as concatemers [30]. The topological status of aberrant viral DNA intermediates and products accumulating when ATM or ATR were inhibited was not examined in these studies nor was the potential redundancy between the two kinases established. To examine viral replication products in detail, DNA was extracted at 48 hours post infection (hpi) from cells treated with ATRi (ATR inhibitor VE-821 [35]), ATMi (ATM inhibitor Ku-55933 [36]), both ATRi and ATMi, or the solvent DMSO from 20 to 48 hpi. DNA was digested with XbaI and SacI, enzymes that cleave frequently within genomic DNA but for which recognition sites are absent in the SV40 viral genome. As previously reported, one-dimensional (1D) agarose gel electrophoresis followed by Southern blotting and hybridization with a probe that recognizes the viral genome primarily detected unit length, monomeric viral genomes of form I (supercoiled) and form II (nicked), with very little monomer form III (linear; Figure 1A, lane 1 and 1B). Upon longer exposure, slower migrating species could be detected that correspond to circular intermediates of bidirectional replication (late Cairns structures [37]) and dimeric genomes [38], [39] (Figure 1A, bottom panel).The total amount of viral DNA produced during infection was quantified using a probe for mitochondrial DNA as an internal standard.

Inhibition of either ATM or ATR alone significantly decreased viral DNA yield as compared with DMSO-treated cells, although the effect was more pronounced for ATRi, as previously reported [30] (Figure 1C). As expected from our previous work [30], treatment with either ATMi or ATRi increased the percent of total viral DNA that migrated at an apparent size of ∼20 kb or failed to enter the gel (Figure 1A, D) and significantly decreased the fraction of viral DNA in monomer form (ATMi treatment only; Figure 1B). We have previously demonstrated that the viral DNA that remains in the well consists of concatemers of the viral genome produced by rolling circle replication (30), however the nature of the product running at an apparent size of ∼20 kb was not previously addressed.
To examine functional overlap between ATM and ATR, cells were treated simultaneously with both inhibitors. While this treatment did not significantly decrease the amount of total viral DNA produced compared to ATR inhibition alone (Figure 1C), the production of unit length monomer was synergistically decreased compared to either single treatment (Figure 1A, B). This defect in form I, II, and III monomer production when ATM and ATR were both inhibited was accompanied by a 2 and 2.3 fold increase in the accumulation of concatemers and the ∼20 kb products, respectively, compared to ATM inhibition alone (Figure 1A, D). This result suggests that ATM and ATR kinase activities are partially redundant in their ability to prevent concatemer and other aberrant product formation during SV40 replication.
To gain insight into the topological status of the observed aberrant products, the DNAs described above were subjected to two-dimensional (2D) agarose gel electrophoresis and Southern blotting (Figure 2). This method can resolve circular (form I and form II) and linear (form III) viral DNA replication products from intact or broken viral replication intermediates and large, unbranched linear forms of the viral genome (Figure 2A) [40], [41]. Analysis of the undigested viral DNA from DMSO-treated SV40-infected cells by 2D gel electrophoresis demonstrated that the majority of viral replication intermediates consisted of intact form II theta molecules (Figure 2B), as expected from the divergence of two forks from a single viral origin of replication. The majority of non-replicating viral DNAs were form I or II monomers (Figure 2B). When infected cells were exposed to ATRi, there was a pronounced increase in the amount of linear viral DNA at greater-than-monomer length (compare Figure 2B and C, linear arc). Furthermore, a partial sigma arc (Figure 2A; pink line marked “rolling circle”) was present upon ATRi treatment and was absent from DNA extracted from DMSO-treated cells (Figure 2B, C, lower panel, arrow). Sigma forms are generated from breakage of one replication fork in theta form DNA, followed by fork arrest or unidirectional, rolling circle replication. In ATRi-treated cells, virtually all viral DNA that migrated at ∼20 kb in the first gel dimension (Figure 2C, arrow) migrated as expected for linear molecules in the second dimension. When infected cells were exposed to ATMi, both intense linear and full sigma arcs were present (Figure 2D, arrow points to sigma arc), consistent with high levels of viral DNA breakage. Notably, the spots corresponding to nicked, catenated, and head to tail dimer intermediates were greatly decreased by ATM inhibition relative to DMSO treatment (Figure 2D).

As predicted from the 1D gel analysis, combined inhibition of ATR and ATM increased all broken forms of viral DNA relative to DMSO, ATRi, or ATMi treated cells (Figure 2E, linear and sigma arcs). The prominent sigma arc, corresponding to unidirectional replication, correlated with a great decrease in fork convergence products (Cat I, CatI/II, and Dimer; Figure 2E, lower panel). Additionally, the ∼20 kb product detected in the first dimension (Figure 2E, arrow) again migrated on the linear arc of the 2D gel (Figure 2E). We conclude that the products migrating at an apparent size of ∼20 kb are aberrant linear forms of the viral genome running at the limiting mobility of the gel. These molecules most likely result from breakage of a rolling circle intermediate during SV40 replication. Taken together, these results suggest that ATM and ATR function to prevent DSB accumulation during viral replication and that each kinase is able to partially substitute for the other in this function.
DNA-PKcs contributes to viral replication fork repair when ATM is inhibited
DNA-PKcs is robustly activated when ATM or ATR are inhibited during SV40 DNA replication [30]. Thus, the NHEJ pathway of DSB repair directed by DNA-PKcs might fulfill some aspect of DNA repair upon ATM or ATR inhibition during SV40 DNA replication. To examine this possibility, SV40-infected cells were treated with a combination the DNA-PKcs inhibitor Nu7026 [42], [43] and DMSO, ATMi, or ATRi from 20 to 48 hpi. DNA was extracted at 48 hpi, separated in a single dimension by gel electrophoresis, and subjected to Southern blotting (Figure 1A). DNA-PKcs inhibition alone with Nu7026 had no effect on any aspect of SV40 DNA replication (Figure 1A–D) [30]. Likewise, inhibition of DNA-PKcs in combination with ATR inhibition did not increase the appearance of aberrant products relative to ATR inhibition alone (Figure 1A, D). Indeed, DNA-PKcs inhibition partially rescued the reduction in total viral replication levels observed upon ATR inhibition alone (Figure 1C). We conclude that DNA-PKcs is unlikely to contribute to the repair of DSBs formed when ATR kinase activity is blocked, but may contribute to the inhibition of viral replication observed in the absence of ATR function.
Simultaneous exposure of cells to ATMi and Nu7026 had only a minimal effect on overall levels of viral DNA replication (Figure 1A, C). However, the percentage of monomeric products decreased from 64% when ATM was solely inhibited to 50% when ATM and DNA-PKcs were inhibited (Figure 1A, B). Concomitant with the decrease in total monomer fraction, the relative fraction of concatemeric forms increased 1.7 fold relative to ATM inhibition alone (Figure 1A, D). These data imply that NHEJ orchestrated by DNA-PKcs may facilitate the repair of a portion of DSBs formed when ATM is inhibited. Alternatively, the inhibited DNA-PKcs bound to DSBs that accumulate upon ATM inhibition could create a barrier to DSB repair by preventing access of DSB termini to 5′ to 3′ end resection proteins [44], [45], [46].
Homology-directed repair factors colocalize with viral replication centers
The pronounced increase in viral replication products indicative of unrepaired DSB observed upon inhibition of ATR and/or ATM suggested that normal viral replication may require the function of DSB repair factors. Several such factors with poorly understood functions in SV40-infected cells have been found to concentrate at or near SV40 DNA replication centers [26], [27]. Many of these factors, including MRN, RAD51, FANCD2, and BRCA1 function in HDR [3] and might act to repair broken DNA resulting from viral DNA replication [30]. To define further which DSB repair activities are recruited to or localized near viral DNA replication centers, the localization of chromatin-bound DSB repair proteins relative to Tag foci was examined in SV40-infected BSC40 and U2OS cells using fluorescence microscopy.
CtIP, a protein critical for HDR due to its role in the initiation of 5′ end resection [8], formed intense foci that colocalized with Tag in SV40-infected BSC40 and U2OS cells (Figure 3A, B). Furthermore, the RAD51 loader BRCA2, the single-strand annealing protein RAD52, and the dHJ dissolvase BTR (BLM, RMI1/2, and Topoisomerase IIIα) colocalized on chromatin with viral DNA replication centers in infected BSC40 and U2OS cells (Figure 3A, B). If these proteins are needed for the completion of normal viral replication, the kinetics with which repair factors arrive at the viral replication center should correlate with the time of DNA replication. To address this possibility, the nuclear dynamics of the BTR component BLM were examined during a 60 h time course of SV40 infection in BSC40 cells. The colocalization of BLM with viral DNA replication centers was greatest at times longer than 24 hpi (Figure 3C). This timeframe of BLM colocalization with Tag correlated well with the reported timing of viral DNA replication [47], [48] and with the observed incorporation of EdU at Tag foci (Figure 3C). The colocalization of BLM and Tag is reminiscent of that found for MRN and Tag in SV40-infected cells [26]. These results suggest that localization of HDR to the viral DNA replication centers marked with Tag is a characteristic of SV40-infected cells.

NHEJ proteins do not localize to Tag foci in SV40-infected cells
Similar to HDR protein recruitment to viral replication centers, SV40 infection induces ATM and ATR DNA damage signaling in a time frame that closely mirrors viral DNA replication (Figure 4A). Thus, a related PIKK, DNA-PKcs, might be activated with similar kinetics during SV40 infection. To assess DNA-PKcs kinase activation and the total DNA-PKcs, KU70, and KU80 protein levels during viral infection, steady-state levels of these proteins were determined by immunoblotting in extracts of SV40-infected BSC40 cells harvested over a 60 h time course (Figure 4A, B). These blots revealed that ATR and ATM began to phosphorylate substrates CHK1 and CHK2, respectively, at 24 hpi (Figure 4A). ATM and ATR activities were greatest from 36 through 48 hpi, after which they began to decline (Figure 4A, compare lanes 4, 5 to lane 6). A second marker of ATM activation, NBS1 pS343, displayed similar kinetics of phosphorylation, although it may peak slightly later than CHK2 pT68 (Figure 4A, B). On the other hand, DNA-PKcs kinase activation was observed most strongly at 48 hpi and more weakly at 36 and 60 hpi, as assessed by DNA-PKcs autophosphorylation at S2056 (Figure 4B, compare lane 1 to lanes 4–6) [44], [49], [50], [51], [52]. The total protein levels of KU70, KU80, and DNA-PKcs were stable throughout 60 h of SV40 infection (Figure 4A, B, compare lanes 1–6). Thus, the timing of DNA-PKcs kinase activation during SV40 infection poorly correlates with viral DNA replication.

To determine whether the limited DNA-PKcs kinase activation observed at 48 hpi was associated with localization of the DNA-PK holoenzyme to viral DNA replication centers, chromatin-bound NHEJ proteins KU70, KU80, and DNA-PKcs were examined by fluorescence microscopy at 48 hpi in SV40-infected BSC40 and U2OS cells. DNA-PKcs, KU70, and KU80 showed little preference for binding at or near viral DNA replication centers in both BSC40 and U2OS cells (Figures 4C, D and S1). DNA-PKcs auto-phosphorylated on S2056 was not abundant on the chromatin of SV40-infected cells at 48 hpi and did not associate with viral DNA replication centers (Figures 4D and S1B). Indeed, close inspection of viral replication centers revealed that DNA-PKcs and KU70/80 appeared to be excluded from Tag foci (Figures 4C, D and S1, arrows, enlarged boxes). Line tracings through viral replication centers confirmed that intense KU70/80 and DNA-PKcs foci did not colocalize with Tag (Figures 4C, D and S1). Taken together, these results are consistent with a correlation between ATM, ATR, and HDR activation and the timing of viral DNA replication. In contrast, DNA-PKcs activation correlates poorly with viral replication.
ATM and ATR contribute differentially to S phase arrest of SV40 infected cells
Having characterized the DNA repair factors that localize at viral replication centers during SV40 infection, we were interested in determining whether ATM and ATR are required for that recruitment. However, ATM kinase activity is known to contribute both to cell cycle arrest and to DNA repair efficiency after DNA damage [3], [53]. ATM inhibition increases aberrant SV40 DNA replication products (Figures 1, 2, and see reference [30]) and results in a dispersed, non-focal Tag staining pattern [26], [30], but the relative contribution of different ATM functions to these phenotypes is unclear. We sought to separate these functions of ATM by varying the time period of viral infection during which ATM inhibitor was applied. SV40-infected BSC40 cells were exposed to ATMi during an early period (from 30 minutes prior to infection through 20 hpi), a late period (from 20 to 48 hpi), or for the duration of the infection (Figure 5A) [30]. The early phase corresponds to the cellular G1 to S transition and viral early gene expression while the late phase encompasses the period of viral DNA replication (Figure 5A). SV40-infected cells exposed to the inhibitor solvent, DMSO, served as a positive control for S phase arrest, and uninfected cells were used to determine cell cycle distribution in an asynchronous population of cells (Figure 5A). Cell cycle stage was determined by measuring the presence of CENPF immunostaining and EdU incorporation into DNA (Figure 5B) [54], [55], [56].

As expected, a substantial percentage of mock-infected cells were in G1 and S phase (Figure 5C). Upon SV40 infection in the presence of DMSO, ∼90% of SV40-infected BSC40 cells arrested in S phase, consistent with checkpoint activation (Figure 5C). Conversely, similar percentages of G1 and S phase cells to those observed in asynchronous, mock-infected BSC40 cells were observed when ATM was inhibited for the duration of infection (Figure 5C). The presence of ATMi during the early phase of infection partially relieved the S phase arrest (Figure 5C). In contrast, inhibition of ATM during the late phase of infection only very weakly attenuated the SV40-induced S phase arrest (Figure 5C). Taken together, the results suggest that the ATM-CHK2 pathway is required during the early phase of infection for efficient S phase arrest.
To determine the minimum time of exposure to ATMi required to induce the characteristic non-focal viral replication centers to form, the chromatin-bound Tag staining pattern was monitored by immunofluorescence microscopy from infected cells when ATMi was added to the medium during the final 4, 8, 16, or 24 h of a 48 h infection. DMSO-treated, SV40-infected cells demonstrated the intense Tag foci that normally accompany productive infection, with a smaller percentage of cells showing a weak staining pattern that lacked chromatin-bound Tag (Figure 5D, 0 h ATMi exposure). The addition of ATMi for 4 h prior to the 48 hpi fixation time point caused the appearance of cells showing an aberrant, dispersed Tag staining pattern (23%; Figure 5D). Incubations equal to or greater than 8 h with ATMi increased the number of cells showing this dispersed Tag staining pattern to ∼49% (Figure 5D). During the time period tested by this experiment, the prevalence of the weak staining pattern was unaffected by the presence of ATMi (Figure 5D). This weak pattern may represent cells that failed to arrest in S phase. Since an 8 h ATMi exposure is sufficient to stably produce aberrant Tag immunostaining, while no effect on the infected cell cycle is observed with ATM inhibition during this time period, an 8 h exposure to ATMi was utilized in subsequent experiments.
Similar to ATM, ATR also modulates DNA repair and cell cycle checkpoint control during S phase [3]. To determine the effect of ATR kinase activity on cell cycle arrest in infected cells, SV40-infected BSC40 cells were exposed to ATRi during periods of SV40 infection as shown in Figure S2A, pulsed with EdU, fixed and immunostained for CENPF to determine the phase of the cell cycle. Inhibition of ATR during any phase of infection compromised cell cycle arrest as indicated by an increased percentage of cells in G1 phase and a decreased percentage of cells in S phase (Figure S2B). In contrast to ATMi treatment, ATRi treatment during the last 8 hours of the 48-hour infection did not induce the formation of the dispersed Tag staining pattern. However, the number of cells showing a weak Tag pattern increased from ∼32% in the DMSO-treated infected control to ∼58% after ATR inhibition (Figure S2C). Only ∼14% of cells with the weak Tag staining pattern incorporated EdU, while ∼96% of the ATRi-treated cells with normal Tag staining pattern showed EdU incorporation (Figure S2C). This result aligns with the observation that the presence of ATRi from 40 to 48 hpi decreased the fraction of cells in S phase from 87% in the DMSO control infection to 46% (Figure S2B). Again, we attribute the increase in “weak” Tag staining cells to those that have escaped S phase arrest. Cells that do remain in S phase appear to retain normal morphology of the viral replication center as measured by Tag staining (Figure S2C). We conclude that ATR contributes to S phase arrest of SV40 infected cells throughout infection, but does not affect viral replication center stability in the fraction of cells that remain arrested in S phase.
We then verified that inhibition of ATM or ATR from 40 to 48 hpi produced similar aberrant DNA replication products to those previously reported when ATM or ATR were inhibited for a longer period (from 20 to 48 hpi Figures 1 and 2 and see reference [30]). Whole cell DNA extracted from SV40-infected BSC40 cells exposed to ATRi or ATMi from 40 to 48 hpi was digested with XbaI and SacI, blotted, and probed for the SV40 genome. Similar to results obtained upon longer inhibitor treatment [30], viral DNA concatemer formation increased by 10 and 2.7 fold for ATMi and ATRi, respectively, compared to DMSO-treated infected cells (Figure 6A, left panel).
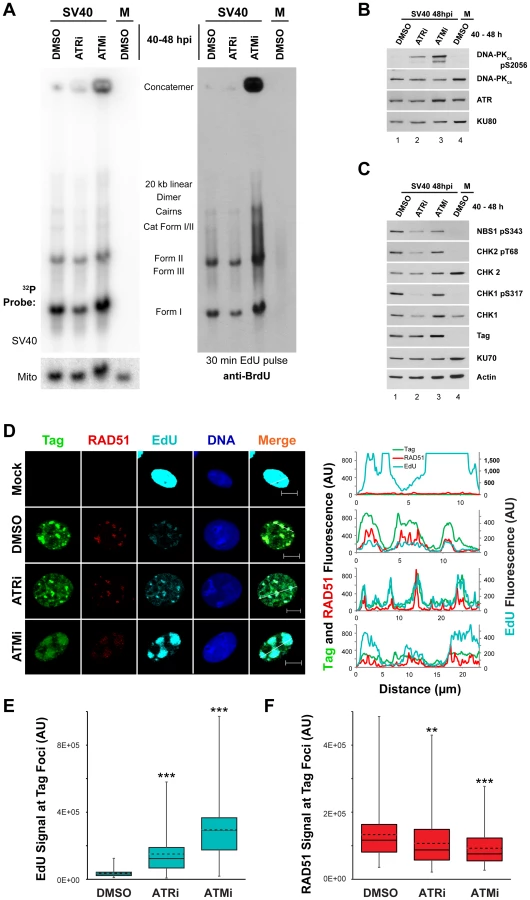
ATM or ATR inhibition late in infection does not affect SV40 DNA replicative capacity in S phase but increases DNA-PK activation
The vigorous activation of ATM and ATR that accompanies viral DNA replication is required to prevent the appearance of aberrant viral DNA replication products that result from replication of broken viral DNA [30]. However, the mechanistic basis of this replication defect is not understood. Since ATM and ATR phosphorylate a plethora of targets including factors that influence HDR and NHEJ [57], [58], [59], we hypothesized that the viral replication defects associated with inactivation of ATM and ATR might correlate with changes in the activation or recruitment of DNA repair factors at the viral replication center. We first used immunoblotting to determine the phosphorylation state of residues on CHK1, CHK2, NBS1, and DNA-PKcs in cell lysates from SV40 - or mock-infected cells exposed from 40 to 48 hpi to DMSO, ATRi, or ATMi. The activation of DNA-PKcs, indicated by autophosphorylation of S2056, was increased upon ATRi or ATMi treatment relative to DMSO-treated SV40-infected cells (Figure 6B, compare lane 1 to lanes 2, 3). The presence of ATRi decreased the phosphorylation of both ATR and ATM substrates (Figure 6C, compare lane 1 to 2). We suggest that the effect of ATRi on phosphorylation of ATM substrates is an indirect consequence of the significantly reduced fraction of cells undergoing viral replication upon ATR inhibition (Figure S2B, C). ATM inhibition only mildly decreased the steady state levels of phosphorylated CHK2 and NBS1 (Figure 6C, compare lane 1 to lane 3). This modest response is not due to ineffectual repression of ATM activity since ATMi effectively blocks the phosphorylation of CHK2 in response to the DSB-inducing radiomimetic compound neocarzinostatin (Figure S3A, compare lane 2, 3 to 4, 5). Furthermore, ATM inhibition from 40 to 48 hpi decreased levels of the hyper-phosphorylated form of CtIP (Figure S3B, compare lane 1 to 2), an ATM and CDK dependent product [59]. Similar to observations in ATM-inhibited or ataxia-telangiectasia cells exposed to DNA damaging agents [60], these results suggest that ATR and DNA-PKcs may cross-phosphorylate substrates of ATM during SV40 infection. Importantly, these results indicate that the brief 40 to 48 h inhibitor exposure is sufficient to produce the DNA-PKcs activation previously observed with longer periods of ATM or ATR inhibition [30].
The accumulation of aberrant replication products observed when ATM or ATR were inhibited (Figure 6A–C) might correlate with a change in the distribution of repair proteins that associate with SV40 DNA replication centers. To examine whether DNA damage signaling during SV40 infection influences the association of DNA repair factors with viral DNA replication centers, the subcellular localization of several HDR and NHEJ proteins was assessed at 48 hpi by immunofluorescence microscopy of SV40 - or mock-infected BSC40 cells exposed to DMSO, ATRi, or ATMi from 40 to 48 hpi. We first verified that treatment with the inhibitors did not interfere with bulk viral DNA replication at Tag foci. Importantly, replication centers are only present in S phase infected cells (Figure S2), and examination of the infected cell population that contains viral replication centers avoids the inclusion of the G1, G2, and M phase cell populations. ATRi-treated cells contain a large portion of non-S phase cells (Figure S2B, C) that decrease total viral DNA replication of ATRi-treated cells relative to DMSO-treated cells (Figures 1 and 6A [30]). Thus to test the general ability of ATRi - or ATMi-treated cells to replicate viral DNA in S phase, we examined the incorporation of EdU at the viral replication center.
As observed previously [27], [30], [61], chromatin-bound RAD51 and EdU incorporated during a 5 minute pulse administered immediately prior to fixation co-localized with Tag at 48 hpi in DMSO-treated, SV40-infected BSC40 cells (Figure 6D). Although the fraction of total cells exhibiting Tag staining decreased upon ATRi treatment (Figure S2C), in those cells containing viral replication foci, the EdU signal associated with Tag was more intense than in DMSO-treated, SV40-infected cells (Figure 6D, E). In contrast, treatment with ATMi led to a dispersion of Tag signal. However, EdU signal at these dispersed Tag foci was actually increased relative to the normal replication centers observed in DMSO-treated infected cells (Figure 6D, E).
To verify that EdU labeling in ATRi or ATMi treated cells was viral, not cellular, DNA replication, DNA was extracted from SV40 - or mock-infected BSC40 cells treated with DMSO, ATRi, or ATMi from 40 to 48 hpi and pulsed with EdU for 5 minutes prior to extraction. DNA was cleaved with XbaI and SacI, blotted, and probed with an antibody that cross-reacts with EdU (Figure 6A, right panel). The EdU-labeled DNA from SV40-infected cells treated with DMSO migrated as a unit-length supercoiled (I) and nicked (II) monomer, converging replication forks (LCI), dimers, and catenated intermediates (Figure 6A, right panel, lane 1), as expected for viral DNA. Replicating cellular DNA from uninfected cells migrated as a weaker smear of signal between 3 and 7 kilobases due to sensitivity of the cellular DNA to XbaI and SacI endonucleases (Figure 6A, right panel, compare lane 1 to 4). EdU-labeled DNA from ATR inhibited cells migrated identically to that observed from the SV40-infected, DMSO-treated cells (Figure 6A, right panel, compare lane 1 to 2). EdU-labeled DNA from ATM-inhibited, SV40-infected BSC40 cells contained all the replication intermediates and products that occur in DMSO-treated SV40-infected cells, but also contained larger products and concatemers (Figure 6A, right panel, compare lanes 1, 4 to lane 3). We conclude that Tag and EdU staining in the presence of ATMi and ATRi mark replicating viral, not cellular, DNA.
RPA is actively loaded onto replicating single-stranded DNA by Tag during SV40 DNA replication [62]. Consistent with ongoing replication, the RPA subunit RPA70 colocalized with Tag when replication foci were observed in ATMi and ATRi-treated SV40-infected cells (Figure S3C). Relative to DMSO-treated SV40-infected cells, RPA70 intensity was unchanged upon ATRi treatment, but was slightly enhanced at the viral replication center when ATM was inhibited (Figure S3D). Independent of DSB formation during cellular DNA replication, single-stranded DNA, produced by helicase unwinding, is also bound by RAD51 [15], [20]. Similar to RPA, RAD51 remained co-localized with Tag regardless of whether ATM or ATR were inhibited (Figure 6D) with a minor, albeit significant, decrease in RAD51 fluorescence signal in both cases (Figure 6F). We conclude that RAD51 and RPA loading onto replicating single-stranded viral DNA are only mildly affected by ATR and ATM kinase activities.
ATM inhibition decreases CtIP and BLM association with viral DNA replication foci
The initiation of 5′ to 3′ end resection, an event required prior to strand invasion during HDR, has been implicated as a process that requires ATR - or ATM-dependent phosphorylations [59]. Therefore, we queried whether the enrichment of NBS1, CtIP, and BLM to SV40 replication centers requires ATM or ATR. The presence of ATRi from 40–48 hpi had no effect on the co-localization of Tag with NBS1, BLM, or CtIP compared to DMSO-treated, SV40-infected cells (Figure 7A, C). Likewise, NBS1 remained associated with Tag foci upon ATM inhibition with slightly decreased fluorescence intensity relative to DMSO - and ATRi-treated SV40-infected cells (Figure 7A, B). However both CtIP and BLM, proteins that function during end resection downstream of initial MRN recruitment [10], no longer colocalized with the majority of Tag when ATM was inhibited (Figure 7C). Additionally, the lack of co-localization with Tag coincided with a large decrease in the total fluorescence intensity of BLM and CtIP (Figure 7D, E). We conclude that ATM, but not ATR, inhibition results in a defect in CtIP and BLM recruitment to the viral replication center.

ATM kinase activity prevents DNA-PKcs activation at the viral replication center
DNA-PKcs auto-phosphorylation is increased by ATM or ATR inhibition during viral DNA replication (Figure 6B and [30]). This change in the activation state of DNA-PKcs could correspond to a difference in the localization of NHEJ factors to viral replication centers. This possibility was tested via immunostaining for chromatin-bound NHEJ components KU70, KU80, and DNA-PKcs in SV40 - or mock - infected BSC40 cells treated with ATRi, ATMi, or DMSO from 40 to 48 hpi prior to fixation. In DMSO-treated SV40-infected BSC40 cells, activated DNA-PKcs (marked by S2056 phosphorylation) and KU70 were excluded from the Tag foci (Figure 8A). Inhibition of ATR from 40–48 hpi had no effect on the localization of KU70 or activated DNA-PKcs (Figure 8A), consistent with the minor effect of ATRi on DNA-PKcs activation (Figure 8B). In contrast, KU70 and activated DNA-PKcs became prominent at the dispersed Tag replication centers following treatment with ATMi from 40–48 hpi (Figure 8A). The fluorescence signal intensity of DNA-PKcs pS2056 was greatly enhanced upon ATMi treatment relative to DMSO or ATRi treatment (Figure 8C).

DNA-PKcs and KU binding to DNA DSBs can be visualized by removing RNA from fixed cells with RNAseA [63]. This technique has been utilized to visualize exact sites of DSB formation in irradiated cells [63]. We employed RNAaseA pre-extraction of RNA-bound and non-chromatin bound protein on SV40-infected BSC40 cells exposed to DMSO, ATRi, or ATMi during the final 8 h of a 48 h SV40 infection. Similar to our previous extraction protocol, further RNAseA extraction of SV40-infected cells exposed to ATRi or DMSO revealed a complete lack of DNA-PKcs and KU80 foci at Tag foci (Figure 8B). In contrast, ATM inhibition in cells infected with SV40 caused strong colocalization of DNA-PKcs and KU80 with Tag foci (Figure 8B), accompanied by an increase in the florescence intensity of DNA-PKcs and KU80 at the viral DNA replication center (Figure 8D, E). Taken together, these results indicate that ATM kinase activity prevents the recruitment of KU and DNA-PK activation at replicating viral DNA in Tag foci.
Discussion
Our data demonstrate that through the action of ATM kinase, SV40 recruits a complex, specific set of DNA repair activities to viral DNA replication centers that likely prevent accumulation of broken viral DNA. Evidence is presented that ATM kinase activity during viral DNA replication modulates the preference for HDR over NHEJ. Supporting the idea that ATR and DNA-PKcs kinase activities aid in the repair of some breaks when ATM is inhibited, we found that inhibition of either ATR or DNA-PK in combination with ATM increased the amount of concatemers formed (Figure 1). However, only ATM and/or ATR inhibition, but not DNA-PKcs inhibition, increased the appearance of long (∼20 kb) linear products (Figures 1 and 2), consistent with a role for ATM and ATR in preventing DSB formation or facilitating efficient DSB repair during SV40 chromatin replication. We demonstrate that SV40 replication centers colocalize with HDR factors (Figure 3) but do not associate with NHEJ proteins (Figures 4 and S1). Importantly, ATM - and ATR-dependent DNA damage signaling are both required for SV40-induced S phase arrest, but ATR kinase inhibition does not affect the stability of viral replication centers (Figures 5 and S2). Furthermore, we present data that ATM kinase activity is required for the colocalization of CtIP and BLM (but not RAD51, NBS1, and RPA) with Tag (Figures 6 and 7), implying that 5′ to 3′ end resection may be the primary aspect of HDR affected by ATM signaling. Although the retention of RPA and RAD51 appears inconsistent with the reduction of 5′ end resection factors [64], ATM and ATR inhibition from 40–48 hpi did not affect the ability of viral DNA to undergo replication as measured by EdU incorporation at the viral replication center (Figure 6). Therefore, the majority of RPA [62] and a portion of RAD51 [15], [20] at Tag foci are likely bound to replicating single-stranded DNA generated by Tag helicase activity. Inhibition of ATM kinase activity increased NHEJ factor association and DNA-PKcs activation at the viral DNA replication center (Figures 8), indicating that ATM kinase activity prevents NHEJ activities from acting upon broken, replicating viral DNA. Collectively, our results are consistent with the hypothesis that ATM promotes viral DNA replication by controlling DSB repair pathway choice (Figure 9).

ATM and ATR kinase activities contribute to DNA repair and cell cycle arrest during infection
ATM and ATR kinases have dynamic roles during SV40 infection in preventing DSB formation on replicating viral DNA (Figures 1, 2, and see reference [30]) and in activating CHK2 and CHK1, respectively, to potentiate S phase arrest (Figures 5 and S2). Our results imply that ATM and ATR kinases contribute differently to the S phase arrest occurring during SV40 infection (Figures 5 and S2) which helps to explain the differential effects on monomer production when ATM or ATR are inhibited (Figure 1 and reference [30]). Whereas ATM inhibition prior to 20 hpi decreased viral genome production [30] and increased the fraction of cells in G1 (Figure 5), the effect of ATMi treatment between 20 and 48 hpi on both cell cycle arrest and monomer production is relatively minor. In contrast, ATR kinase is needed throughout infection to enforce the S phase checkpoint (Figures 5 and S2) and, as a consequence, is required for efficient viral genome replication (Figure 1 and reference [30]). It is possible that the small number of replicating viral genomes present early during viral infection (0–20 hpi) [47], [48] provides insufficient replication stress or DNA breakage (Figure 2 and reference [65]) to elicit the ATR-CHK1 kinase activity required to arrest cells in S phase [66], [67]. Similarly, the amount of Tag present on chromatin during the early phase might be insufficient to deplete essential DNA replication factors away from host replisomes inside the infected cell [68] resulting in an inefficient ATR-CHK1 activation. The fact that ATM function is dispensable at later time points suggests that robust viral DNA replication activates ATR-CHK1 [30] sufficiently to maintain cell cycle arrest in S phase.
Prior to 20 hpi, the interplay of ATM-CHK2 and ATR-CHK1 kinase signaling during SV40 infection is reminiscent of that observed following ionizing radiation [69]. After a sufficient amount of ionizing radiation is administered during S phase [70], the ATM-CHK2 pathway is quickly activated to arrest the cell cycle, and DSB termini are processed to create 3′ tails. The 3′ tails subsequently serve as substrates for ATR kinase activation, and thereafter the ATR-CHK1 kinase pathway can maintain the checkpoint [69]. Such interplay may occur during SV40 infection as well when viral and cellular DNA suffers replication-associated breaks that efficiently activate ATM [27], [30]. Resection of these broken DNAs could activate ATR [69], providing for efficient S phase arrest of SV40 infected cells early during infection. Consistent with resection mediating ATR activation early during infection, ATM inhibition prior to 20 hpi decreased ATR activation whereas inhibition from 20 to 48 hpi did not affect ATR kinase function [30]. Notably, the proteins that CHK2 and CHK1 phosphorylate to arrest the cell cycle of infected cells are poorly understood. DNA damage signaling in uninfected cells during S phase arrests the cell cycle via CHK1 and CHK2-dependent phosphorylation of CDC25 family phosphatases [71], and other mechanisms have been suggested to medicate cell cycle arrest during SV40 infection [61]. We suggest that phosphorylation of CDC25 family phosphatases by CHK1 and CHK2 might represent a mechanism that contributes to cell cycle arrest during small DNA tumor virus infection.
ATM activity promotes HDR and inhibits DNA-PKcs activation at viral DNA replication centers
The large number of repair factors that associate with SV40 replication compartments was initially unexpected given the simplicity of the in vitro replication system [1]. However, cellular studies of other small DNA tumor viruses, including BKV [32], polyomavirus [72], MCV [73], JCV [31], [74], and HPV [29], have demonstrated that viral DNA replication can recruit numerous proteins that appear to fill roles that are dispensable in vitro. Prevention of DSB accumulation during viral DNA replication (Figures 1, 2, 9 and see reference [30]) appears to be the major function of the repair factors that accumulate at viral DNA replication foci. In spite of the collective activities of HDR, ATM, and ATR at replicating SV40 DNA, one ended DSBs in the form of rolling circles have been observed at low percentages in viral DNA extracted from unperturbed SV40-infected cells by both electron microscopy [47], [65] and 2D gel electrophoresis [30]. DSBs associated with cellular DNA replication are repaired by HDR [12]. Thus, the ATM-directed HDR activities at the viral replication center (Figures 3, 7) likely suppress the formation of rolling circles by promoting efficient repair of one-ended DSBs on replicating viral chromatin (Figure 9A–E). Conversely when ATM is inhibited, DNA-PK stably binds and is activated at DSB termini (Figure 8). In this circumstance, HDR fails to efficiently repair the replication-associated DSB, resulting in increased levels of rolling circles and linear viral products (Figures 1, 2, 6, 9F–G and reference [30]).
The prevention of DNA-PK recruitment to viral DNA replication centers and kinase activation during SV40 DNA replication is reminiscent of the effect of the cell cycle on NHEJ function [49], [75]. DNA-PKcs kinase function is markedly decreased during S phase in response to several DNA-damaging agents [49]. This decrease in DNA-PK function results from the activation of 5′ to 3′ end resection during S phase [8], [9], [76], thereby resulting in an increased competition between MRN/CtIP and KU for binding to and processing of exposed ends of the DSB (Figure 9B). As both MRN/CtIP and KU possess activities that promote repair of DSBs by either HDR or NHEJ, respectively [58], binding one protein (e.g. KU) prior to the other (e.g., MRN/CtIP) might channel repair in one direction or another.
ATM is not required for homologous recombination [77], but its activity influences the repair kinetics of DSBs [78] and the extent of 5′ to 3′ end resection following treatment with exogenous DNA damaging agents [59], [75], [79]. Therefore, a plausible model for how ATM activity mediates DNA repair at viral DNA replication centers may be that ATM kinase activity activates or promotes stable association of 5′ to 3′ end resection proteins, including CtIP, EXO1, and BLM/DNA2 [10], with damaged viral DNA (Figures 7 and 9C). Importantly, ATM directly phosphorylates CtIP [59], EXO1 [80], and BLM [81], and ATM phosphorylation of CtIP influences the recruitment of BLM and EXO1, factors required for processive DSB end resection [10], to DSBs [59]. Such a mechanism might in turn prevent DNA-PK kinase activation by displacing KU from the DSB termini, allowing subsequent RAD51 binding to viral DNA and repair by HDR (Figure 9D, E).
In contrast, when ATM is inhibited during SV40 DNA replication, failure of ATM to phosphorylate CtIP, EXO1, and BLM/DNA2 might prevent processive end resection, resulting in inefficient digestion by chromatin-bound MRN at the viral DNA replication center [8] (Figure 7A, B, NBS1). By this model, KU then stably binds the one-ended replication-associated DSB (Figure 8). The KU-bound DSB can recruit DNA-PKcs, activate the DNA-PKcs kinase (Figure 8), and create a potential barrier to efficient replication associated DSB repair (Figure 9F, G). The DNA-bound, active DNA-PK would search for a cognate DSB bound by another heterotrimer of DNA-PK. The kinetics of NHEJ at a replication-associated DSB are likely slow and inefficient, allowing rolling circle replication to generate numerous concatemers before DNA repair (Figures 8 and 9G). We suggest that the primary function of ATM at the SV40 DNA replication center is to promote 5′ to 3′ end resection of viral DNA. Determining how other proteins including ATR act to prevent DSB formation (Figure 1, 2) or promote DNA repair (Figure 9) during SV40 DNA replication will be an interesting endeavor. Future examination of repair pathways used during SV40 infection might reveal components of DNA repair or damage signaling that could be manipulated to prevent viral DNA replication during SV40 or other small DNA tumor virus infections.
Materials and Methods
Cells and SV40 infection
Complete Dulbecco's modified Eagle's medium (DMEM supplemented with 10% fetal bovine serum) was used to culture both BSC40 (ATCC) and U2OS (ATCC) cells. Plating for immunofluorescence and SV40 infection were as previously described [30].
Plasmid transfection
For expression of HA-Topoisomerase IIIα and Flag-RMI2, 24 h prior to infection pCMV-HA-Topoisomerase IIIα or pIRES-neo-Flag-RMI2 [82] were transfected into BSC40 or U2OS cells using Fugene HD (Promega) using the manufacturer's protocol.
DNA isolation, agarose gel electrophoresis, and Southern blotting
Total intracellular DNA was extracted from SV40 - and mock-infected cells and subjected to agarose gel electrophoresis and Southern blotting as described [30]. In Figure 1, the same amount of SV40 DNA was loaded into each lane. In all other Southern blots, loading was normalized to cell number. In both cases, mitochondrial DNA was probed as an internal loading control. Loading determined by cell number or by mitochondrial DNA signal was highly correlated. 2D gel electrophoresis was as described by [30] with the following modification: the second dimension of the 2D gel was run for 7 h through a 0.95% 1×TBE agarose gel containing 0.5 ng/mL ethidium bromide. Southern blotting probes and data analysis using ImageQuant 5.2 were as previously described [30].
For DNA extractions from cells exposed to 20 µM EdU for 30 minutes (Figure 6A), DNA was isolated as per [30], but dissolved in 1 µL of 10 mM Tris pH 8.0 with 0.1 mM EDTA per 20,000 cells. A rat anti-BrdU antibody ([BU1/75 (ICR1)], Abcam), anti-rat conjugated to HRP (Jackson ImmunoResearch), and ECL-plus reagent (Perkin Elmer) were used to detect EdU label on the Southern blot as described in [83].
Immunoblots
Cells were lysed as previously described [30]. The following antibodies that follow were used: anti-Tag (Pab101, [84]), anti-actin (I-19, Santa Cruz), anti-CHK1 (G-4, Santa Cruz), anti-CHK1 pS317 (Cell Signaling), anti-CHK2 pT68 (Y171, Epitomics), anti-CHK2 (EPR4325, Epitomics), anti-NBS1 pS343 (EP178, Epitomics), anti-NBS1 (A301-289A, Bethyl Laboratories), anti-ATR (N-19, Santa Cruz), anti-DNA-PKcs (G-4, Santa Cruz), anti-KU80 (C-20, Santa Cruz), anti-KU70 (M-19, Santa Cruz), anti-DNA-PKcs pS2056 (EPR5670, Epitomics), anti-CtIP (A300-488A, Bethyl Laboratories), and anti-GAPDH (0411, Santa Cruz).
Immunofluorescence microscopy
Chromatin-associated proteins were visualized via pre-extraction of cells, prior to fixation and immunostaining as detailed by [26]. For RNAseA pre-extraction, 0.3 mg/mL RNAseA was included in pre-extraction buffer before fixation with 4% paraformaldehyde as previously described [63] and immunostaining was performed as before [30]. For EdU labeling of DNA, unless otherwise state, 20 µM EdU nucleoside in complete DMEM was added to cells for 5 minutes before fixation of cells and application of the click reaction at 48 hpi using the manufacturer's protocol (Invitrogen). An AxioObserver Z1 (Zeiss) equipped with a 63× Plan Apochromat (NA 1.4) oil objective (Zeiss) and an apotome (0.6 µm z slice) (Zeiss) was used to take all micrographs with two exceptions. For BLM and RMI1 in Figure 3A and B, a picture with a 1 µm z slice was taken using a LSM 510 META inverted microscope (Zeiss) equipped with a 63× Plan Apochromat (NA 1.4) oil objective (Zeiss).
To determine cell cycle phases of SV40-infected cells exposed to ATMi or ATRi (Figures 5C and S2B), the staining pattern of CENPF, EdU, and DAPI [56] in each cell was recorded from at least 300 cells. The average values were graphed in Figures 5C and S2B. Aberrant and normal Tag staining patterns from 3 independent experiments, each with at least 200 cells, were quantified from ATMi and ATRi treated cells and the average values were graphed in Figure 5D and tabulated in Figure S2C. Fluorescence intensity values at Tag focus were obtained using a 2.7 by 2.7 µm box in ImageJ. An equal area outside the nucleus near each Tag foci was used to obtain background intensity values. Fluorescence signal intensity values outside the replication center were subtracted from values at the viral replication center to correct for background. The corrected fluorescence intensities for a minimum of 70 viral replication centers are graphed in Figures 6 (E, F), 7 (B, D, E), 8 (C–E), and S3 (D).
Primary antibodies used for immunostaining were anti-Tag (Pab101), rabbit anti-Tag (in house), anti-CtIP (H-300, Santa Cruz), anti-CtIP (A300-488A, Bethyl Laboratories), anti-BRCA2 (ab-1, Calbiochem), anti-RAD52 (H-300, Santa Cruz), anti-BLM (C-18, Santa Cruz), anti-RMI1 (Novus), anti-Flag (M-2, Sigma), anti-HA (abcam), anti-CENPF (abcam), anti-DNA-PKcs (G-4, Santa Cruz), anti-KU80 (C-20, Santa Cruz), anti-KU70 (M-19, Santa Cruz), anti-DNA-PKcs pS2056 (EPR5670, Epitomics), anti-NBS1 (A301-289A, Bethyl Laboratories), anti-RAD51 (H-92, Santa Cruz), anti-RPA70B (In house), anti-PAR ([10H], Millipore), and anti-XRCC1 ([EPR4389(2)], abcam). Secondary antibodies used for immunostaining were anti-mouse conjugated to Alexa Fluor 488 (Invitrogen), anti-rabbit conjugated to Alexa Fluor 555 (Invitrogen), anti-goat conjugated to Alexa Fluor 555 (Invitrogen), and anti-goat conjugated to Alexa Fluor 647 (Jackson Immuno Research).
Use of inhibitors
ATMi [36], ATRi [35], and Nu7026 [42] were as previously described [30] and used at 10, 5, and 10 µM, respectively. Olaparib [85] (AZD2281) (Selleck Chemicals) was dissolved in DMSO to 3 mM and used at a final concentration of 3 µM. An equal concentration of DMSO solvent was used as a control vehicle for each drug. Hydroxyurea (MP Biochemicals) was dissolved in water at a final concentration of 1 M and used at 0.5 and 10 mM in complete DMEM for 18 and 2 h, respectively.
To treat cells with inhibitors during the final 28 h or 8 h of a 48 h SV40 infection, the media was removed from the cells at 20 and 40 hpi, respectively, and replaced with fresh media containing either inhibitor or DMSO. Exposure of cells to inhibitors during periods of a 48 h SV40 infection was as per [30].
Statistics
For Southern blotting data shown in Figure 1, the data analysis package of Microsoft Excel was used to perform statistics on appropriate data samples. A single factor ANOVA analysis was performed before t test. A two sample t-test assuming unequal variances was performed when the p<0.05 for the ANOVA test. In Figure 1, Bonferroni correction was applied when appropriate for multiple comparisons. One-tailed p values from t test are shown in Figure 1 (except for the ATRi to ATRi and Nu7026 t test which was two-tailed in Figure 1C). Unless otherwise indicated, bar graphs present the average of 3 or greater experiments and error bars represent standard deviation.
Comparisons of fluorescence intensity for Figures 6 (E, F), 7 (B, D, E), 8 (C–E), and S3B were performed using JMP 10.0.1. Normality and variance were evaluated by Shapiro-Wilk and Levene's test, respectively. Data that did not match a normal distribution were transformed. For data meeting the assumptions for a parametric test, single-factor ANOVA was used to test for a group effect. Significant ANOVA results (p<0.05) were followed by pair-wise comparisons by Tukey-Cramer. For data that violated the assumptions, a non-parametric Kruskal-Wallis test was used to test for a group effect. Significant Kruskal-Wallis results (p<0.05) were followed by pair-wise comparisons using the Mann-Whitney U test with a Bonferroni correction to allow for multiple comparisons.
p values are denoted by the number of asterisk(s): * p ≤0.05 ** p≤0.01 *** p≤0.001 **** p≤0.0001.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. WagaS, StillmanB (1998) The DNA replication fork in eukaryotic cells. Annu Rev Biochem 67 : 721–751.
2. MasaiH, MatsumotoS, YouZ, Yoshizawa-SugataN, OdaM (2010) Eukaryotic chromosome DNA replication: where, when, and how? Annu Rev Biochem 79 : 89–130.
3. CicciaA, ElledgeSJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40 : 179–204.
4. ZouL, ElledgeSJ (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300 : 1542–1548.
5. GoodarziAA, JeggoPA (2013) The repair and signaling responses to DNA double-strand breaks. Adv Genet 82 : 1–45.
6. NealJA, MeekK (2011) Choosing the right path: does DNA-PK help make the decision? Mutat Res 711 : 73–86.
7. StrackerTH, PetriniJH (2011) The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol 12 : 90–103.
8. SartoriAA, LukasC, CoatesJ, MistrikM, FuS, et al. (2007) Human CtIP promotes DNA end resection. Nature 450 : 509–514.
9. HuertasP, JacksonSP (2009) Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem 284 : 9558–9565.
10. NimonkarAV, GenschelJ, KinoshitaE, PolaczekP, CampbellJL, et al. (2011) BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 25 : 350–362.
11. PierceAJ, JohnsonRD, ThompsonLH, JasinM (1999) XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev 13 : 2633–2638.
12. MoynahanME, JasinM (2010) Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol 11 : 196–207.
13. PetermannE, HelledayT (2010) Pathways of mammalian replication fork restart. Nat Rev Mol Cell Biol 11 : 683–687.
14. DaviesSL, NorthPS, HicksonID (2007) Role for BLM in replication-fork restart and suppression of origin firing after replicative stress. Nat Struct Mol Biol 14 : 677–679.
15. SchlacherK, ChristN, SiaudN, EgashiraA, WuH, et al. (2011) Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145 : 529–542.
16. ChanKL, NorthPS, HicksonID (2007) BLM is required for faithful chromosome segregation and its localization defines a class of ultrafine anaphase bridges. EMBO J 26 : 3397–3409.
17. ChanKL, Palmai-PallagT, YingS, HicksonID (2009) Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol 11 : 753–760.
18. LaulierC, ChengA, StarkJM (2011) The relative efficiency of homology-directed repair has distinct effects on proper anaphase chromosome separation. Nucleic Acids Res 39 : 5935–5944.
19. FranchittoA, PichierriP (2011) Understanding the molecular basis of common fragile sites instability: role of the proteins involved in the recovery of stalled replication forks. Cell Cycle 10 : 4039–4046.
20. HashimotoY, PudduF, CostanzoV (2012) RAD51 - and MRE11-dependent reassembly of uncoupled CMG helicase complex at collapsed replication forks. Nat Struct Mol Biol 19 : 17–24.
21. WechslerT, NewmanS, WestSC (2011) Aberrant chromosome morphology in human cells defective for Holliday junction resolution. Nature 471 : 642–646.
22. AldertonGK, JoenjeH, VaronR, BorglumAD, JeggoPA, et al. (2004) Seckel syndrome exhibits cellular features demonstrating defects in the ATR-signalling pathway. Hum Mol Genet 13 : 3127–3138.
23. CasperAM, DurkinSG, ArltMF, GloverTW (2004) Chromosomal instability at common fragile sites in Seckel syndrome. Am J Hum Genet 75 : 654–660.
24. O'DriscollM, Ruiz-PerezVL, WoodsCG, JeggoPA, GoodshipJA (2003) A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet 33 : 497–501.
25. QvistP, HuertasP, JimenoS, NyegaardM, HassanMJ, et al. (2011) CtIP Mutations Cause Seckel and Jawad Syndromes. PLoS Genet 7: e1002310.
26. ZhaoX, Madden-FuentesRJ, LouBX, PipasJM, GerhardtJ, et al. (2008) Ataxia telangiectasia-mutated damage-signaling kinase - and proteasome-dependent destruction of Mre11-Rad50-Nbs1 subunits in Simian virus 40-infected primate cells. J Virol 82 : 5316–5328.
27. BoichukS, HuL, HeinJ, GjoerupOV (2010) Multiple DNA damage signaling and repair pathways deregulated by simian virus 40 large T antigen. J Virol 84 : 8007–8020.
28. MoodyCA, LaiminsLA (2009) Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog 5: e1000605.
29. GillespieKA, MehtaKP, LaiminsLA, MoodyCA (2012) Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. J Virol 86 : 9520–9526.
30. SowdG, LiN, FanningE (2013) ATM and ATR Activities Maintain Replication Fork Integrity during SV40 Chromatin Replication. PLoS Pathog 9: e1003283.
31. OrbaY, SuzukiT, MakinoY, KubotaK, TanakaS, et al. (2010) Large T antigen promotes JC virus replication in G2-arrested cells by inducing ATM - and ATR-mediated G2 checkpoint signaling. J Biol Chem 285 : 1544–1554.
32. JiangM, ZhaoL, GamezM, ImperialeMJ (2012) Roles of ATM and ATR-mediated DNA damage responses during lytic BK polyomavirus infection. PLoS Pathog 8: e1002898.
33. SakakibaraN, MitraR, McBrideAA (2011) The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J Virol 85 : 8981–8995.
34. SowdGA, FanningE (2012) A wolf in sheep's clothing: SV40 co-opts host genome maintenance proteins to replicate viral DNA. PLoS Pathog 8: e1002994.
35. ReaperPM, GriffithsMR, LongJM, CharrierJD, MaccormickS, et al. (2011) Selective killing of ATM - or p53-deficient cancer cells through inhibition of ATR. Nat Chem Biol 7 : 428–430.
36. HicksonI, ZhaoY, RichardsonCJ, GreenSJ, MartinNM, et al. (2004) Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res 64 : 9152–9159.
37. TapperDP, DePamphilisML (1978) Discontinuous DNA replication: accumulation of Simian virus 40 DNA at specific stages in its replication. J Mol Biol 120 : 401–422.
38. GoffSP, BergP (1977) Structure and formation of circular dimers of simian virus 40 DNA. J Virol 24 : 295–302.
39. SundinO, VarshavskyA (1980) Terminal stages of SV40 DNA replication proceed via multiply intertwined catenated dimers. Cell 21 : 103–114.
40. PohlhausJR, KreuzerKN (2006) Formation and processing of stalled replication forks–utility of two-dimensional agarose gels. Methods Enzymol 409 : 477–493.
41. Martin-ParrasL, LucasI, Martinez-RoblesML, HernandezP, KrimerDB, et al. (1998) Topological complexity of different populations of pBR322 as visualized by two-dimensional agarose gel electrophoresis. Nucleic Acids Res 26 : 3424–3432.
42. VeugerSJ, CurtinNJ, RichardsonCJ, SmithGC, DurkaczBW (2003) Radiosensitization and DNA repair inhibition by the combined use of novel inhibitors of DNA-dependent protein kinase and poly(ADP-ribose) polymerase-1. Cancer Res 63 : 6008–6015.
43. WillmoreE, de CauxS, SunterNJ, TilbyMJ, JacksonGH, et al. (2004) A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood 103 : 4659–4665.
44. MeekK, DouglasP, CuiX, DingQ, Lees-MillerSP (2007) trans Autophosphorylation at DNA-dependent protein kinase's two major autophosphorylation site clusters facilitates end processing but not end joining. Mol Cell Biol 27 : 3881–3890.
45. NealJA, DangV, DouglasP, WoldMS, Lees-MillerSP, et al. (2011) Inhibition of homologous recombination by DNA-dependent protein kinase requires kinase activity, is titratable, and is modulated by autophosphorylation. Mol Cell Biol 31 : 1719–1733.
46. ZhouY, PaullTT (2013) DNA-dependent protein kinase regulates DNA end resection in concert with Mre11-Rad50-Nbs1 (MRN) and ataxia telangiectasia-mutated (ATM). J Biol Chem 288 : 37112–37125.
47. RigbyPW, BergP (1978) Does simian virus 40 DNA integrate into cellular DNA during productive infection? J Virol 28 : 475–489.
48. SchirmbeckR, DeppertW (1987) Specific interaction of simian virus 40 large T antigen with cellular chromatin and nuclear matrix during the course of infection. J Virol 61 : 3561–3569.
49. ChenBP, ChanDW, KobayashiJ, BurmaS, AsaithambyA, et al. (2005) Cell cycle dependence of DNA-dependent protein kinase phosphorylation in response to DNA double strand breaks. J Biol Chem 280 : 14709–14715.
50. ChenBP, UematsuN, KobayashiJ, LerenthalY, KremplerA, et al. (2007) Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J Biol Chem 282 : 6582–6587.
51. UematsuN, WeteringsE, YanoK, Morotomi-YanoK, JakobB, et al. (2007) Autophosphorylation of DNA-PKCS regulates its dynamics at DNA double-strand breaks. J Cell Biol 177 : 219–229.
52. YajimaH, LeeKJ, ChenBP (2006) ATR-dependent phosphorylation of DNA-dependent protein kinase catalytic subunit in response to UV-induced replication stress. Mol Cell Biol 26 : 7520–7528.
53. McKinnonPJ (2012) ATM and the molecular pathogenesis of ataxia telangiectasia. Annu Rev Pathol 7 : 303–321.
54. LiaoH, WinkfeinRJ, MackG, RattnerJB, YenTJ (1995) CENP-F is a protein of the nuclear matrix that assembles onto kinetochores at late G2 and is rapidly degraded after mitosis. J Cell Biol 130 : 507–518.
55. ZhuX, ManciniMA, ChangKH, LiuCY, ChenCF, et al. (1995) Characterization of a novel 350-kilodalton nuclear phosphoprotein that is specifically involved in mitotic-phase progression. Mol Cell Biol 15 : 5017–5029.
56. LobrichM, ShibataA, BeucherA, FisherA, EnsmingerM, et al. (2010) gammaH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle 9 : 662–669.
57. MatsuokaS, BallifBA, SmogorzewskaA, McDonaldER3rd, HurovKE, et al. (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316 : 1160–1166.
58. ChapmanJR, TaylorMR, BoultonSJ (2012) Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 47 : 497–510.
59. WangH, ShiLZ, WongCC, HanX, HwangPY, et al. (2013) The interaction of CtIP and Nbs1 connects CDK and ATM to regulate HR-mediated double-strand break repair. PLoS Genet 9: e1003277.
60. TomimatsuN, MukherjeeB, BurmaS (2009) Distinct roles of ATR and DNA-PKcs in triggering DNA damage responses in ATM-deficient cells. EMBO Rep 10 : 629–635.
61. RohalyG, KorfK, DehdeS, DornreiterI (2010) Simian virus 40 activates ATR-Delta p53 signaling to override cell cycle and DNA replication control. J Virol 84 : 10727–10747.
62. JiangX, KlimovichV, ArunkumarAI, HysingerEB, WangY, et al. (2006) Structural mechanism of RPA loading on DNA during activation of a simple pre-replication complex. EMBO J 25 : 5516–5526.
63. BrittonS, CoatesJ, JacksonSP (2013) A new method for high-resolution imaging of Ku foci to decipher mechanisms of DNA double-strand break repair. J Cell Biol 202 : 579–595.
64. BeucherA, BirrauxJ, TchouandongL, BartonO, ShibataA, et al. (2009) ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J 28 : 3413–3427.
65. TapperDP, AndersonS, DePamphilisML (1982) Distribution of replicating simian virus 40 DNA in intact cells and its maturation in isolated nuclei. J Virol 41 : 877–892.
66. ShimadaK, PaseroP, GasserSM (2002) ORC and the intra-S-phase checkpoint: a threshold regulates Rad53p activation in S phase. Genes Dev 16 : 3236–3252.
67. CobbJA, ShimadaK, GasserSM (2004) Redundancy, insult-specific sensors and thresholds: unlocking the S-phase checkpoint response. Curr Opin Genet Dev 14 : 292–300.
68. BanerjeeP, DeJesusR, GjoerupO, SchaffhausenBS (2013) Viral interference with DNA repair by targeting of the single-stranded DNA binding protein RPA. PLoS Pathog 9: e1003725.
69. KousholtAN, FuggerK, HoffmannS, LarsenBD, MenzelT, et al. (2012) CtIP-dependent DNA resection is required for DNA damage checkpoint maintenance but not initiation. J Cell Biol 197 : 869–876.
70. LobrichM, JeggoPA (2007) The impact of a negligent G2/M checkpoint on genomic instability and cancer induction. Nat Rev Cancer 7 : 861–869.
71. BoutrosR, LobjoisV, DucommunB (2007) CDC25 phosphatases in cancer cells: key players? Good targets? Nat Rev Cancer 7 : 495–507.
72. EricksonKD, Bouchet-MarquisC, HeiserK, Szomolanyi-TsudaE, MishraR, et al. (2012) Virion assembly factories in the nucleus of polyomavirus-infected cells. PLoS Pathog 8: e1002630.
73. TsangSH, WangX, LiJ, BuckCB, YouJ (2014) Host DNA damage response factors localize to merkel cell polyomavirus DNA replication sites to support efficient viral DNA replication. J Virol 88 : 3285–3297.
74. DarbinyanA, WhiteMK, AkanS, RadhakrishnanS, Del ValleL, et al. (2007) Alterations of DNA damage repair pathways resulting from JCV infection. Virology 364 : 73–86.
75. ShibataA, ConradS, BirrauxJ, GeutingV, BartonO, et al. (2011) Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J 30 : 1079–1092.
76. YunMH, HiomK (2009) CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 459 : 460–463.
77. RassE, ChandramoulyG, ZhaS, AltFW, XieA (2013) Ataxia telangiectasia mutated (ATM) is dispensable for endonuclease I-SceI-induced homologous recombination in mouse embryonic stem cells. J Biol Chem 288 : 7086–7095.
78. BerkovichE, MonnatRJJr, KastanMB (2007) Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol 9 : 683–690.
79. ShibataA, MoianiD, ArvaiAS, PerryJ, HardingSM, et al. (2014) DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell 53 : 7–18.
80. BoldersonE, TomimatsuN, RichardDJ, BoucherD, KumarR, et al. (2010) Phosphorylation of Exo1 modulates homologous recombination repair of DNA double-strand breaks. Nucleic Acids Res 38 : 1821–1831.
81. AbabouM, DutertreS, LecluseY, OnclercqR, ChattonB, et al. (2000) ATM-dependent phosphorylation and accumulation of endogenous BLM protein in response to ionizing radiation. Oncogene 19 : 5955–5963.
82. SinghTR, AliAM, BusyginaV, RaynardS, FanQ, et al. (2008) BLAP18/RMI2, a novel OB-fold-containing protein, is an essential component of the Bloom helicase-double Holliday junction dissolvasome. Genes Dev 22 : 2856–2868.
83. PootRA, BozhenokL, van den BergDL, SteffensenS, FerreiraF, et al. (2004) The Williams syndrome transcription factor interacts with PCNA to target chromatin remodelling by ISWI to replication foci. Nat Cell Biol 6 : 1236–1244.
84. DeppertW, GurneyEG, HarrisonRO (1981) Monoclonal antibodies against simian virus 40 tumor antigens: analysis of antigenic binding sites, using adenovirus type 2-simian virus 40 hybrid viruses. J Virol 37 : 478–482.
85. MenearKA, AdcockC, BoulterR, CockcroftXL, CopseyL, et al. (2008) 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin - 1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J Med Chem 51 : 6581–6591.
86. LucasI, GermeT, Chevrier-MillerM, HyrienO (2001) Topoisomerase II can unlink replicating DNA by precatenane removal. EMBO J 20 : 6509–6519.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 12
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Plasma Membrane-Located Purine Nucleotide Transport Proteins Are Key Components for Host Exploitation by Microsporidian Intracellular Parasites
- Rubella Virus: First Calcium-Requiring Viral Fusion Protein
- Emergence of MERS-CoV in the Middle East: Origins, Transmission, Treatment, and Perspectives
- Neutral Sphingomyelinase in Physiological and Measles Virus Induced T Cell Suppression
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy