
A Natural Genetic Variant of Granzyme B Confers Lethality to a Common Viral Infection
Granzymes (Gzm) are serine proteases expressed by cytotoxic T cells and natural killer cells, and are important for the destruction of virally infected cells. To date, the function of these molecules has been assessed exclusively in common laboratory mouse strains that express identical granzyme proteins. In wild mouse populations, variants of granzyme B have been identified, but how these function, especially in the context of infections, is unknown. We have generated a novel mouse strain expressing a granzyme B variant found in wild mice (GzmBW), and exposed these mice to viral infections. The substrates cleaved by GzmBW were found to differ significantly from those cleaved by the GzmBP protein, which is normally expressed by laboratory mice. Alterations in substrate specificity resulted in GzmBW mice being significantly more susceptible to infection with murine cytomegalovirus, a common mouse pathogen. Our findings demonstrate that polymorphisms in granzyme B can profoundly affect the outcome of infections with some viral pathogens.
Published in the journal:
. PLoS Pathog 10(12): e32767. doi:10.1371/journal.ppat.1004526
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1004526
Summary
Granzymes (Gzm) are serine proteases expressed by cytotoxic T cells and natural killer cells, and are important for the destruction of virally infected cells. To date, the function of these molecules has been assessed exclusively in common laboratory mouse strains that express identical granzyme proteins. In wild mouse populations, variants of granzyme B have been identified, but how these function, especially in the context of infections, is unknown. We have generated a novel mouse strain expressing a granzyme B variant found in wild mice (GzmBW), and exposed these mice to viral infections. The substrates cleaved by GzmBW were found to differ significantly from those cleaved by the GzmBP protein, which is normally expressed by laboratory mice. Alterations in substrate specificity resulted in GzmBW mice being significantly more susceptible to infection with murine cytomegalovirus, a common mouse pathogen. Our findings demonstrate that polymorphisms in granzyme B can profoundly affect the outcome of infections with some viral pathogens.
Introduction
Cytotoxic lymphocytes, such as natural killer (NK) cells and CD8 T cells, are essential for the elimination of tumour cells or cells infected with intracellular pathogens. One mechanism cytotoxic lymphocytes utilize to initiate the destruction of target cells is the exocytosis of granules containing perforin (Pfp) and a family of serine proteases known as granzymes (Gzms) [1]. Pfp facilitates the entry of Gzms into the cytoplasm of target cells, where the Gzms cleave specific proteins triggering death of the target. Multiple Gzms have been identified in both humans and the mouse, with GzmA and GzmB being the most abundant and best characterized in both species. While non-cytotoxic functions of Gzms have been described, inducing target cell death appears to be a major function of GzmA and GzmB, and the increased sensitivity of mice lacking these proteins to infection with ectromelia virus (ECTV) and murine cytomegalovirus (MCMV) has been attributed to the role of the Gzms in the killing of infected cells [2]–[4]. Unlike GzmB, which is universally agreed to induce apoptosis [5], the mechanism employed by GzmA to induce cell death remains controversial [6]–[8]; however, it is agreed that this mechanism does not require activated caspases.
Human and mouse GzmB share extensive sequence homology and thus were predicted to kill cells by the same mechanism. However, amino acids that influence substrate binding differ between human and mouse GzmB, with the two proteins now recognized to have different substrate preferences [9]–[11]. A significant difference between the two proteins is that human, but not mouse GzmB, efficiently cleaves the BH3-only protein Bid [10], [12], [13]. Once cleaved, tBid is capable of inducing permeabilization of the mitochondrial outer membrane (MOMP) resulting in the release of pro-apoptotic mediators that ultimately activate a caspase cascade. The finding that cells lacking Bid or overexpressing Bcl-2 survive treatment with human GzmB is consistent with the theory that human GzmB indirectly activates caspases [12], [14], [15]. By contrast, mouse GzmB appears to mediate its effects by directly processing pro-caspases to their active form, and does not require MOMP in order to induce apoptosis [9], [10]. Thus, while both human and mouse GzmB efficiently induce the death of target cells, they achieve this by different mechanisms.
Many pathogens inhibit apoptotic pathways as a means of survival. The differences in mouse and human GzmB substrate specificity may therefore have arisen in response to pathogens targeting different apoptotic pathways in humans and mice. Alternatively, the need to directly target proteins produced by species-specific pathogens could have driven the divergence in GzmB substrate specificities. For example, GzmB inhibits the reactivation of HSV-1 by cleaving the virally encoded ICP4 protein [16]. Similarly, GzmH and GzmB cooperate to suppresses the spread of human adenovirus V by degrading viral proteins essential for replication [17]. Further evidence that selective pressure from pathogens has contributed to changes in GzmB has come from the finding that GzmB polymorphisms exist. In humans, a limited degree of GzmB polymorphism has been described [18], however, the significance of this finding is unclear as there is no difference in the proteolytic activities of the two common alleles and both have equivalent biochemical and cytotoxic functions, at least in vitro [19]. In the mouse, 13 common inbred laboratory mouse strains that were tested show no GzmB sequence variation, a finding that probably reflects the limited gene pool from which laboratory strains were derived [20]. By contrast, significant variation in the GzmB sequence was noted in wild mice or wild-derived inbred mouse strains, including in some of the residues that line the substrate cleft. Interestingly, the single allele found in all the inbred mouse strains was relatively uncommon in the wild, found in <20% of isolates [20].
Responses to pathogen challenges have been investigated almost exclusively using inbred mouse strains including knock-out mice rendered genetically deficient in GzmB, but the important question as to whether polymorphisms in GzmB influence the outcome of infections with common pathogens has not been addressed. Here, we have characterized a GzmB allele present in wild mice and found that although its substrate specificity differs from that of GzmB encoded by C57BL/6 (B6) mice, its in vitro cytotoxic potential is identical to that of the B6 allele. Nevertheless, substitution of the GzmB allele encoded by B6 mice with the GzmB allele from wild mice led to the inability to effectively control MCMV infection. These data provide novel insights about the relevance of GzmB polymorphisms and demonstrate that polymorphisms in GzmB significantly influence the response to infection with a common, natural viral pathogen.
Results
GzmB alleles have different substrate preferences
We had previously shown that the mouse GzmB gene is highly polymorphic amongst outbred mouse populations and that some of the polymorphic residues are predicted to impinge on the substrate binding pocket and to potentially influence fine protease specificity [20]. To investigate this further, we selected a wild (w) mouse GzmB allele that is markedly divergent from the allele common to B6 mice as well as the 13 inbred mouse strains we previously typed. For clarity we will refer to the prototype B6 inbred allele as GzmBP, and the outbred wild allele as GzmBW. GzmBW encodes 13 differences in amino acid sequence from GzmBP or 94.7% amino acid identity over the entire 247 amino acid sequence (Fig. 1A).

We expressed and purified recombinant GzmBW and GzmBP in Pichia pastoris yeast cells, as previously described [21]. Both forms were indistinguishable in their ability to cleave the generic GzmB substrate AAD-SBzl (Fig. 1B), indicating that GzmBW possesses classic Asp-ase activity. Both forms also bound Serpinb9, the intracellular inhibitor of GzmB [22]–[24](Fig. 1C), suggesting that GzmBP and GzmBW are subject to similar regulation in vivo. Inhibition of any serine proteases by its cognate serpin can only occur if the protease can correctly recognize and cleave the relatively unstructured reactive site loop of the serpin. This further confirmed the proteolytic activity of GzmBW, and its cleavage after aspartate [22].
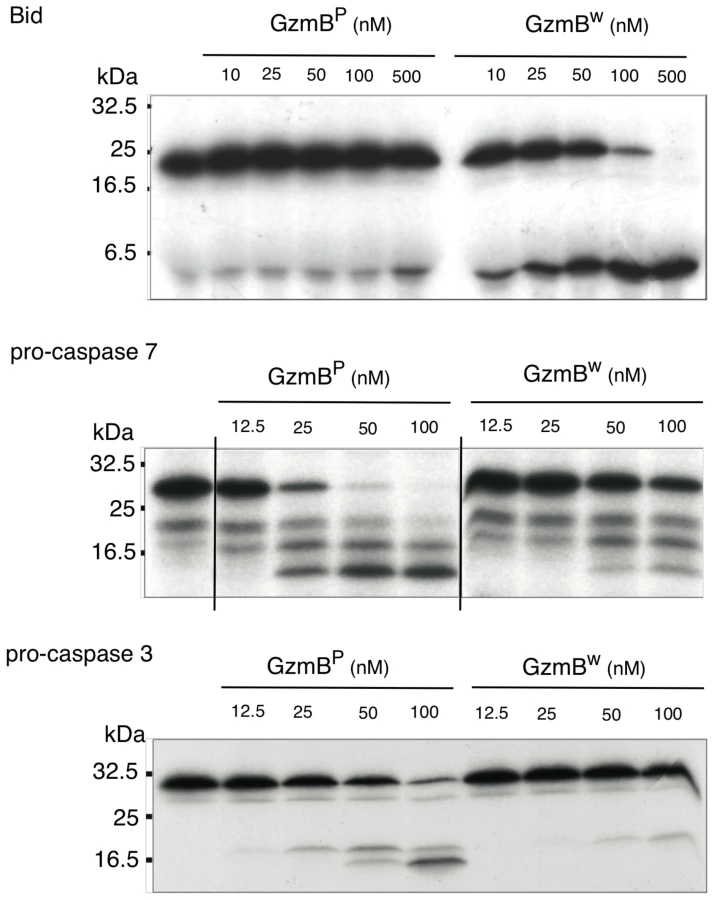
We have previously shown that GzmBP differs from human GzmB in that it cleaves peptide substrates based on Bid (e.g. Abz-IEPDSESQK-dnp) very poorly, and prefers substrates with an aromatic P2 residue and glycine at P2′ ([10] and Table 1). Strikingly, GzmBW cleaves peptide substrates based on Bid over 100 times more efficiently than GzmBP, and three-fold more efficiently than human GzmB (Table 1). Like GzmBP, GzmBW prefers substrates with glycine at P2′, but cleaves substrates with an aromatic P2 residue four - to five-fold less efficiently. To confirm these differences in relation to broader substrate specificity, both forms were used in a substrate phage display experiment on a library with Asp fixed at the P1 position [10]. The results indicated very similar requirements at the P4 position (I/L), as well as a requirement for glycine at P2′ (Figure S1). However, the strong preference of GzmBP for aromatic residues at P2 was not conserved in GzmBW, consistent with the peptide substrate results (Table 1).

We next examined whether the differences in turnover of Bid (IEPD) substrates is also reflected in a different capacity to cleave intact Bid or effector pro-caspases 3 and 7. We found that GzmBW is 50–100 fold more efficient than GzmBP at cleaving recombinant Bid, but far less efficient at activating pro-caspase-3 or pro-caspapse-7 (Fig. 2). To determine whether these changes in substrate preference translate into a variable capacity to kill target cells, we exposed P815 (mouse mastocytoma), EL-4 (mouse thymoma), HeLa (human cervical cancer) and Jurkat (human T lymphoma) cells to graded doses of each GzmB form and very low (‘sublytic’) quantities of purified recombinant Pfp (Fig. 3). For each cell line, we found no significant difference in susceptibility to apoptosis. Overall, we concluded that there is no significant difference in the intrinsic pro-apoptotic activity of GzmB expressed by the inbred laboratory mice and outbred wild mice.

GzmBW from T and NK cells retains full functionality
To examine the role of the GzmBW allele in more physiological settings, we generated a congenic mouse strain carrying the w allele. This was achieved by backcrossing the w/w mouse for >20 generations with B6, at each generation selecting for the w allele. This strain was also crossed with B6.OT1 mice to create the GzmBW/W.OT1 strain, so that antigen restricted CTLs could be studied in both strains (see below). An alternative and probably less laborious approach to deriving the congenic line over so many generations would have been to develop a ‘knock in’ of the W allele on P strain. However, we and other groups have not been successful in this approach, due to the large number of highly homologous granzyme gene sequences closely linked in the GzmB locus.
Using whole exome DNA sequencing we confirmed that the GzmB locus on chromosome 14 including the GzmB gene, all 6 linked Gzm genes and the gene encoding Cathepsin G (expressed in neutrophils, but not cytotoxic lymphocytes) were derived from w. The genetic interval derived from w comprised approximately 18% of the chromosome 14 DNA. Overall, the genetic content of the w/w mouse was >99.1% derived from B6. Having derived the DNA sequence of the 0.9% of the genome that remained from the w strain, we were able to assess whether the high degree of polymorphism identified for GzmB was also the case for the other granzymes (GzmC-G and N). This was not the case: whereas >5.3% of the amino acids of GzmB differed between the two allotypes, the corresponding figure across the other 6 genes was <0.3% (a total of four polymorphic residues out of 1362 across the 6 coding regions; P<0.0001) (Table 2).

We wished to establish beyond doubt that our in vitro enzymatic findings, which were derived with purified recombinant proteases, would be replicated in bona fide antigen restricted CTLs, which, along with NK cells, are the authentic physiological context for granzyme expression. We therefore generated OVA257 specific activated T cells from the spleens of B6 and GzmBW/W congenic OT1 mice and tested the resultant cell lysates on peptide substrates. The generic Asp-ase substrate AAD-SBzl was cleaved with similar efficiency by B6 and w/w T cell lysates (Fig. 4A). By contrast, the tetrapeptide substrate IEPD was cleaved more efficiently by w/w lysate (with >3 times the maximum velocity of p/p, p<0.05), confirming differences in the fine specificity of the two alloforms of GzmB observed with in vitro-generated proteins (Fig. 4B). Structural predictions for the granzymes whose genes are closely linked to GzmB, indicate that these proteases should have chymotrypsin-like (‘chymase’) activity and cleave after hydrophobic P1 residues [25] as has been clearly demonstrated for the human orthologue GzmH [26]. As expected, the respective CTL lysates from the w and p mice had indistinguishable chymase activity (Figure 4C), further confirming that the minimal changes in amino acid sequence among the chymase granzymes had no impact on substrate preference. There was no turnover in AAD or IEPD in T cells derived from GzmAB-null mice, while B6 mice deficient in perforin (whose gene maps to Chr 10) showed similar activity to wild type B6 mice (Fig. 4A and C). Western blot of cell lysates also showed no quantitative difference in GzmB expression across the various strains tested (Fig. 4D).

GzmBW/W mice show increased sensitivity to MCMV infection
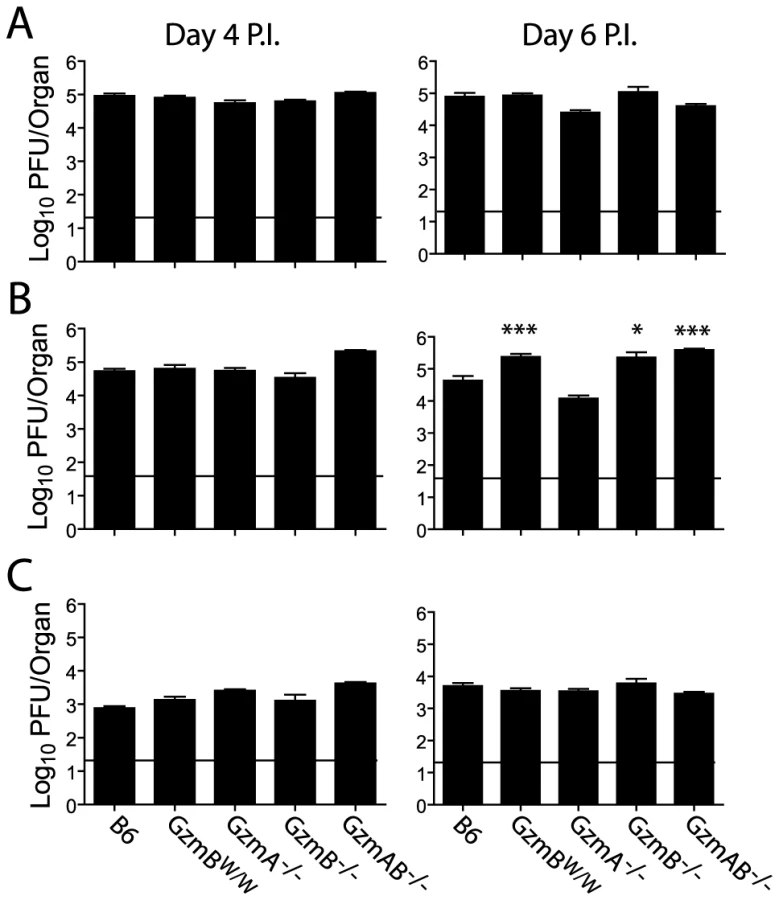
Our in vitro analyses determined that the substrate specificity of GzmBW differs from that of the B6 allele. Since the Gzms have been shown to play a critical role in viral infections, we next examined whether the differences in substrate specificity observed in vitro and ex vivo can lead to functional differences after infection with bona fide mouse pathogens. MCMV, a natural pathogen of mice, is partly controlled by activities mediated by GzmB [4]. Thus, the effect of GzmBW on the ability of the host to control MCMV infection was investigated. In B6 mice, NK cells rapidly control MCMV infection via activation mediated by engagement of the Ly49H activating NK cell receptor. However, in the wild most (≥80%) MCMV variants encode m157 proteins that are unable to activate NK cells [27] and indeed, the frequency of Ly49H-resistance is rare in outbred wild mice [28]. These findings indicate that B6-like Ly49H-m157 interactions are not a feature of host–MCMV interactions in the wild. Thus, to examine the role of GzmBW/W in a setting that reflects the situation in wild mouse populations, we utilized a virus lacking the m157 viral protein (Δm157). In the absence of m157, MCMV replicates to high titers in the visceral organs of B6 mice, and is eventually controlled primarily by cytotoxic CD8 T cells. Unlike B6 mice (GzmBP/P), infection of the GzmBW/W mice with Δm157 MCMV resulted in rapid mortality (Fig. 5A). At day 7 post-infection, viral loads in the spleens and lungs of GzmBW/W mice were not significantly different from those observed in B6 mice (Fig. 5B). By contrast, the livers of GzmBW/W mice contained approximately 10 fold more virus than livers of B6 mice (Fig. 5B). Histological analysis of GzmBW/W livers harvested at day 6 post-infection revealed significant areas of focal necrosis and diffuse cellular infiltrates, while in B6 mice no significant damage was evident in liver sections (Fig. 5C). The advanced liver damage observed in GzmBW/W mice by histological analysis was also confirmed by measuring circulating liver transaminase levels. Serum levels of the liver enzymes alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in GzmBW/W mice were significantly elevated at day 6 post-infection (Fig. 5D). These data indicate that GzmBW/W mice have an impaired response to Δm157 MCMV infection that manifests as significantly higher viral loads within the liver, and tissue damage to this organ, which markedly increases mortality.

GzmB, along with GzmA, produced by cytotoxic CD8 T cells is essential for host defense against the poxvirus ECTV [2], [29]. ECTV is a large DNA virus that is the causative agent of mousepox. GzmBW/W mice infected with 105 pfu of the Moscow strain of ETCV succumbed to infection at rate equivalent to that of B6 mice (Figure S2A), and viral load in the blood of GzmBW/W mice at day 8 post-infection was not significantly different to that of B6 mice (Figure S2B). Since mice that lack GzmB are 100-fold more susceptible to ECTV infection [2], these data indicate that GzmBW can substitute for the B6 allele of GzmB in the context of ECTV infection, and provide independent evidence that the GzmBW allele is functional not only in vitro (Table 1), but also in vivo.
The response of GzmBW/W mice to MCMV infection mirrors that of GzmB-deficient mice
The outcome of infection with the Δm157 MCMV virus was then compared in GzmBW/W mice and mice lacking GzmA, GzmB or both GzmA and B. Viral loads were measured in target organs (spleen, liver and lungs) at days 4 and 6 post-infection by plaque assay. Experiments were terminated at day 6 post-infection as GzmBW/W mice become highly sensitive to infection after this time. Viral loads in mice deficient for GzmA were equivalent to those of B6 mice in all organs tested, at both day 4 and 6 (Fig. 6), suggesting that, at least during the acute phase of infection, GzmA is not required for viral control. By contrast, viral loads in the livers of mice deficient in GzmB, either alone, or in combination with GzmA, were significantly higher than those observed in B6 mice at day 6 post-infection (Fig. 6B). Thus, GzmB is essential for effective control of MCMV Δm157 in the liver. Furthermore, replication of the Δm157 virus in the livers of GzmBW/W mice was equivalent to that of GzmB−/− and GzmA/B−/− mice, indicating that GzmBW/W cannot substitute for the B6 allele during the anti-viral response to MCMV.
Deregulated cytokine production does not contribute to liver damage in MCMV infected GzmBW/W mice
The effect of the Δm157 virus on GzmBW/W mice is reminiscent of the effects of MCMV infection on pfp-deficient mice. Mice lacking pfp exhibit increased mortality after MCMV infection [30]. While MCMV replication in the liver of pfp−/− mice is significantly higher than that observed in B6 mice, uncontrolled viral replication is not the cause of mortality [4]. Rather, a fatal hemophagocytic lymphohistiocytsis (HLH)-like syndrome develops in pfp−/− mice due to the uncontrolled production of TNFα by accumulating activated macrophages [4]. To determine if GzmBW/W mice exhibited any signs of an HLH-like syndrome, we examined lymphocyte populations and cytokine production after MCMV infection. Following infection with the Δm157 MCMV mutant, the livers of GzmBW/W mice contained significantly more leukocytes at day 6 post-infection (Fig. 7A). Analysis of these leukocytes by FACS revealed that the number of inflammatory monocytes (CD11b+ Ly6C+ Ly6G−) in the liver of GzmBW/W mice were significantly increased (Fig. 7A). While a similar trend was observed for granulocytes (CD11b+ Ly6G+), this did not reach statistical significance (Fig. 7A). Given the increased numbers of inflammatory monocytes in the livers of GzmBW/W mice, we measured pro-inflammatory cytokine production to determine if this may be contributing to the observed mortality. The levels of TNFα and IFNγ in the livers of GzmBW/W mice were not significantly different from those observed in B6 mice (Fig. 7B). These findings indicate that GzmBW/W mice do not develop an HLH-like syndrome following MCMV infection.

In order to better characterize the effects of MCMV infection in GzmBW/W mice, immunohistochemistry (IHC) staining of liver sections was performed. IHC staining of liver sections with an antibody specific for the IE1 protein of MCMV revealed a stark difference between B6 and the GzmBW/W mice. In B6 mice inflammatory foci were evident at day 6 post-infection, consisting of small numbers of infected hepatocytes (brown stain), typically surrounded by a large number of lymphocytes (Fig. 7C). In GzmBW/W mice, the inflammatory foci were larger in size and contained significantly more infected hepatocytes (Fig. 7C). Furthermore, areas of necrosis and cell debris within the centre of the foci were apparent (Fig. 7C). Together the data indicate that the liver damage observed in GzmBW/W mice was the direct result of uncontrolled viral replication, rather than the outcome of immune-mediated pathology.
CD8 T cells from GzmBW/W mice are unable to kill virally infected cells
The inability of GzmBW/W mice to control MCMV in the liver suggests that these mice may not be generating an appropriate anti-viral CTL response. Serpinb9 is a potent inhibitor of GzmB that is expressed by CTL [24]. Expression of Serpinb9 is required to prevent the premature apoptosis of CTL generated in response to lymphocytic choriomenigitis virus (LCMV) or Listeria monoctogenes infection [31]. We found that the GzmBW protein effectively bound Serpinb9 in an in vitro assay (Fig. 1C), but this finding does not preclude the possibility that Serpinb9 is unable to efficiently inhibit GzmBW in CTL in vivo. We therefore infected Serpinb9−/− mice with Δm157 MCMV and quantified viral replication. Viral titers in the spleen, liver, and lungs of Serpinb9−/− mice were not significantly different from those of B6 mice (Figure S3). Thus, the effects observed in GzmBW/W mice are not the result of an inability of Serpinb9 to inhibit GzmBW.
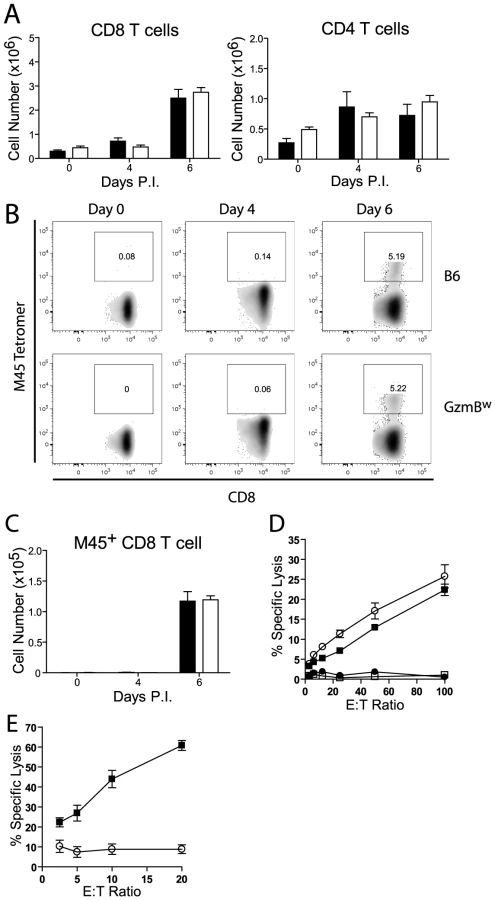
Next, we assessed the generation and effectiveness of anti-viral T cell responses in GzmBW/W mice. The total numbers of CD8 and CD4 T cells localizing to the livers of GzmBW/W mice after MCMV infection were not significantly different from those observed in MCMV-infected B6 mice (Fig. 8A). A peptide derived from the M45 protein of MCMV is the immunodominant epitope recognized by CD8 T cells in B6 mice [32]. The percentage of CD8 T cells stained by an M45 tetramer in B6 mice following MCMV infection was similar to that observed in GzmBW/W mice (Fig. 8B), and there were no differences in the total number of M45-specific CD8 T cells generated (Fig. 8C). The capacity of M45-specific T cells to kill target cells was also assessed. Splenocytes isolated from B6 mice at day 6 post-infection efficiently lysed M45 pulsed target cells, while no significant lysis was apparent when splenocytes from uninfected mice were used (Fig. 8D). The capacity of GzmBW/W splenocytes to lyse M45 pulsed target cells was similar to that of B6 cells (Fig. 8D). Hence, GzmBW/W mice generate an effective CD8 T cell response following infection with Δm157 MCMV and these T cells are able to efficiently kill model target cells. In addition to peptide pulsed targets, we tested the capacity of GzmBW/W and GzmBP/P CD8 T cells to kill MCMV-infected cells. CD8 T cells were purified from the spleen of B6 mice or GzmBW/W mice 6 days after infection, co-cultured with MCMV-infected IC-21 macrophages and macrophage viability assessed 18 h later. GzmBP/P CD8 T efficiently lysed the MCMV-infected target cells in a dose dependent manner, whereas GzmBW/W CD8 T cells were almost completely ineffective (Fig. 8E). Collectively, the data suggested that CD8 T cells expressing the w allele of GzmB are elicited and activated in response to MCMV infection, but are unable to kill MCMV-infected targets, accounting of the susceptibility of these mice to the virus.
Discussion
Murine GzmB is highly polymorphic in wild mice, but the physiological relevance of this finding has been unclear. Here, we have characterized the biochemical and physiological function of an allelic variant of GzmB, GzmBW, found in wild mice. We have found that GzmB polymorphism affects the substrates cleaved by the protease in vitro. GzmBW efficiently cleaved peptide substrates based on the Bid sequence, and Bid itself, and as such, has an activity that is distinct from the GzmBP allele expressed by inbred mouse strains. Despite having different substrate preferences, GzmBP and GzmBW induced apoptosis of uninfected target cells with similar efficiency in vitro, and GzmBW/W mice were as efficient as wild type B6 mice in controlling ECTV infection. These data clearly show that the GzmBW isoform is bioactive in vitro and, more importantly, in vivo, at least as far as the requirements for its critical role in inducing the death of “generic” targets cells and in controlling infection with ECTV.
A striking finding of the study is that B6 mice expressing GzmBW are extremely sensitive to MCMV infection demonstrating that polymorphism in GzmB can have a significant impact on the capacity of the host to control specific pathogens. We found that GzmB was essential for control of Δμ157 MCMV (the most common MCMV variant found in the wild), and that the GzmBW allele was unable to substitute for GzmBP in this setting. These results argue for a defect in cytotoxicity, however, several non-cytotoxic roles have been ascribed to various Gzms. For example, GzmA and GzmM have a role in the production/release of pro-inflammatory cytokines [33]–[35]. We found that production of the pro-inflammatory cytokines IFNγ and TNFα by GzmBW/W mice following MCMV infection was equivalent to that of B6 mice. Furthermore, GzmBW/W mice generated CD8 T cell effectors at the expected frequency with localisation of these cells to the liver equivalent to that observed in B6 mice. Thus, the inability of GzmBW/W mice to control MCMV infection was not the result of defects in the production of pro-inflammatory cytokines, nor was it due to defective CD8 T cell numbers.
The initiation of apoptosis by activating caspases is the best-characterized function of GzmB. Human GzmB initiates apoptosis by activating caspases via two distinct pathways [36]. A mitochondrial-dependent pathway is activated when human GzmB cleaves Bid resulting in MOMP and the release of pro-apoptotic mediators [13], [37]. Mitochondrial-independent pathways operate via direct pro-caspase activation when sufficiently high concentrations of GzmB are delivered to the target cell cytosol [36]. Given the substrate specificity of the w allele, apoptosis induced by this form of GzmB is likely to mirror that of human GzmB. In vitro, GzmBW was as effective as GzmBP at inducing apoptosis in uninfected target cells, and anti-viral specific CD8 T cells isolated from GzmBW/W mice killed peptide pulsed target cells efficiently, indicating that there is no intrinsic defect in the direct cytotoxic capacity of GzmBW. However, CD8 T cells isolated from GzmBW/W mice were unable to kill MCMV-infected target cells in vitro. MCMV encodes potent inhibitors of both Bax and Bak that together prevent MOMP in response to multiple stimuli [38]–[41]. Collectively, the data strongly suggest that as GzmBW preferentially cleaves Bid, rather than directly activating caspases, it is susceptible to inhibition by MCMV-encoded proteins that block the intrinsic cell death pathway.
Our previous work has studied some of the key residues that dictate the species-specific substrate preferences of human and mouse GzmB. We found that two residues whose side-chains impinge on the substrate cleft, 180Arg and 222Lys are important in this regard, as substitution of the corresponding human or rat residue conferred a greater capacity to cleave Bid [10]. Residue 222 was invariant in all of the mouse GzmB alleles we previously sequenced and is thus common to both the mouse w and p alleles. However at position 180, the w allele encodes His, rather than Arg (present in the p allele), or Tyr which is found in human GzmB. His is also present at the same position in M. casteneus and M. spretus, mouse subspecies commonly found in certain parts of Asia. Given that 222Lys is invariant, it is extremely likely that the altered substrate preferences that result in the w allele having a far greater ability to cleave mouse Bid than to activate pro-caspases directly must rely on 180His. We found that in contrast to GzmBP, we found that GzmBW is unable to effectively activate some pro-caspases directly. Thus, apoptosis induced by GzmBW is reliant on activating the intrinsic pathway. Overall, a combination of changes in GzmB's fine substrate specificity, together with the expression of MCMV-encoded inhibitors of MOMP accounts for the failure of GzmBW expressing CD8 T cells to kill virally infected cells, resulting in uncontrolled viral replication.
In summary, we have demonstrated that a GzmB polymorphism commonly found in the wild has a profound influence on the ability of mice to control a natural pathogen. Importantly, the devastating effects elicited in hosts carrying the GzmBW allele by what is a common viral infection suggest that this allele has been maintained in the population because it confers a survival advantage in a setting yet to be defined. Furthermore, the function of GzmB in the response to pathogens and tumours has been investigated almost exclusively using inbred mouse strains all of which express the same allele of GzmB. The results of this study suggest that the use of mouse strains expressing alternative alleles of GzmB will be important for gaining a full understating of the role played by GzmB during an immune response. These findings also raise the possibility that alleles of GzmB identified in humans could impact on the control of some human pathogens.
Materials and Methods
Ethics statement
This study was performed in accordance with the recommendations in the Australian code of practice for the care and use of animals for scientific purposes and the Australian National Health and Medical Research Council Guidelines and Policies on Animal Ethics. Experiments were approved by the Animal Ethics and Experimentation Committee of the University of Western Australia (Protocol # RA3/100/1094), the Animal Ethics and Experimentation Committee of the Peter MacCallum Cancer Centre (Protocol #E381) and the Animal Ethics and Experimentation Committee of the Australian National University (Protocol # A2012/041).
Protein production
Recombinant granzymes were produced as artificial zymogens in Pichia pastoris, activated using enterokinase following purification, and assessed for the ability to cleave the synthetic peptide thiobenzylester (Boc-Ala-Ala-Asp (AAD)-SBzl [10], [21]. A GzmBw cDNA with optimized mouse codon usage was synthesized in vitro (GenScript). Specific activity of purified mouse granzymes was assessed by SDS-stable binding to an enhanced form of Serpinb9 (Cys339Asp) produced as described [42]. Preparations were routinely >95% active.
Production of granzyme substrates in vitro
35S-labeled mouse procaspase 3 or mouse Bid was produced from cDNAs in the expression vector pSVTf via in vitro transcription and translation [19].
Phage display
Recombinant granzymes were used to probe a P1 Asp-anchored library, as described [43].
Generation of wild GzmB mouse lines
A wild mouse colony (B1–6) maintained at the Animal Resource Centre (Canning Vale, WA), which expressed the outbred GzmB allele (GzmBw/w) [20] was crossed with C57BL/6 mice. Mice homozygous for GzmBw/w were backcrossed to C57BL/6 for 22 generations. These F22 mice were also crossed with B6.OT1 (ovalbumin-specific, H-2b -restricted T-cell receptor transgenic) mice to generate a GzmBw/w.OT1 line.
Mice
Inbred C57BL/6J (B6) mice were obtained from the Animal Resources Centre (Perth, Western Australia, Australia), orWalter and Eliza Hall Institute (Melbourne). B6.granzymeA−/− (GzmA−/−), B6.granzymeB−/− (GzmB−/−), B6.granzymeAB−/− (GzmAB−/−), B6.perforin−/− (Pfp−/−) B6.OT1, B6.Gzm.AB−/−.OT1, and B6.pfp.OT1 mice were bred and maintained at the Peter MacCallum Cancer Centre, Melbourne. B6 mice carrying the Serpinb9tm1.1/Pib allele (Serpinb9−/− mice) were generated and maintained at Monash University [44]. Mice were used at 6–10 weeks of age.
Generation of primary mouse CTL
OVA257 specific activated T cells were generated from the spleens of the various OT1 mice (B6, B6.Pfp−/−, B6.GzmAB−/−, B6.GzmBw/w) as previously described [45]. The cytotoxic activity of the CTL was verified in 51Cr release assays using H-2b-peptide pulsed target cells (EL-4), as previously described [46].
Granule enzyme activity assays
Whole cell lysates were prepared from CTL cultures and normalized for protein content. Granule enzyme activity was determined as previously described [47] using synthetic peptide thiobenzylester (Boc-Ala-Ala-Asp (AAD)-SBzl and Suc-Phe-Leu-Phe SBzl) and paranitroanilide (Acetyl-Ile-Glu-Pro-Asp-paranitroanilide, Ac-IEPD-pNA) substrates (SM Biochemicals, CA, USA).
Western blot
Ten µg of whole cell lysate was separated on NuPAGE 4–12% Bis Tris gels (Life Technologies, CA, USA), transferred and probed for mouse GrB protein (rat anti-mouse GrB, clone 16G6, eBioscience, CA, USA), as previously described [12]. Equal protein loading was confirmed by re-probing the blot with an anti-mouse β-actin antibody (Sigma-Aldrich, USA).
Viral infections
Murine cytomegalovirus
For pathogenesis studies, mice were inoculated intraperitoneally (ip) with 2×104 plaque-forming units (pfu) of salivary gland propagated (SGV) MCMV-Δm157 mutant virus [48]. SGV stocks were diluted in phosphate-buffered saline-0.05% fetal bovine serum. A dose of 5×103 pfu was used for phenotypic analysis of lymphocytes.
Ectromelia virus
Mice were inoculated with the Moscow strain of ECTV subcutaneously (sc) with 105 pfu of virus in the flank of the left hind limb (hock) of mice under avertin anesthesia. All animals were monitored daily for clinical signs of disease, weighed every 2–3 days and euthanized if they lost 25% of their original body weight and recorded as dead the following day.
Viral quantification
MCMV viral titers in organs were determined by plaque assay using M210B4 cells as previously described [49].
ETCV genome copies in blood were measured by quantitative real time PCR (qRT-PCR) to amplify the target sequence of ECTV-Mos-156 gene, as described elsewhere [50]. Oligonucleotide primers used were, forward: CGCTACACCTTATCCTCAGACAC, and reverse: GGAATTGGGCTCCTTATACCA. Viral DNA was prepared using QiaAmp DNA Mini Kit (Qiagen Pty Ltd, Victoria, Australia) as per manufacturer's instructions. Serial dilutions of a plasmid encoding ECTV-Mos-156 were used as the standard. The qRT-PCR reaction was carried out in SYBR iQ Supermix (Bio-Rad Laboratories), in a total volume of 20 µl using the iQ5 cycler (Bio-Rad Laboratories Pty Ltd, New South Wales, Australia).
Flow cytometry
Single-cell suspensions were prepared by perfusing the liver via the portal vein with phosphate buffered saline (PBS). The liver was then digested with collagenase buffer (RPMI/2% FCS, 1% (w/v) collagenase IV (GIBCO) for 20 min before being passing through steel mesh. The resulting preparation was resuspended in in a 37.5% isotonic Percoll solution (Pharmacia) and centrifuged at 690 g for 12 min to separate lymphocytes from hepatocytes. Red blood cells were osmotically lysed using NH4Cl and cells washed in FACS buffer. Antibodies used Antibodies used for analysis (TCRβ, CD8, CD4, CD11b, CD11c, Ly6C, Ly6G) were purchased from BD Biosciences and the M45 tetramer was obtained from ImmunoID Tetramers, University of Melbourne, Australia. Dead cells were excluded from analysis using propidium iodide.
Cytokine quantification
IFNγ and TNFα levels in the liver were measured by standard sandwich enzyme-linked immunosorbent assay (ELISA) with antibodies from BD Biosciences. Detection was achieved with poly-horseradish peroxidase (poly-HRP) conjugated to streptavidin (CBL, Amsterdam, Netherlands) and K-Blue (Elisa Systems, Brisbane, Australia).
Histological analysis
Organs were removed from mice at the designated times and fixed in 10% buffered formal saline. Organs were then embedded in paraffin, tissue sections prepared and sections stained with haematoxylin and counter stained with eosin. IE1 protein was detected by staining slides with an anti-IE1 monoclonal antibody (Clone Chroma 101), and detected with goat anti mouse HRPO and metal enhanced DBA substrate (Thermo Fisher).
Cell death assays
Cell death induced by perforin (0.135–1.3 nM) and the recombinant mouse inbred and the outbred wild GzmB (12.5–25 nM) was assessed by 51Cr release assays as previously described [51].
Cytotoxic activity of M45 specific T cells was assessed by preparing a single-cell suspension from the spleens. Splenocytes were diluted 2-fold on 96-well plates starting with 1×106 cells/well and 51Cr-labeled EL4 cells pulsed with M45 peptide (1×104 cells/well) were added. Each assay was performed in triplicate. Chromium release was measure after 4 h incubation. Data are presented as percentage of specific lysis, calculated by the following formula: percentage specific lysis = (experimental c.p.m.−spontaneous release c.p.m.)/(total c.p.m.−spontaneous release c.p.m.)×100.
The capacity of activated CD8 T cells to kill MCMV infected cells was assessed using IC-21 macrophages. B6 or GzmBW/W mice were infected with Δm157 virus and spleens removed at day 6 post-infection. CD8 T cells were purified from the spleen using a CD8a positive selection kit (Stem Cell Technologies) according to the manufacturers instructions. IC-21 macrophages were infected with MCMV 12 h prior to co-culture with the purified CD8 T cells. Cell viability was quantified after 16 h of co-culture by MTT assay.
Statistical analysis
In vitro assays were assessed using a one way ANOVA with Tukey's multiple comparison test. Statistical analysis of survival curves were performed using the Log Rank test. Differences in viral replication within organs were assessed using a two-tailed Mann-Whitney test. Statistical tests were performed using the statistical software package InStat (GraphPad Software, San Diego California USA).
Supporting Information
{kind=link}
Zdroje
1. RussellJH, LeyTJ (2002) Lymphocyte-mediated cytotoxicity. Ann Rev Immunol 20 : 323–370.
2. MullbacherA, WaringP, Tha HlaR, TranT, ChinS, et al. (1999) Granzymes are the essential downstream effector molecules for the control of primary virus infections by cytolytic leukocytes. Proc Natl Acad Sci U S A 96 : 13950–13995.
3. RieraL, GariglioM, ValenteG, MullbacherA, MuseteanuC, et al. (2000) Murine cytomegalovirus replication in salivary glands is controlled by both perforin and granzymes during acute infection. European journal of immunology 30 : 1350–1355.
4. van DommelenSL, SumariaN, SchreiberRD, ScalzoAA, SmythMJ, et al. (2006) Perforin and granzymes have distinct roles in defensive immunity and immunopathology. Immunity 25 : 835–848.
5. VoskoboinikI, SmythMJ, TrapaniJA (2006) Perforin-mediated target-cell death and immune homeostasis. Nature reviews Immunology 6 : 940–952.
6. BeresfordPJ, XiaZ, GreenbergAH, JL (1999) Granzyme A loading induces rapid cytolysis and a novel form of DNA damage independently of caspase activation. Immunity 10 : 585–594.
7. MartinvaletD, ZhuP, LiebermanJ (2005) Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity 22 : 355–370.
8. SusantoO, StewartSE, VoskoboinikI, BrasacchioD, HagnM, et al. (2013) Mouse granzyme A induces a novel death with writhing morphology that is mechanistically distinct from granzyme B-induced apoptosis. Cell death and differentiation 20 : 1183–1193.
9. CullenSP, AdrainC, LuthiAU, DuriezPJ, MartinSJ (2007) Human and murine granzyme B exhibit divergent substrate preferences. The Journal of cell biology 176 : 435–444.
10. KaisermanD, BirdCH, SunJ, MatthewsA, UngK, et al. (2006) The major human and mouse granzymes are structurally and functionally divergent. The Journal of cell biology 175 : 619–630.
11. Casciola-RosenL, Garcia-CalvoM, BullHG, BeckerJW, HinesT, et al. (2007) Mouse and human granzyme B have distinct tetrapeptide specificities and abilities to recruit the bid pathway. The Journal of biological chemistry 282 : 4545–4552.
12. SuttonVR, DavisJE, CancillaM, JohnstoneRW, RuefliAA, et al. (2000) Initiation of apoptosis by granzyme B requires direct cleavage of bid, but not direct granzyme B-mediated caspase activation. The Journal of experimental medicine 192 : 1403–1414.
13. SuttonVR, WowkME, CancillaM, TrapaniJA (2003) Caspase activation by granzyme B is indirect, and caspase autoprocessing requires the release of proapoptotic mitochondrial factors. Immunity 18 : 319–329.
14. WaterhouseNJ, SedeliesKA, BrowneKA, WowkME, NewboldA, et al. (2005) A central role for Bid in granzyme B-induced apoptosis. The Journal of biological chemistry 280 : 4476–4482.
15. WaterhouseNJ, SedeliesKA, SuttonVR, PinkoskiMJ, ThiaKY, et al. (2006) Functional dissociation of DeltaPsim and cytochrome c release defines the contribution of mitochondria upstream of caspase activation during granzyme B-induced apoptosis. Cell death and differentiation 13 : 607–618.
16. KnickelbeinJE, KhannaKM, YeeMB, BatyCJ, KinchingtonPR, et al. (2008) Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 322 : 268–271.
17. AndradeF, FellowsE, JenneDE, RosenA, YoungCS (2007) Granzyme H destroys the function of critical adenoviral proteins required for viral DNA replication and granzyme B inhibition. EMBO J 26 : 2148–2157.
18. McIlroyD, CartronP-F, TufferyP, DudoitY, SamriA, et al. (2003) A triple-mutated allele of granzyme B incapable of inducing apoptosis. Proceedings of the National Academy of Sciences of the United States of America 100 : 2562–2567.
19. SunJ, BirdCH, ThiaKY, MatthewsAY, TrapaniJA, et al. (2004) Granzyme B encoded by the commonly occurring human RAH allele retains pro-apoptotic activity. The Journal of biological chemistry 279 : 16907–16911.
20. ThiaKYT, TrapaniJA (2007) The granzyme B gene is highly polymorphic in wild mice but essentially invariant in common inbred laboratory strains. Tissue antigens 70 : 198–204.
21. SunJ, BirdCH, BuzzaMS, McKeeKE, WhisstockJC, et al. (1999) Expression and purification of recombinant human granzyme B from Pichia pastoris. Biochemical and biophysical research communications 261 : 251–255.
22. SunJ, BirdCH, SuttonV, McDonaldL, CoughlinPB, et al. (1996) A cytosolic granzyme B inhibitor related to the viral apoptotic regulator cytokine response modifier A is present in cytotoxic lymphocytes. The Journal of biological chemistry 271 : 27802–27809.
23. SunJ, OomsL, BirdCH, SuttonVR, TrapaniJA, et al. (1997) A new family of 10 murine ovalbumin serpins includes two homologs of proteinase inhibitor 8 and two homologs of the granzyme B inhibitor (proteinase inhibitor 9). The Journal of biological chemistry 272 : 15434–15441.
24. BirdCH, SuttonVR, SunJR, HirstCE, NovakA, et al. (1998) Selective regulation of apoptosis - the cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme-B-mediated apoptosis without perturbing the Fas cell death pathway. Mol & Cell Biol 18 : 6387–6398.
25. AnthonyDA, AndrewsDM, WattSV, TrapaniJA, SmythMJ (2010) Functional dissection of the granzyme family: cell death and inflammation. Immunological Reviews 235 : 73–92.
26. EdwardsKM, KamCM, PowersJC, TrapaniJA (1999) The human cytotoxic T cell granule serine protease granzyme H has chymotrypsin-like (chymase) activity and is taken up into cytoplasmic vesicles reminiscent of granzyme B-containing endosomes. Journal of Biological Chemistry 274 : 30468–30473.
27. VoigtV, ForbesCA, TonkinJN, Degli-EspostiMA, SmithHR, et al. (2003) Murine cytomegalovirus m157 mutation and variation leads to immune evasion of natural killer cells. Proc Natl Acad Sci U S A 100 : 13483–13488.
28. ScalzoAA, ManzurM, ForbesCA, BrownMG, ShellamGR (2005) NK gene complex haplotype variability and host resistance alleles to murine cytomegalovirus in wild mouse populations. Immunology & Cell Biology 83 : 144–149.
29. KeesU, BlandenRV (1976) A single genetic element in H-2K affects mouse T-cell antiviral function in poxvirus infection. Journal of Experimental Medicine 143 : 450–455.
30. LohJ, ChuDT, O'GuinAK, YokoyamaWM, VirginHWt (2005) Natural killer cells utilize both perforin and gamma interferon to regulate murine cytomegalovirus infection in the spleen and liver. J Virol 79 : 661–667.
31. ZhangM, ParkS-M, WangY, ShahR, LiuN, et al. (2006) Serine protease inhibitor 6 protects cytotoxic T cells from self-inflicted injury by ensuring the integrity of cytotoxic granules. Immunity 24 : 451–461.
32. GoldMC, MunksMW, WagnerM, KoszinowskiUH, HillAB, et al. (2002) The murine cytomegalovirus immunomodulatory gene m152 prevents recognition of infected cells by M45-specific CTL but does not alter the immunodominance of the M45-specific CD8 T cell response in vivo. Journal of Immunology 169 : 359–365.
33. MetkarSS, MenaaC, PardoJ, WangB, WallichR, et al. (2008) Human and mouse granzyme A induce a proinflammatory cytokine response. Immunity 29 : 720–733.
34. AnthonyDA, AndrewsDM, ChowM, WattSV, HouseC, et al. (2010) A role for granzyme M in TLR4-driven inflammation and endotoxicosis. Journal of immunology (Baltimore, Md: 1950) 185 : 1794–1803.
35. BaschukN, WangN, WattSV, HalseH, HouseC, et al. (2014) NK cell intrinsic regulation of MIP-1alpha by granzyme M. Cell death & disease. 5: e1115.
36. SedeliesKA, CicconeA, ClarkeCJP, OliaroJ, SuttonVR, et al. (2008) Blocking granule-mediated death by primary human NK cells requires both protection of mitochondria and inhibition of caspase activity. Cell death and differentiation 15 : 708–717.
37. PinkoskiMJ, WaterhouseNJ, HeibeinJA, WolfBB, KuwanaT, et al. (2001) Granzyme B-mediated apoptosis proceeds predominantly through a Bcl-2-inhibitable mitochondrial pathway. The Journal of biological chemistry 276 : 12060–12067.
38. JurakI, SchumacherU, SimicH, VoigtS, BruneW (2008) Murine cytomegalovirus m38.5 protein inhibits Bax-mediated cell death. J Virol 82 : 4812–4822.
39. ManzurM, FlemingP, HuangDC, Degli-EspostiMA, AndoniouCE (2009) Virally mediated inhibition of Bax in leukocytes promotes dissemination of murine cytomegalovirus. Cell Death Differ 16 : 312–320.
40. CamM, HandkeW, Picard-MaureauM, BruneW (2010) Cytomegaloviruses inhibit Bak - and Bax-mediated apoptosis with two separate viral proteins. Cell Death Differ 17 : 655–665.
41. FlemingP, KvansakulM, VoigtV, KileBT, KluckRM, et al. (2013) MCMV-mediated inhibition of the pro-apoptotic Bak protein is required for optimal in vivo replication. PLoS Pathogens 9: e1003192.
42. SunJ, CoughlinP, SalemHH, BirdP (1995) Production and characterization of recombinant human proteinase inhibitor 6 expressed in Pichia pastoris. Biochimica et biophysica acta 1252 : 28–34.
43. SongJ, MatthewsAY, ReboulCF, KaisermanD, PikeRN, et al. (2011) Predicting serpin/protease interactions. Methods in enzymology 501 : 237–273.
44. RizzitelliA, MeuterS, Vega RamosJ, BirdCH, MinternJD, et al. (2012) Serpinb9 (Spi6)-deficient mice are impaired in dendritic cell-mediated antigen cross-presentation. Immunology and cell biology 90 : 841–851.
45. LopezJA, JenkinsMR, Rudd-SchmidtJA, BrennanAJ, DanneJC, et al. (2013) Rapid and unidirectional perforin pore delivery at the cytotoxic immune synapse. Journal of immunology (Baltimore, Md: 1950) 191 : 2328–2334.
46. KonjarS, SuttonVR, HovesS, RepnikU, YagitaH, et al. (2010) Human and mouse perforin are processed in part through cleavage by the lysosomal cysteine proteinase cathepsin L. Immunology. 131 : 257–267.
47. SuttonVR, WaterhouseNJ, BrowneKA, SedeliesK, CicconeA, et al. (2007) Residual active granzyme B in cathepsin C-null lymphocytes is sufficient for perforin-dependent target cell apoptosis. The Journal of cell biology 176 : 425–433.
48. CorbettAJ, CoudertJD, ForbesCA, ScalzoAA (2011) Functional consequences of natural sequence variation of murine cytomegalovirus m157 for Ly49 receptor specificity and NK cell activation. J Immunol 186 : 1713–1722.
49. OngML, WikstromME, FlemingP, EstcourtMJ, HertzogPJ, et al. (2013) CpG pretreatment enhances antiviral T-cell immunity against cytomegalovirus. Blood 122 : 55–60.
50. TahilianiV, ChaudhriG, EldiP, KarupiahG (2013) The orchestrated functions of innate leukocytes and T cell subsets contribute to humoral immunity, virus control, and recovery from secondary poxvirus challenge. Journal of Virology 87 : 3852–3861.
51. SuttonVR, VauxDL, TrapaniJA (1997) bcl-2 prevents apoptosis induced by perforin and granzyme B, but not that mediated by whole cytotoxic lymphocytes. J Immunol 158 : 5783–5790.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 12
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Plasma Membrane-Located Purine Nucleotide Transport Proteins Are Key Components for Host Exploitation by Microsporidian Intracellular Parasites
- Rubella Virus: First Calcium-Requiring Viral Fusion Protein
- Emergence of MERS-CoV in the Middle East: Origins, Transmission, Treatment, and Perspectives
- Neutral Sphingomyelinase in Physiological and Measles Virus Induced T Cell Suppression
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy