
TREM-1 Deficiency Can Attenuate Disease Severity without Affecting Pathogen Clearance
Triggering receptor expressed on myeloid cells-1 (TREM-1) is a potent amplifier of pro-inflammatory innate immune reactions. While TREM-1-amplified responses likely aid an improved detection and elimination of pathogens, excessive production of cytokines and oxygen radicals can also severely harm the host. Studies addressing the pathogenic role of TREM-1 during endotoxin-induced shock or microbial sepsis have so far mostly relied on the administration of TREM-1 fusion proteins or peptides representing part of the extracellular domain of TREM-1. However, binding of these agents to the yet unidentified TREM-1 ligand could also impact signaling through alternative receptors. More importantly, controversial results have been obtained regarding the requirement of TREM-1 for microbial control. To unambiguously investigate the role of TREM-1 in homeostasis and disease, we have generated mice deficient in Trem1. Trem1−/− mice are viable, fertile and show no altered hematopoietic compartment. In CD4+ T cell - and dextran sodium sulfate-induced models of colitis, Trem1−/− mice displayed significantly attenuated disease that was associated with reduced inflammatory infiltrates and diminished expression of pro-inflammatory cytokines. Trem1−/− mice also exhibited reduced neutrophilic infiltration and decreased lesion size upon infection with Leishmania major. Furthermore, reduced morbidity was observed for influenza virus-infected Trem1−/− mice. Importantly, while immune-associated pathologies were significantly reduced, Trem1−/− mice were equally capable of controlling infections with L. major, influenza virus, but also Legionella pneumophila as Trem1+/+ controls. Our results not only demonstrate an unanticipated pathogenic impact of TREM-1 during a viral and parasitic infection, but also indicate that therapeutic blocking of TREM-1 in distinct inflammatory disorders holds considerable promise by blunting excessive inflammation while preserving the capacity for microbial control.
Published in the journal:
. PLoS Pathog 10(1): e32767. doi:10.1371/journal.ppat.1003900
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003900
Summary
Triggering receptor expressed on myeloid cells-1 (TREM-1) is a potent amplifier of pro-inflammatory innate immune reactions. While TREM-1-amplified responses likely aid an improved detection and elimination of pathogens, excessive production of cytokines and oxygen radicals can also severely harm the host. Studies addressing the pathogenic role of TREM-1 during endotoxin-induced shock or microbial sepsis have so far mostly relied on the administration of TREM-1 fusion proteins or peptides representing part of the extracellular domain of TREM-1. However, binding of these agents to the yet unidentified TREM-1 ligand could also impact signaling through alternative receptors. More importantly, controversial results have been obtained regarding the requirement of TREM-1 for microbial control. To unambiguously investigate the role of TREM-1 in homeostasis and disease, we have generated mice deficient in Trem1. Trem1−/− mice are viable, fertile and show no altered hematopoietic compartment. In CD4+ T cell - and dextran sodium sulfate-induced models of colitis, Trem1−/− mice displayed significantly attenuated disease that was associated with reduced inflammatory infiltrates and diminished expression of pro-inflammatory cytokines. Trem1−/− mice also exhibited reduced neutrophilic infiltration and decreased lesion size upon infection with Leishmania major. Furthermore, reduced morbidity was observed for influenza virus-infected Trem1−/− mice. Importantly, while immune-associated pathologies were significantly reduced, Trem1−/− mice were equally capable of controlling infections with L. major, influenza virus, but also Legionella pneumophila as Trem1+/+ controls. Our results not only demonstrate an unanticipated pathogenic impact of TREM-1 during a viral and parasitic infection, but also indicate that therapeutic blocking of TREM-1 in distinct inflammatory disorders holds considerable promise by blunting excessive inflammation while preserving the capacity for microbial control.
Introduction
Innate immune cells express several cell surface receptors and intracellular sensing molecules that allow for autonomous recognition of pathogen - and danger-associated molecular patterns (PAMPs and DAMPs) and initiation of pro-inflammatory anti-microbial reponses. Toll-like receptors (TLR) and nucleotide-binding oligomerization domain (NOD)-like receptors, which recognize a diverse group of highly conserved microbial structures, represent only two examples of large innate immune receptor families with activating functions. Over the last decade, an additional family of evolutionary conserved innate immune receptors has been identified and characterized, the so-called triggering receptors expressed on myeloid cells (TREMs). TREMs belong to the immunoglobulin (Ig) superfamily of receptors and contain both inhibitory and activating receptors [1], [2], [3]. In contrast to the fairly ubiquitously expressed TLRs and NOD-like receptors, expression of TREMs is restricted to cells of the myeloid lineage [4]. Moreover, based on their capacity to integrate and potently modulate TLR - and NOD-induced signals, TREMs appear to mainly act as fine-tuners rather than initiators of inflammatory responses [3], [5]. While TREM-1, TREM-2, TREM-3 (in the mouse) receptors [4], [6], and the TREM-1 like transcripts TLT-1 and TLT-2 have been described [7], [8], TREM-1 is the first identified and best characterized receptor of the TREM family with activating functions. TREM-1 consists of an ectodomain, composed of a single Ig V-type domain, a transmembrane region and a short cytoplasmic tail that recruits DAP12 for signaling [4]. TREM-1 is constitutively expressed on neutrophils and on subsets of monocytes and macrophages, and TREM-1 expression is further upregulated upon exposure of cells to microbial products [4]. Whereas crosslinking of TREM-1 with agonistic antibodies alone induces only modest cellular activation, TREM-1 potently synergizes with distinct TLR ligands for a substantial amplification of oxidative burst and production of pro-inflammatory mediators such as TNF, IL-1β, IL-6, IL-8, MCP-1 and Mip-1α [4], [9], [10].
In vivo, the role of TREM-1 has been mostly characterized in experimental models of endotoxin-induced shock or microbial sepsis where blockade of TREM-1 signaling conferred significant protection [9], [11], [12]. The detection of TREM-1 in inflammatory lesions caused by bacterial or fungal agents, but not in psoriasis or immune-mediated vasculitis [9], has further led to the general concept that TREM-1 is primarily involved in microbial diseases, particularly, since elevated levels of the serum soluble form of the shed TREM-1 surface receptor (sTREM-1) also appear to associate with bacterial infections in patients with pneumonia or suspected sepsis [13], [14].
However, increasing evidence is now emerging that TREM-1 may additionally play a role in non-infectious inflammatory conditions. Thus, expression of TREM-1 can also be induced by the non-microbial agent monosodium urate monohydrate crystals (MSU) or by hypoxic cell culture conditions in vitro [15], [16]. Augmented sTREM-1 levels have been reported for patients with rheumatoid arthritis, acute pancreatitis, chronic obstructive pulmonary disease and cardiac arrest [17], [18], [19], [20]. Furthermore, we have previously described an involvement of TREM-1 in human inflammatory bowel diseases (IBD) and in models of experimental colitis [21], [22], [23].
Investigations on the precise function of TREM-1 in distinct diseases have so far been complicated by the still unidentified ligand(s) for TREM-1. Putative ligands for TREM-1 have been described on the surface of human platelets and on murine granulocytes during experimental peritonitis and endotoxaemia [12], [24], [25]. In addition, necrotic cell lysates also appear to stimulate pro-inflammatory responses in a TREM-1-dependent manner, which may relate to association of TREM-1 with the High Mobility Group Box 1 (HMGB1) protein [26], [27]. Hence, it can be speculated that not only PAMPs but also DAMPs induce signaling via TREM-1 and that several ligands for TREM-1 may exist.
In the absence of clearly defined ligands for TREM-1, studies addressing the impact of TREM-1 in disease have so far mostly relied on the use of TREM-1/Ig fusion proteins or synthetic peptides mimicking part of the extracellular domain of TREM-1. Although by the use of these agents substantial protection from endotoxin-induced shock, microbial sepsis or experimental colitis could be conferred [9], [11], [12], [22], several aspects regarding the true biological role of TREM-1 remain unclear. First, considering the redundancy of innate immune receptor-ligand interactions, the possibility exists that in these previous studies not only signaling through TREM-1 but through additional, potentially more relevant receptors was prevented. Second, controversial findings have been obtained with respect to the impact of impaired TREM-1 signaling on microbial control [9], [28], [29], [30].
In order to investigate the role of TREM-1 in homeostasis and disease, we have generated a TREM-1-deficient (Trem1−/−) mouse by targeted deletion of exon 2. Here we show, employing distinct inflammation and infection models ranging from experimental colitis to infections with Leishmania major, influenza virus and Legionella pneumophila, that complete absence of TREM-1 significantly attenuates morbidity and immune-mediated pathologies while microbial control remains unimpaired. These findings not only demonstrate an unanticipated clear role for TREM-1 in chronic inflammatory disorders, parasitic and viral infections, but also illustrate the potential for a novel therapeutic intervention in various disease settings.
Results
Deletion of Trem1 has no apparent impact under homeostatic conditions
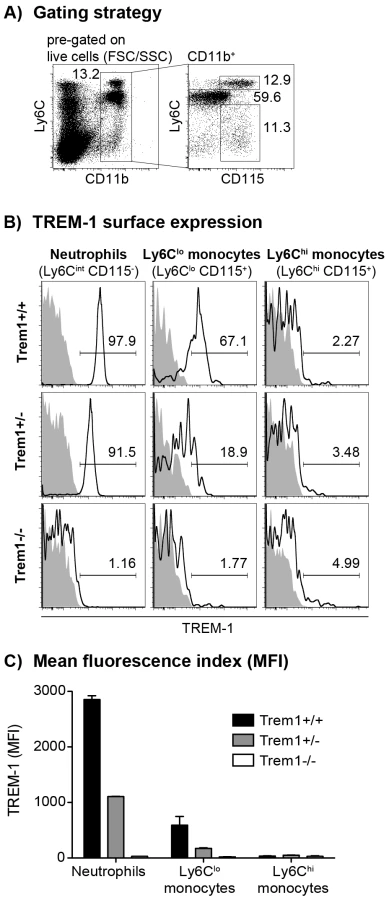
To account for potential embryonically lethal effects of a total deletion of the Trem1 gene, a targeting vector was designed for conditional deletion of exon 2 (Fig. S1). Exon 2 encodes the extracellular domain of TREM-1 and also contains the putative ligand binding site [31]. Breeding of Trem1+/flox chimeric offspring mice with deleter mice that expressed Cre ubiquitously yielded viable Trem1+/− x Cre+/− offspring. Moreover, interbreeding of Trem1+/− mice gave rise to Trem1−/− mice at the expected Mendelian frequencies, and Trem1−/− mice were equal in size, weight and fertility to littermate Trem1+/+ controls. We thus continued to characterize Trem1−/− mice with a ubiquitously deleted Trem1 gene by elementary flow cytometry analyses. Deletion of Trem1 resulted in a gene-dose-dependent loss of TREM-1 surface expression by peripheral blood neutrophils and Ly6Clo monocytes (Fig. 1). Accordingly, TREM-1 was still expressed at ∼2-fold reduced levels in Trem1+/− mice, while surface TREM-1 expression was absent on myeloid cells in Trem1−/− mice (Fig. 1). Absence of Trem1 did not appear to affect the composition of various immune compartments, since numbers of distinct myeloid and lymphoid cell subsets isolated from the peripheral blood, bone marrow (BM) and spleen of Trem1−/− and Trem1+/+ mice were identical (Fig. S2). However, to formally exclude a potential effect of TREM-1 on hematopoiesis, the BM of Trem1−/− and Trem1+/+ mice was analysed in more depth with respect to hematopoietic stem cell and myeloid progenitor numbers following lineage depletion and depletion of lymphoid progenitors (Fig. 2). Stem cell-enriched cells were identified by their lineage− (lin−) Sca-1+ c-kithi phenotype (LSK cells) while common myeloid progenitors (CMP), granulocyte/macrophage progenitors (GMP) and megakaryocyte/erythrocyte precursors (MEP) were discriminated within the Sca-1− c-kithi population according to their differential expression of FcγR and CD34, respectively (Fig. 2a). Compared to Trem1+/+ mice, Trem1−/− mice exhibited equal numbers of LSK cells, CMP, GMP and MEP (Fig. 2B). Moreover, similar numbers of colony forming units could be observed in lineage-depleted (lin−) BM cells isolated from Trem1−/− mice (Fig. 2B). Although these analyses indicated again that TREM-1 is unlikely to play a substantial role in hematopoietic processes, we were intrigued by the selective expression of surface TREM-1 by GMP, but not by CMP (Fig. 2A). As a final measure, we therefore established mixed bone marrow chimeras with either Trem1−/− (x GFP−/−) and Trem1+/+ x GFP+/+ BM cells or, as a control, Trem1+/+ (x GFP−/−) and Trem1+/+ x GFP+/+ BM cells. Analysis of chimeric mice at 10 and 31 weeks post reconstitution and calculation of the respective ratios of GFP− to GFP+ peripheral blood neutrophil, Ly6Chi or Ly6Clo monocyte numbers demonstrated an equal capacity of Trem1−/− BM to give rise to distinct myeloid subsets as Trem1+/+ BM. Thus, while the potential role of TREM-1 expression by GMP still remains to be explored, deficiency in Trem1 does not appear to affect hematopoietic processes under homeostatic conditions.

We next addressed whether absence of Trem1 could affect other receptors that use DAP12 for signaling, either by the potential presence of increased levels of intracellularly available DAP12 or by the lack of counterregulatory signals conferred by TREM-1. Indeed, the hyperresponsive phenotype of DAP12-deficient macrophages is largely ascribed to a lack of inhibitory signals by TREM-2 which also employs DAP12 [32]. Due to the important role of TREM-2 in osteoclast formation and function [33], [34], we reasoned that lack of TREM-1 expression in Trem1−/− mice could possibly manifest in altered osteoclastogenesis. However, as determined by Xray and MicroCT analyses, no differences in bone density could be detected between Trem1−/− mice and their age - and sex-matched Trem1+/+ controls (data not shown).
Taken together, these analyses revealed no apparent phenotype of Trem1−/− mice under homeostatic conditions.
Trem1−/− x Rag2−/− mice are largely protected from a CD4 T cell-induced colitis
We have previously demonstrated a substantial accumulation of TREM-1 expressing macrophages in the inflamed, but not healthy intestinal mucosa of patients with IBD and of mice with experimental colitis [22], [23]. Hence, one of our major interests in the characterization of the Trem1−/− mouse was to unambigously investigate the role of Trem1 in the pathogenesis of IBD. To this end, CD4+ CD25− CD45RBhi T cells were adoptively transferred into Helicobacter-positive Trem1+/+ x Rag2−/− and Trem1−/− x Rag2−/− recipient mice and animals were monitored regularly for clinical signs of colitis. Importantly, whereas Trem1+/+ x Rag2−/− mice lost ∼20% of their initial body weight at the end of the observation period, weight loss in Trem1−/− x Rag2−/− mice was minimal and only transient (Fig. 3A). Furthermore, shortening of the colon was substantially attenuated in Trem1−/− x Rag2−/− mice compared to controls (Fig. 3B). While some of the Trem1−/− x Rag2−/− mice still exhibited moderate histopathological signs of intestinal inflammation, individual parameters of the histopathological scoring as well as the overall histopathological score were significantly reduced (Fig. 3C–E).

In order to gain insight in the potential underlying mechanism of the highly attenuated colitis in Trem1−/− x Rag2−/− mice, colonic lamina propria cells that were isolated from both groups of mice in the absence of an adoptive CD4+ T cell transfer (healthy colon) or 12–13 days post colitis induction were analysed by FACS. As depicted in Fig. 4A, the colonic lamina propria of healthy Trem1+/+ x Rag2+/+ mice and Trem1−/− x Rag2−/− mice contained a similar proportion of CD11b+ MHCIIhi cells and Gr1+ cells were virtually absent. In contrast, Gr1+ cells, representing infiltrating Ly6Chi Gr1int monocytes and Ly6Cint Gr1hi neutrophils, were readily detected in Trem1+/+ x Rag2+/+ mice and Trem1−/− x Rag2−/− mice at 12–13 days post colitis induction (Fig. 4A). Notably, the relative frequency of Gr1+ cells among CD45+ CD11b+ colonic LP cells was ∼5-fold lower in Trem1−/− x Rag2−/− mice (Fig. 4A). In further contrast to the colonic LP of healthy mice, among LP MHCII+ Gr1− cells of colitic mice two populations of MHCIIhi Ly6Clo and MHCIIint Ly6Chi cells could be discriminated, likely representing intestinal macrophages and monocytes in the process of differentiation (Fig. 4A) [35], [36]. Also within this gate of MHCII+ Gr1− cells, substantial differences could be detected between the two groups of mice. Accordingly, the relative frequency of MHCIIint Ly6Chi cells was increased in colitic Trem1+/+ x Rag2−/− mice whereas Trem1−/− x Rag2−/− mice exhibited a larger proportion of MHCIIhi Ly6Clo macrophages (Fig. 4A).

When colonic LP cells of n = 9 mice of both groups were systematically analysed at 12–13 days post colitis induction, substantially reduced numbers of various cell subsets could be seen in Trem1−/− x Rag2−/− mice (Fig. 4B). Thus, Trem1−/− x Rag2−/− mice not only exhibited reduced infiltrating CD4+ T cells but also significantly decreased numbers of neutrophils, Ly6Chi monocytes and MHCIIint Ly6Chi cells (Fig. 4B). These differences were not apparent for the colonic LP of healthy Trem1+/+ x Rag2−/− and Trem1−/− x Rag2−/− mice which mainly contained MHCIIhi Ly6Clo cells anyway (Fig. 4A and 4C).
To gain more insight which myeloid TREM-1-expressing cell subset could potentially be involved in driving intestinal inflammation in Trem1+/+ x Rag2−/− mice, TREM-1 surface expression was analysed on colonic LP neutrophils, Ly6Chi monocytes as well as CD11b+ Ly6C+ Gr1− and CD11b+ Ly6C− Gr1− cells. As reported previously [23], [37], in the healthy colonic LP TREM-1 expression was hardly detectable owing to the absence of infiltrating neutrophils and Ly6C+ cells (Fig. 4A and 4D). In colitic Trem1+/+ x Rag2−/− mice, TREM-1 expression was observed on neutrophils, Ly6Chi monocytes and CD11b+ Ly6C+ Gr1− cells (Fig. 4D). Moreover, CD11b+ Ly6C− Gr1− cells, likely representing intestinal macrophages, that were isolated from colitic Trem1+/+ x Rag2−/− mice exhibited a ∼3-fold upregulated expression of surface TREM-1 (Fig. 4D).
In line with the reduced infiltrating cell numbers, mRNA expression for various innate and adaptive pro-inflammatory chemokines and cytokines was significantly decreased in the lamina propria of Trem1−/− x Rag2−/− compared to Trem1−/− x Rag2−/− mice (Fig. 4E).
Dextran sodium sulfate (DSS)-induced colitis is attenuated in Trem1−/− mice
While protection from colitis in Trem1−/− x Rag2−/− mice was associated with reduced expression of several pro-inflammatory mediators, previous data generated in our laboratory have demonstrated that TNF produced by nonlymphoid cells plays a non-redundant pathogenic role in the CD4+ T cell transfer model of colitis since Tnf−/− x Rag2−/− mice are completely protected from colitis induction [38]. However, in acute models of intestinal inflammation such as the DSS-induced colitis, Tnf−/− mice exhibit aggrevated disease [39], [40], presumably, because early anti-microbial and repair responses following DSS-induced breaching of the epithelial barrier are fundamentally impaired. Due to the central function of TREM-1 in amplifying pro-inflammatory cytokine production and oxidative burst, we hypothesized that during acute intestinal inflammation complete absence of Trem1 could also prove detrimental. Intriguingly, although upon administration of 3% DSS Trem1−/− mice initially lost weight to a similar extent as Trem1+/+ mice, weight loss was considerably attenuated at 7 days post colitis induction and at 9 days Trem1−/− mice had already improved again (Fig. 5A). In Trem1−/− mice, shortening of the colon was markedly attenuated and total histopathological colitis scores were significantly decreased (Fig. 5B and 5C–E). Furthermore, analogous to the CD4 T cell-induced colitis (Fig. 4E), TREM-1 deficiency resulted in reduced colonic mRNA expression of several pro-inflammatory mediators (Fig. 5F). Hence, in contrast to Tnf−/− mice [39], [40], Trem1−/− mice still showed an adequate host response to DSS-induced epithelial injury while exhibiting reduced immune-mediated pathologies.

Infection with Leishmania major leads to smaller inflammatory lesions with decreased neutrophilic cellular infiltrates in Trem1−/− mice
The observations made in the acute DSS model of colitis raised our interest whether Trem1−/− mice would also be able to control bona fide microbial infections, in particular, since maximal silencing of TREM-1 by a siRNA approach had proven deleterious in a fecal peritonitis model [29]. Since the rapid kinetics of this model hardly allowed to simultaneously look at beneficial effects of the Trem1 deficiency on immune-mediated tissue damage or to assess potential adverse consequences for the priming of adaptive immune responses, we chose the Leishmania major infection model. Following infection with L. major, C57BL/6 mice develop local cutaneous lesions that spontaneously resolve within 4–8 weeks. Central to the resolution is the TNF-mediated control of the early inflammatory response or the clearance of neutrophils and the later IFNγ-mediated and Th1-driven elimination of the parasite by infected macrophages [41], [42].
Upon injection of 3×106 L. major promastigotes s.c. in the footpad of Trem1+/+ and Trem1−/− mice, an attenuation in lesion development was apparent in Trem1−/− mice already at 14 days post infection. From thereof, Trem1−/− mice showed a significantly decreased lesion size (Fig. 6A). Notably, however, parasite counts did not differ between Trem1−/− and Trem1+/+ mice (Fig. 6B). We further analysed the cellular composition of infected footpads from Trem1−/− and Trem1+/+ mice at 21 days post infection. While the overall cell counts were comparable, the cellular infiltrate in Trem1−/− mice exhibited ∼3-fold reduced neutrophil numbers (Fig. 6C). To look at the potential impact of the Trem1-deficiency on the priming of Th1 responses, expression of IFNγ was analysed in cells isolated from the draining lymph node.

The frequency of IFNγ-secreting CD4+ T cells was similar in Trem1−/− and Trem1+/+ mice (Fig. 6D). In addition, comparable levels of IFNγ were detected in cells of both groups of mice upon re-stimulation in vitro with the parasitic antigen (Fig. 6D). These data are in line with comparable parasite killing observed in Trem1−/− and Trem1+/+ mice. Thus, in the L. major infection model, absence of Trem1 does not appear to have adverse consequences on the priming of adaptive immune responses and on parasite control while neutrophil-mediated inflammatory lesion development is substantially reduced.
Stimulation via TREM-1 induces TNF secretion and resistance to apoptosis in SCF-condHoxb8 progenitor-derived neutrophils
The reduced neutrophil numbers at L. major-infected sites and the decreased lesion size in Trem1−/− mice agree with the notion that neutrophils play a central role in inflammatory lesion development. Indeed, the presence of non-healing lesions in L. major susceptible BALB/c strains is associated with elevated numbers of neutrophils [43]. Since neutrophils constitutively express high levels of TREM-1 (Fig. 1B, 4D), we aimed to investigate the consequences of TREM-1-mediated stimulation on their functional responses in more detail. Analysis of mouse neutrophils has so far been complicated by the limited numbers of cells that can be retrieved from the peripheral blood, their short life span or the distinct differentiation stages of BM-derived neutrophils. Hence, we made use of a recently described system by which neutrophils can be differentiated ex vivo in large numbers using conditional Hoxb8 [44]. Using a slightly modified protocol, BM-derived progenitors were lentivirally transduced with conditional Hoxb8 in the presence of SCF, resulting in immortalized myeloid progenitor lines, termed SCF-condHoxb8. Upon shutdown of Hoxb8 expression by withdrawal of 4-OHT, cells differentiate into mature neutrophils in the presence of SCF. As shown in Figure 7A and 7B, withdrawal of 4-OHT in fact induced the appearance of cells bearing the characteristic phenotype of mouse neutrophils after in vitro differentiation for 5 days. Moreover, Trem1+/+ SCF-condHoxb8-derived mature neutrophils also expressed distinct levels of surface TREM-1 (Fig. 7B). Stimulation of Trem1+/+, but not of Trem1−/−, SCF-condHoxb8-derived mature neutrophils with a plate-bound agonistic anti-TREM-1 antibody resulted in pronounced mRNA expression of iNOS and TNF (Fig. 7C) and induced rapid secretion of TNF at levels comparable to those triggered by LPS (Fig. 7D).

Since neutrophil survival versus apoptosis could represent a deciding factor in the control of inflammation not only in the L. major infection model but also in the pathogenesis of experimental colitis [45], we compared the susceptibility of Trem1+/+ and Trem1−/− neutrophils to spontaneous apoptosis. Following prolonged culture of fully differentiated SCF-condHoxb8-derived neutrophils in vitro, an increasing and comparable frequency of AnnexinV+DAPI+ cells was detected for Trem1+/+ and Trem1−/− neutrophils (Fig. 7E). However, in the presence of TREM-1-mediated stimulation a reduced apoptosis rate based on diminished Caspase 3/7 activity could be observed for Trem1+/+ neutrophils (Fig. 7F). Furthermore, agonistic anti-TREM-1 stimulation of Trem1+/+ but not Trem1−/− neutrophils resulted in ∼2-fold upregulated mRNA expression of Myeloid Cell Leukemia-1 (Mcl-1) (Fig. 7G), analogously to what has recently been described for human TREM-1-stimulated monocytes [46].
Thus, TREM-1-mediated activation of mouse neutrophil appears to contribute to their survival.
Influenza virus-infected Trem1−/− mice exhibit reduced morbidity but an equal capacity for viral clearance
Intrigued by the substantially diminished inflammatory lesions, yet intact parasite clearance in L. major-infected Trem1−/− mice, we aimed to substantiate these findings in an altogether different infection model. Since high expression of TREM-1 by alveolar macrophages and previously published data [28], [30], [47], [48] suggest a potential role for TREM-1 in lung inflammatory responses, we infected Trem1+/+ and Trem1−/− mice intratracheally with 50 PFU influenza A virus PR8. Hypothermia and body weight loss, which are characteristically associated with experimental influenza virus infection, were observed in Trem1+/+ mice at 7 days post infection (Fig. 8A and 8B). While the body temperature also dropped in Trem1−/− mice, a quicker recovery from hypothermia was seen in the Trem1−/− group (Fig. 8A). Moreover, weight loss in Trem1−/− mice was significantly attenuated and Trem1−/− mice further exhibited reduced levels of IL-6 in bronchoalveolar lavage fluid (BALF) at 10 days post infection (Fig. 8B and 8C). Notably, in spite of the reduced morbidity observed, Trem1−/− mice were equally competent in clearing the influenza virus infection as Trem1+/+ controls (Fig. 8D).

Trem1−/− mice are equally capable of clearing L. pneumophila as Trem1+/+ controls
After having established that deficiency in TREM-1 attenuates disease but does not impair pathogen control during a parasitic and viral infection, we last sought to address the significance of TREM-1 in a bacterial infection model. Indeed, controversial results have been obtained with respect to the importance of TREM-1 in microbial control following infection of experimental animals with Pseudomonas aeruginosa [28], [30]. Here, we employed a Legionella pneumophila infection model which also causes severe upper airway inflammation in permissive mice and critically depends on neutrophil-mediated control [49], [50], [51]. As shown in Figure 9, at 3 days post infection with 5×106 CFU of L. pneumophila, CFU and neutrophil numbers in the BALF did not significantly differ between Trem1+/+ and Trem1−/− mice. Furthermore, at day 5 post infection CFU were reduced to < 500/BALF in both groups of mice with no significant differences observed between Trem1+/+ and Trem1−/− mice (data not shown).

Discussion
The significance of TREM-1 as a central amplifier of acute pro-inflammatory responses during endotoxin-induced shock and microbial sepsis is well established. However, increasing evidence, including the recently reported association of TREM-1 with the DAMP protein HMGB1 [26], [27], also suggests a potential role for TREM-1 during non-infectious and chronic inflammatory conditions. In line with this notion, we have previously described a crucial involvement of TREM-1 in IBD as based on the significant amelioration of experimental colitis upon blockade of TREM-1 with the antagonistic LP17 peptide [22]. Blocking TREM-1 signaling by daily administration of TREM-1-Ig fusion proteins or synthetic analogues in chronic disease models is not only costly and straining but also fails to cover for the possibility that the yet unidentified TREM-1 ligand may signal through alternative receptors.
Here, we have generated a Trem1−/− mouse to unambiguously investigate the impact of a complete TREM-1-deficiency on the pathogenesis of experimental colitis but also of two other distinct sub-acute disease settings where the role of TREM-1 has so far not been addressed in vivo, i.e. inflammation induced by a parasitic and viral infection. Our findings demonstrate that Trem1−/− mice not only show a highly attenuated CD4+ T cell - and DSS-induced colitis but also display significantly reduced lesion size and diminished morbidity during infections with L. major and influenza virus, respectively.
The substantial attenuation of illness and immune-mediated pathologies in Trem1−/− mice across these distinct models suggests a common mechanism by which TREM-1 signaling promotes inflammation irrespective of the original trigger. Several non-exclusive scenarios can be considered that may account for the attenuated disease in Trem1−/− mice: A priori reduced chemotactic recruitment of Trem1−/− neutrophils and monocytes, diminished pro-inflammatory activities, and/or reduced life-span of myeloid cell subsets. Although we observed significantly decreased numbers of distinct myeloid cell subsets in the LP of colitic Trem1−/− x Rag2−/− mice and at L. major-infected sites in Trem1−/− mice, we consider it unlikely that deficiency in TREM-1 causes an intrinsic primary defect in chemotaxis. When we analysed L. major infected sites at an early time-point, i.e. 3 days post infection, no differences in cellularity were detected between Trem1+/+ and Trem1−/− mice (data not shown). Moreover, a recent study clearly demonstrated that TREM-1/3 proteins are not required for transendothelial migration of neutrophils [30]. Nonetheless, the markedly decreased expression of mRNA for monocyte - (CCL2), granulocyte - (CXCL1, CXCL2, CXCL5) but also T cell (CXCL9) chemoattractants in the LP of Trem1−/− x Rag2−/− mice will in a secondary manner certainly have contributed to the decreased accumulation of inflammatory cells. Besides the diminished expression of chemotactic mediators, the colonic LP of colitic Trem1−/− x Rag2−/− mice also exhibited substantially reduced mRNA levels of several innate cytokines, including IL-1β, IL-6 and TNF. The reduced expression of pro-inflammatory mediators in the entire colonic LP in Trem1−/− x Rag2−/− mice at the late stage of colitis induction certainly also mirrors the decreased cellular infiltration. However, considering the capacity of TREM-1 to augment the production of several chemotactic mediators either directly in infiltrating myeloid cells or likely also indirectly, in a paracrine manner (e.g. through secretion of IL-1β) [4], [22], [52], we believe that TREM-1-amplified production of pro-inflammatory cytokines represents a key early pathogenic event that will ultimately determine the later disease course and may largely account for the attenuated disease in Trem1−/− mice. In this respect, it is noteworthy that the colonic LP of colitic Trem1−/− mice contained markedly fewer MHCIIint Ly6Chi cells or inflammatory macrophages with the capacity for expression of pro-inflammatory mediators [35], [36].
As we have employed a CD4+ T cell-dependent colitis model and indeed observed considerably reduced CD4+ T cell numbers and correspondingly also mRNA levels for IFNγ and IL-17 in the colonic LP of transferred Trem1−/− x Rag2−/− mice, the question arises whether deficiency in TREM-1 may directly impact the priming of adaptive immune responses. Whereas in the colitis model we have not analysed CD4+ T cell responses in more detail, CD4+ T cells isolated from L. major-infected and CD8+ T cells retrieved from influenza virus-infected Trem1−/− mice, respectively, exhibited an unimpaired capacity for IFNγ production compared to T cells from Trem1+/+ mice.
The deciding role of neutrophils in the L. major infection model [42] and the substantially decreased lesion size in Trem1−/− mice have prompted us to investigate the impact of TREM-1 on neutrophil-mediated functions in more detail. In particular, we were interested in the potential modulatory effect of TREM-1 ligation on neutrophil survival as delayed neutrophil apoptosis could also represent a critical pathogenic factor in intestinal inflammation [45]. In agreement with a previous report [30], deficiency in TREM-1 caused no intrinsic predisposition for increased spontaneous neutrophil apoptosis. However, agonistic TREM-1 stimulation significantly promoted survival of SCF-condHoxb8 progenitor-derived neutrophils in vitro. We have also sought to assess the frequency of apoptotic neutrophils in Trem1+/+ versus Trem1−/− mice in situ by staining colonic tissue sections from colitic mice for cleaved caspase 3 (data not shown). However, due to the likely very rapid clearance of apoptotic neutrophils by intestinal phagocytes, cleaved caspase 3 positive cells were rare in Trem1+/+ mice and even more scarce in Trem1−/− mice. Moreover, the substantially decreased cellular infiltrate in Trem1−/− mice further precluded an objective comparison and quantification of apoptotic cells in situ. While we cannot present definitive evidence that TREM-1 prolongs neutrophil survival in vivo, we still believe that the reduced presence of neutrophils in Trem1-deficient mice in the CD4 T cell induced colitis and L. major infection model relates to both: A reduced secondary recruitment based on the diminished expression of neutrophil chemotactic mediators and to a reduced life-span in the absence of TREM-1 ligation, with the former mechanism numerically perhaps being more relevant.
Considering the various effector cell types and mechanisms by which TREM-1-mediated stimulation could contribute to disease, it may appear intriguing that deficiency in TREM-1 did not completely protect from disease. Accordingly, the degree of protection from colitis was not much higher in Trem1−/− mice compared to mice that were treated with the antagonistic LP17 peptide in our previous study [22]. We hypothesize that the absence of complete protection in Trem1−/− mice may primarily relate to the role of TREM-1 as an amplifier but not inducer of pro-inflammatory reactions [3], [5]. While we cannot rule out a potential participation of TREM-3, we believe that the protective effects seen in Trem1−/− mice are too substantial for a major involvement of TREM-3 in the inflammatory models analysed.
One of the most striking findings of the present study was the observation that microbial control in the models analysed was apparently not impaired in Trem1−/− mice in spite of the blunted inflammatory responses. Hence, while Tnf−/− or anti-TNF-treated mice exhibit an aggrevated acute DSS-induced colitis [39], [40] and also show enhanced parasite and bacterial burdens upon infection with L. major and L. pneumophila, respectively [41], [53], Trem1−/− mice appeared equally capable of controlling a parasitic, viral and bacterial infection as Trem1+/+ controls. This observation is in line with the main function of TREM-1 as an inflammatory fine-tuner which still allows for pro-inflammatory TLR or NOD-like receptor-induced reactions in its absence. Moreover, TREM-1 does not appear to be involved in phagocytic or direct antimicrobial activity of myeloid cells [30], [54], [55]. Still, conflicting data on the effect of a TREM-1 blockade on microbial control have been reported from various bacterial challenge models. Injection of a TREM-1/IgG fusion protein allowed for sufficient control of an E. coli-induced peritoneal infection and conferred protection [9] whereas maximal but not half-dose siRNA silencing of TREM-1 increased mortality in a fecal peritonitis model [29]. Similarly, administration of the antagonistic LP17 peptide protected rats from a P. aeruginosa-induced pneumonia [28], whereas complete deficiency in TREM-1/3 led to markedly increased mortality in Pseudomonas aeruginosa-challenged mice due to defective transepithelial migration of neutrophils [30]. It has been proposed that the degree of TREM-1 blockade was a likely critical parameter to account for these disparant findings [29], [30]. However, our findings demonstrate that microbial control must not necessarily be impaired in mice with a complete deficiency in TREM-1, even when employing a L. pneumophila infection model where early neutrophil accumulation is also crucial for bacterial clearance [49], [50], [51]. Thus, we speculate that analogous to the divergent results obtained for endotoxin-challenged DAP12-deficient mice [56], [57], possibly the infection dose, the nature of the microbial agent and/or the kinetics of the infection may be critical parameters regarding the requirement for TREM-1. Accordingly, in the presence of low abundance and/or low affinity TLR ligands or fast replicating agents, the inflammatory response may more stringently depend on TREM-1-amplified signaling. Alternatively, the expression of TREM-1 and its association with DAP12 may only preferentially be induced in situations of high abundance and/or high affinity TLR ligands whereas low level signaling would favour the engagement of TREM-2.
In summary, while the impact of TREM-1 on microbial control still needs further investigations across different experimental models, our extensive characterisation of Trem1−/− mice shows an unanticipated prominent role for TREM-1 in parasitic and viral infections. Our findings thus suggest that therapeutic targeting of TREM-1 holds considerable promise for distinct non-infectious and infectious inflammatory disorders and may bypass the increased risk for impaired microbial control which is associated with the general targeting of TNF.
Materials and Methods
Mice
Breedings and cohort maintenance were performed under SPF conditions in isolated ventilated cages in the central animal facility of the Medical School, University of Bern. All studies were conducted with age - and sex-matched animals. Trem1+/+ mice that were used as wildtype controls were originally derived from the interbreeding of Trem1+/− x Cre+/− mice, thus carrying the identical C57BL/6 genetic background and representing former littermates of Trem1−/− mice. To nonetheless adjust for potential differences in the microflora composition in Trem1−/− vs. Trem1+/+ mice, the bedding of Trem1+/+ and Trem1−/− mice was regularly exchanged between cages three weeks prior to the start of the experiments.
Ethics statement
All animal experiments were approved by the Veterinary Offices of the Cantons of Bern, Lausanne and Zurich and performed in compliance with Swiss laws for animal protection.
Generation of Trem1-deficient mice
The generation of Trem1-deficient mice was designed and carried out in collaboration with the TaconicArtemis GmbH (Köln, Germany). To account for potential lethal effects of a total deletion of the Trem1 gene and to allow for a possible cell-specific ablation of Trem1 expression, a targeting vector was designed for conditional deletion of exon 2, which encodes the extracellular part of Trem1 and also contains the putative ligand binding site [31]. As illustrated in the supplemental material Fig. S1, the targeting vector was constructed on the basis of the cloning vector KS loxP ftr Neo BS to flank exon 2 with loxP sites, to comprise additional restriction sites (AseI and AvaI) and to contain PuroR (flanked by F3 sites) and Neomycin (flanked by FRT sites) positive selection marker cassettes to control for homologous recombination upstream and downstream of exon 2, respectively. For counterselection, a Tk cassette was included downstream of exon 4. As a template for the PCR reactions a BAC-based plasmid containing the entire genomic mouse Trem1 locus (RP23-32N8) was obtained from BACPAC Resources Center BPRC (Oakland, USA). The targeting vector was electroporated into a C57BL/6N.tac embryonic stem cell line (TaconicArtemis). On day 2, cells were selected with Puromycin and G418 and on day 8 counterselection with Gancyclovir was initiated. Isolated and expanded ES clones were screened for complete integration of the targeted allele by standard Southern blotting analyses with probes located upstream of exon 1 (5e2) or exon 3 (ila1) (Fig. S1). Primer sequences for generation of the 5e2 Southern probes were: CGGATTTGACCAGGAATGACAG(sense) and CTTCCAGTTCATTCATGGACAGC (antisense) and for the ila1 Southern probe: AGCTCCTCTTGTCTGCCATTCAAGGC (sense) and GGCTACAACCTTGTTCTGCAG (antisense). Eight positive clones could be identified and the ES clone A-A5 was subsequently injected into Balb/c derived blastocytes which were then transferred to pseudopregnant NMRI females. Chimeric offspring were bred to C57BL/6 female mice (C57BL/6-Tg(ACTB-Flpe)2Arte, TaconicArtemis) transgenic for Flp recombinase to achieve deletion of the FRT and F3 flanked selection cassettes PuroR and Neomycin, respectively. Germline transmission of the targeted Trem1 allele was identified by coat color contribution and by PCR using oligo 1_sense (GTGCTCAGAGAATGTCTTTGTATCC) and oligo 4_antisense (CCCTGGTCAGACCATTTACC) which either yield a 1.3 kb fragment for the wildtype (WT) allele or a 1.6 kb fragment for the conditional Trem1flox allele. (Fig. S1). Cycling conditions were: 5′ at 95°C followed by 35 cycles consisting of 30″ at 95°C, 30″ at 60°C, 1′ at 72°C, followed by 10′ at 72°C. Thus identified Trem1+/flox mice (C57BL/6-TREM-1tm1821_33.1Arte) were mated with male mice carrying the Cre recombinase under the control of the ROSA26 locus (C57BL/6-Gt(ROSA)26Sortm16(Cre)arte, TaconicArtemis) to obtain systemic deletion of one Trem1 allele (Trem1+/−). Trem1+/− x Cre+/− mice were interbred to achieve deletion of Cre and to obtain wildtype controls (Trem1+/+) and heterozygous (Trem1+/−) and homozygous (Trem1−/−) Trem1-deficient mice. Deletion of exon 2 in Trem1+/− and Trem1−/− mice was assessed by the genotyping PCR strategy described above and depicted in Fig. S1. Trem1+/+ and Trem1−/− mice were subsequently expanded for experiments. For the CD4 adoptive transfer model of colitis, Trem1−/− x Rag2−/− mice were generated by crossing Trem1−/− mice with Helicobacter+ Rag2−/− mice and interbreeding of the F1 offspring. The Helicobacter+ status of the Trem-1−/− x Rag2−/− offspring was confirmed by PCR testing (Microbios GmbH, Reinach, Switzerland).
Flow cytometry (FACS)
The following mAbs were used: anti-mouse CD11b-Pacific Blue (M1/70), CD45-Pacific Blue (30-F11), CD45-Brilliant Violet570 (30-F11), CD4-APC-Cy7 (RM4-5), Gr1-PE (RB6-8C5), NK1.1-PE-Cy7 (PK136), Ly6G-APC-Cy7 (1A8) and F4/80-biotin (BM8) IL-7Rα-biotin (A7R34), CD3ε-biotin (145-2C11) CD19-biotin (6D11), Gr1-biotin (RB6-8C5) and Ter119-biotin (TER119), all purchased from Biolegend; anti-mouse CD115-PE (AFS98), Gr1-APC (RB6-8C5), CD45-eFluor605 (30-F11), CD45.1-PE (A20), MHCII-APC (M5/114.15.2), CD11c-PE (N418), CD11b-eFluor450 (M1/70), Streptavidin-PE-Cy7, were purchased from eBioscience (San Diego, USA); anti-mouse Ly6C-FITC (AL-21) from BD Pharmingen (San Diego, USA) and anti-mouse TREM-1-APC (174031) from R&D Systems. DAPI (Invitrogen) was used in a final concentration of 0.5 µg/ml to exclude dead cells. Prior to FACS stainings, Fc receptors were blocked using supernatant from the hybridoma 2.4G2. Cells were acquired on a LSRII SORP (BD Biosciences, San Diego, USA) and analysed using FlowJo cytometric analysis program (Tree Star, Ashland, USA).
Impact of TREM-1 on hematopoiesis
Analysis of hematopoietic stem cells and progenitors
For FACS analysis of stem cell enriched LSK cells and myeloid progenitors, and also for determination of colony-forming units, a prior lineage depletion was performed using biotinylated mAbs against red blood cell precursors (α-Ter119), B cells (α-CD19), T cells (α-CD3ε), myeloid cells (α-Gr1), MACS α-biotin beads, and LS columns (Miltenyi Biotec). Lymphoid progenitors were further removed by adding anti-IL-7Rα-biotin.
Determination of colony forming units
3.33×103 lin− cells were transferred into methocult base medium (M3134; Stemcell Technologies) supplemented with 15% FCS, 20% BIT (50 mg/ml BSA in IMDM, 1.44 U/ml recombinant-human (rh) insulin (Actrapid, Novo Nordisk) and 250 ng/ml human holo transferrin [Prospec]), 100 µM 2-β-mercaptoethanol, 2 mM l-glutamine, penicillin/streptomycin, and 50 ng/ml recombinant-mouse SCF (rmSCF), 10 ng/ml rm–IL-3, 10 ng/ml rh-IL-6 and 50 ng/ml rm-Flt3-ligand (all from Prospec). Colonies and cells were enumerated after 7 days (≥30 cells/colony).
Generation of mixed bone marrow chimeras
Congenic (CD45.1+) recipient mice were irradiated in two split doses with 650 cGy in a 4 h interval in a Gammacell 40 exactor (Best Theratronics). Total donor bone marrow (BM) was collected from Trem1−/−, Trem1+/+ and Trem1+/+ x GFP+/+ mice as described below. Donor BM was mixed 1∶1 and 15×106 total cells of either mixed Trem1−/− and Trem1+/+ x GFP+/+ BM cells or mixed Trem1+/+ and Trem1+/+ x GFP+/+ BM cells were transferred in 200 µl PBS i.v. into the irradiated recipient mice. After transfer, the recipients were treated with antibiotics (Baitryl and Bactrim) in the drinking water for two weeks. After 10 weeks, the grade of chimerism was controlled in the peripheral blood by calculating the CD45.2+/CD45.1+ ratio. In all chimeras, the grade of chimerism among the circulating myeloid cells was at least 99%.
Generation and analysis of SCF-dependent conditional Hoxb8-immortalised progenitor cells and neutrophils
Generation of Hoxb8 progenitor lines and differentiation of neutrophils
The protocol was adapted from the method described by Wang et al. [44] with the major modification of using a different inducible expression system [58]. In brief, bone marrow-derived haematopoietic progenitors were isolated from Trem1+/+ and Trem1−/− mice by magnetic bead-based lineage depletion (BD IMag™ mouse hematopoietic progenitor cell enrichment set-DM, BD Biosciences) following the manufacturer's instructions. 2–5×105 lineage− cells were incubated for 36 h in complete RPMI Medium (RPMI 1640/Glutamax supplemented with 10% FCS, 1% penicillin/streptomycin solution and 50 µM 2-mercaptoethanol). Cells were then transduced with pF-5×UAS-Hoxb8(mm)-SV40-puro-GEV16 lentiviral particles by spin-infection (1000 rpm, 2 h, 30°C) in presence of 8 µg/ml polybrene. Cells were subsequently cultured in complete RPMI medium supplemented with SCF (added as 10% of CHO/SCF(mm)-conditioned supernatant) and Hoxb8 expression was induced by addition of 100 nM 4-OHT. Transduced cells were positively selected with 1.0 µg/ml puromycin for a minimum of one month. The resulting immortalized cell lines were termed SCF-condHoxb8. For in vitro differentiation of SCF-condHoxb8 cells into mature neutrophils, cells were washed twice in PBS to remove all traces of 4-OHT and were then replated at 20'000 cells/ml in complete RPMI medium containing SCF (but no 4-OHT). Mature neutrophils were thus obtained after 5–6 days as judged by morphology and surface expression of CD11b and Gr-1, as well as loss of c-kit (CD117) as determined by flow cytometry.
Apoptosis measurements
Spontaneous apoptosis of mature Trem1+/+ versus Trem1−/− neutrophils was assessed at 5 days post differentiation in SCF-containing medium and 8 h, 24 h, and 48 h later. Immediately upon removal from plates, neutrophils were washed in cold PBS and AnnexinV Binding Buffer and stained with FITC-conjugated AnnexinV (BD Pharmingen) according to the manufacturer's instructions. DAPI (Sigma-Aldrich) was added immediately prior to aquisition by flow cytometry. Only AnnexinV and DAPI double-positive cells (AnnexinV+ DAPI+) were considered in the analysis of apoptotic cells. For determination of TREM-1-mediated effects on apoptosis induction, 5 days differentiated neutrophils were stimulated in vitro in 96-well U-bottom plates at 2×105 cells/well in complete RPMI medium lacking SCF in the presence of 10 µg/ml plate-bound anti-TREM-1 mAb (MAB1187; R&D) or a respective isotype control (RTK2758, Biolegend) for 24 h. Since the relative stickiness of neutrophils activated by plate-bound anti-TREM-1 did not allow for removal from the plates and FACS-based analysis of AnnexinV+ DAPI+ cells without causing a substantial bias by the selective analysis of non-adherent cells, Caspase 3/7 activity was determined by the ApoTox-Glo assay (Promega) according to manufacturer's instructions.
Experimental animal models
CD4 T cell adoptive transfer model of colitis
Colitis was induced in (Helicobacter-positive) Rag2−/− and Trem1−/− x Rag2−/− mice by adoptive transfer of 2×105 CD4+ CD25− CD45RBhi FACS-sorted T cells as described previously [59]. Mice were sacrificed at 12–13 days post CD4 T cell transfer at the onset of clinical signs of colitis (diarrhea, weight loss, symptoms of abdominal pain) in Trem1+/+ mice.
Dextran sodium-sulfate (DSS)-induced colitis
Colitis was induced in (Helicobacter-negative) Trem1+/+ and Trem1−/− mice by administration of 3% 36'000–50'000 MW DSS (MP Biomedicals, Solon, USA) in the drinking water for 5 days followed by 4 days of regular tap water and euthanization at 9 days.
Leishmania major (L. major) infections
L. major LV39 (MRHO/Sv/59/P strain) parasites were maintained in vivo in DBA/2J mice and further cultured in vitro in M199 medium supplemented with 10% FCS, 4% HEPES and 2% antibiotics (penicillin, streptomycin, neomycin). Mice were infected with 3×106 parasites s.c. in the hind footpad in a final volume of 50 µl as previously described [41]. Footpad lesion size was measured with a Vernier caliper. The number of parasites in lesions were evaluated by limiting dilution analysis [60].
Influenza virus infections
Influenza virus strain PR8 (A/Puerto Rico/34 H1N1) was originally provided by J. Pavlovic (University of Zurich, Switzerland). For infections, mice were anaesthetized and inoculated intratracheally with 50 PFU influenza virus in 50 µl endotoxin-free PBS. For collection of bronchoalveolar lavage (BAL) fluid, lungs were flushed with 3×400 µl PBS. To determine influenza viral titers in the lungs, lungs were collected at the indicated time-points, homogenized and serially diluted with MDCK cells as previously described [61].
Legionella pneumophila infections
L. pneumophila strain JR32 (Philadelphia-1; sg1) (reference: PMID: 8225610) was grown for 3 days on charcoal yeast extract (CYE) agar plates at 37°C and resuspended in PBS prior to infection. Mice were anesthetized by i.p. injection of 5 µg xylazine/100 µg ketamine per gram body weight and infected intranasally with 5×106 L. pneumophila. Three days post infection, mice were sacrificed and perfused and bronchoalveolar lavage fluid (BALF) was extracted with 1 ml PBS, 5 mM EDTA. CFU were quantified by plating serial dilutions on CYE agar plates and counted after 3 days incubation at 37°C. BALF neutrophils were quantified by flow-cytometry.
Cell isolations
Spleen and peripheral blood
Blood was collected by tail vain incision or by cardiac puncture into heparinised PBS. Spleen cells were released by homogenization of spleens between the frosted ends of two glass slides into PBS containing 5% horse serum. Erythrocytes were depleted by brief incubation with ACK lysis buffer.
Bone marrow
Femurs and tibiae were removed and placed in ice-cold PBS. Remaining flesh was removed and bones were rinsed with sterile PBS. After opening of the bones, the BM was flushed with a 26 GA 3/8 needle into RPMI 10% FCS. Erythrocyte-depleted BM cells were either used for FACS-based characterizations with or without prior lineage depletion or for the generation of mixed BM chimeras.
Colonic lamina propria
Colons were opened longitudinally and cut into small pieces. The epithelium was removed by incubation in HBSS/HEPES containing 5% horse serum, 5 mM EDTA and 2 mM DTT at 37°C for 3×30 min under magnetic stirring. Lamina propria (LP) cells were obtained by subsequent digestion with 200 U/ml collagenase (Type IV; Sigma-Aldrich) and 50 U/ml DNase (Type I, grade II; Roche) for 2×45 min. The LP fraction was filtered through a 40 µM cell strainer, counted by Trypan Blue staining and further characterized by FACS.
Footpads
Footpads were digested using 1 mg/ml collagenase D in HBSS.
Histopathological analysis of mouse intestinal tissue sections
To assess the presence of histopathological alterations on formalin-fixed, paraffin-embedded and hematoxylin-eosin-stained colonic tissue sections, a scoring system ranging from 0 (no alterations) to 15 (most severe signs of colitis) was established, including the following parameters: cellular infiltration (0–3), loss of goblet cells (0–3), crypt abscesses (0–3), epithelial erosions (0–1), hyperemia (0–2), thickness of the colonic mucosa (0–3). Histological scoring was performed by a pathologist (V.G.) blinded to sample identity.
RNA extractions and quantitative RT-PCR analyses
RNA was isolated using RNA isolation reagent (Tri-Reagent, Molecular Research Center). DNA was digested using DNase I (Ambion), and cDNA was generated using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Expression of genes was analysed using Qiagen Quantitect Primer Assays on a 7500 Real-time PCR System (AB Biosystems). The house keeping gene GAPDH was used for normalization of gene expression.
Analysis of cytokines in supernatants and bronchoalveolar lavage (BAL)
Cell-free supernatants derived from SCF−cond Hoxb8 neutrophils and BAL fluid from influenza virus infected mice were analysed by ELISA (Biolegend).
Analysis of bone density
X-ray
High resolution X-ray analyses on anesthetized mice were performed with the MX-20 Faxitron (X-ray Corporation, Edimex, LePlessis, France).
MicroCT
For high resolution microcomputed tomography (MicroCT) tissues were fixed in 4% paraformaldehyde in PBS for 24 h and subsequently transferred to 70% EtOH for μCT analysis (MicroCT40, Scanco, Bruettisellen, Switzerland).
Statistical analyses
All data were analysed with GraphPad Prism software using the Student's t-test or 2-way ANOVA.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. AllcockRJ, BarrowAD, ForbesS, BeckS, TrowsdaleJ (2003) The human TREM gene cluster at 6p21.1 encodes both activating and inhibitory single IgV domain receptors and includes NKp44. Eur J Immunol 33 : 567–577.
2. FordJW, McVicarDW (2009) TREM and TREM-like receptors in inflammation and disease. Curr Opin Immunol 21 : 38–46.
3. Klesney-TaitJ, TurnbullIR, ColonnaM (2006) The TREM receptor family and signal integration. Nat Immunol 7 : 1266–1273.
4. BouchonA, DietrichJ, ColonnaM (2000) Cutting edge: inflammatory responses can be triggered by TREM-1, a novel receptor expressed on neutrophils and monocytes. J Immunol 164 : 4991–4995.
5. ArtsRJ, JoostenLA, van der MeerJW, NeteaMG (2013) TREM-1: intracellular signaling pathways and interaction with pattern recognition receptors. J Leukoc Biol 93 : 209–15.
6. ChungDH, SeamanWE, DawsMR (2002) Characterization of TREM-3, an activating receptor on mouse macrophages: definition of a family of single Ig domain receptors on mouse chromosome 17. Eur J Immunol 32 : 59–66.
7. BarrowAD, AstoulE, FlotoA, BrookeG, RelouIA, et al. (2004) Cutting edge: TREM-like transcript-1, a platelet immunoreceptor tyrosine-based inhibition motif encoding costimulatory immunoreceptor that enhances, rather than inhibits, calcium signaling via SHP-2. J Immunol 172 : 5838–5842.
8. KingRG, HerrinBR, JustementLB (2006) Trem-like transcript 2 is expressed on cells of the myeloid/granuloid and B lymphoid lineage and is up-regulated in response to inflammation. J Immunol 176 : 6012–6021.
9. BouchonA, FacchettiF, WeigandMA, ColonnaM (2001) TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature 410 : 1103–1107.
10. RadsakMP, SalihHR, RammenseeHG, SchildH (2004) Triggering receptor expressed on myeloid cells-1 in neutrophil inflammatory responses: differential regulation of activation and survival. J Immunol 172 : 4956–4963.
11. GibotS, Kolopp-SardaMN, BeneMC, BollaertPE, LozniewskiA, et al. (2004) A soluble form of the triggering receptor expressed on myeloid cells-1 modulates the inflammatory response in murine sepsis. J Exp Med 200 : 1419–1426.
12. GibotS, BuonsantiC, MassinF, RomanoM, Kolopp-SardaMN, et al. (2006) Modulation of the triggering receptor expressed on the myeloid cell type 1 pathway in murine septic shock. Infect Immun 74 : 2823–2830.
13. GibotS, CravoisyA, LevyB, BeneMC, FaureG, et al. (2004) Soluble triggering receptor expressed on myeloid cells and the diagnosis of pneumonia. N Engl J Med 350 : 451–458.
14. GibotS, Kolopp-SardaMN, BeneMC, CravoisyA, LevyB, et al. (2004) Plasma level of a triggering receptor expressed on myeloid cells-1: its diagnostic accuracy in patients with suspected sepsis. Ann Intern Med 141 : 9–15.
15. MurakamiY, AkahoshiT, HayashiI, EndoH, KawaiS, et al. (2006) Induction of triggering receptor expressed on myeloid cells 1 in murine resident peritoneal macrophages by monosodium urate monohydrate crystals. Arthritis Rheum 54 : 455–462.
16. BoscoMC, PierobonD, BlengioF, RaggiF, VanniC, et al. (2011) Hypoxia modulates the gene expression profile of immunoregulatory receptors in human mature dendritic cells: identification of TREM-1 as a novel hypoxic marker in vitro and in vivo. Blood 117 : 2625–2639.
17. CollinsCE, LaDT, YangHT, MassinF, GibotS, et al. (2009) Elevated synovial expression of triggering receptor expressed on myeloid cells 1 in patients with septic arthritis or rheumatoid arthritis. Ann Rheum Dis 68 : 1768–1774.
18. YasudaT, TakeyamaY, UedaT, ShinzekiM, SawaH, et al. (2008) Increased levels of soluble triggering receptor expressed on myeloid cells-1 in patients with acute pancreatitis. Crit Care Med 36 : 2048–2053.
19. RadsakMP, TaubeC, HaselmayerP, TenzerS, SalihHR, et al. (2007) Soluble triggering receptor expressed on myeloid cells 1 is released in patients with stable chronic obstructive pulmonary disease. Clin Dev Immunol 2007 : 52040.
20. Adib-ConquyM, MonchiM, GoulenokC, LaurentI, ThuongM, et al. (2007) Increased plasma levels of soluble triggering receptor expressed on myeloid cells 1 and procalcitonin after cardiac surgery and cardiac arrest without infection. Shock 28 : 406–410.
21. SaurerL, RihsS, BirrerM, Saxer-SeculicN, RadsakM, et al. (2012) Elevated levels of serum-soluble triggering receptor expressed on myeloid cells-1 in patients with IBD do not correlate with intestinal TREM-1 mRNA expression and endoscopic disease activity. J Crohns Colitis 6 : 913–923.
22. SchenkM, BouchonA, SeiboldF, MuellerC (2007) TREM-1-expressing intestinal macrophages crucially amplify chronic inflammation in experimental colitis and inflammatory bowel diseases. J Clin Invest 117 : 3097–3106.
23. WeberB, SaurerL, SchenkM, DickgreberN, MuellerC (2011) CX3CR1 defines functionally distinct intestinal mononuclear phagocyte subsets which maintain their respective functions during homeostatic and inflammatory conditions. Eur J Immunol 41 : 773–779.
24. HaselmayerP, Grosse-HovestL, von LandenbergP, SchildH, RadsakMP (2007) TREM-1 ligand expression on platelets enhances neutrophil activation. Blood 110 : 1029–1035.
25. ZanzingerK, SchellackC, NauschN, CerwenkaA (2009) Regulation of triggering receptor expressed on myeloid cells 1 expression on mouse inflammatory monocytes. Immunology 128 : 185–195.
26. El MezayenR, El GazzarM, SeedsMC, McCallCE, DreskinSC, et al. (2007) Endogenous signals released from necrotic cells augment inflammatory responses to bacterial endotoxin. Immunol Lett 111 : 36–44.
27. WuJ, LiJ, SalcedoR, MivechiNF, TrinchieriG, et al. (2012) The Proinflammatory Myeloid Cell Receptor TREM-1 Controls Kupffer Cell Activation and Development of Hepatocellular Carcinoma. Cancer Res 72 : 3977–3986.
28. GibotS, AlauzetC, MassinF, SennouneN, FaureGC, et al. (2006) Modulation of the triggering receptor expressed on myeloid cells-1 pathway during pneumonia in rats. J Infect Dis 194 : 975–983.
29. GibotS, MassinF, MarcouM, TaylorV, StidwillR, et al. (2007) TREM-1 promotes survival during septic shock in mice. Eur J Immunol 37 : 456–466.
30. Klesney-TaitJ, KeckK, LiX, GilfillanS, OteroK, et al. (2013) Transepithelial migration of neutrophils into the lung requires TREM-1. J Clin Invest 123 : 138–149.
31. RadaevS, KattahM, RostroB, ColonnaM, SunPD (2003) Crystal structure of the human myeloid cell activating receptor TREM-1. Structure 11 : 1527–1535.
32. TurnbullIR, GilfillanS, CellaM, AoshiT, MillerM, et al. (2006) Cutting edge: TREM-2 attenuates macrophage activation. J Immunol 177 : 3520–3524.
33. HumphreyMB, DawsMR, SpustaSC, NiemiEC, TorchiaJA, et al. (2006) TREM2, a DAP12-associated receptor, regulates osteoclast differentiation and function. J Bone Miner Res 21 : 237–245.
34. ColonnaM, TurnbullI, Klesney-TaitJ (2007) The enigmatic function of TREM-2 in osteoclastogenesis. Adv Exp Med Biol 602 : 97–105.
35. TamoutounourS, HenriS, LelouardH, de BovisB, de HaarC, et al. (2012) CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur J Immunol 42 : 3150–3166.
36. BainCC, ScottCL, Uronen-HanssonH, GudjonssonS, JanssonO, et al. (2013) Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6C(hi) monocyte precursors. Mucosal Immunol 6 : 498–510.
37. SchenkM, BouchonA, BirrerS, ColonnaM, MuellerC (2005) Macrophages expressing triggering receptor expressed on myeloid cells-1 are underrepresented in the human intestine. J Immunol 174 : 517–524.
38. CorazzaN, EichenbergerS, EugsterHP, MuellerC (1999) Nonlymphocyte-derived tumor necrosis factor is required for induction of colitis in recombination activating gene (RAG)2(−/−) mice upon transfer of CD4(+)CD45RB(hi) T cells. J Exp Med 190 : 1479–1492.
39. NaitoY, TakagiT, HandaO, IshikawaT, NakagawaS, et al. (2003) Enhanced intestinal inflammation induced by dextran sulfate sodium in tumor necrosis factor-alpha deficient mice. J Gastroenterol Hepatol 18 : 560–569.
40. NotiM, CorazzaN, MuellerC, BergerB, BrunnerT (2010) TNF suppresses acute intestinal inflammation by inducing local glucocorticoid synthesis. J Exp Med 207 : 1057–1066.
41. AllenbachC, LaunoisP, MuellerC, Tacchini-CottierF (2008) An essential role for transmembrane TNF in the resolution of the inflammatory lesion induced by Leishmania major infection. Eur J Immunol 38 : 720–731.
42. CharmoyM, AudersetF, AllenbachC, Tacchini-CottierF (2010) The prominent role of neutrophils during the initial phase of infection by Leishmania parasites. J Biomed Biotechnol 2010 : 719361.
43. Tacchini-CottierF, ZweifelC, BelkaidY, MukankundiyeC, VaseiM, et al. (2000) An immunomodulatory function for neutrophils during the induction of a CD4+ Th2 response in BALB/c mice infected with Leishmania major. J Immunol 165 : 2628–2636.
44. WangGG, CalvoKR, PasillasMP, SykesDB, HackerH, et al. (2006) Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat Methods 3 : 287–293.
45. FournierBM, ParkosCA (2012) The role of neutrophils during intestinal inflammation. Mucosal Immunol 5 : 354–366.
46. CaiM, ChenQ, ChenC, LiuX, HouJ, et al. (2013) Activation of triggering receptor expressed on myeloid cells-1 protects monocyte from apoptosis through regulation of myeloid cell leukemia-1. Anesthesiology 118 : 1140–1149.
47. RicheldiL, MarianiM, LosiM, MaselliF, CorbettaL, et al. (2004) Triggering receptor expressed on myeloid cells: role in the diagnosis of lung infections. Eur Respir J 24 : 247–250.
48. LaglerH, SharifO, HaslingerI, MattU, StichK, et al. (2009) TREM-1 activation alters the dynamics of pulmonary IRAK-M expression in vivo and improves host defense during pneumococcal pneumonia. J Immunol 183 : 2027–2036.
49. TatedaK, MooreTA, DengJC, NewsteadMW, ZengX, et al. (2001) Early recruitment of neutrophils determines subsequent T1/T2 host responses in a murine model of Legionella pneumophila pneumonia. J Immunol 166 : 3355–3361.
50. SporriR, JollerN, HilbiH, OxeniusA (2008) A novel role for neutrophils as critical activators of NK cells. J Immunol 181 : 7121–7130.
51. LeibundGut-LandmannS, WeidnerK, HilbiH, OxeniusA (2011) Nonhematopoietic cells are key players in innate control of bacterial airway infection. J Immunol 186 : 3130–3137.
52. DowerK, EllisDK, SarafK, JelinskySA, LinLL (2008) Innate immune responses to TREM-1 activation: overlap, divergence, and positive and negative cross-talk with bacterial lipopolysaccharide. J Immunol 180 : 3520–3534.
53. SkerrettSJ, BagbyGJ, SchmidtRA, NelsonS (1997) Antibody-mediated depletion of tumor necrosis factor-alpha impairs pulmonary host defenses to Legionella pneumophila. J Infect Dis 176 : 1019–1028.
54. BleharskiJR, KiesslerV, BuonsantiC, SielingPA, StengerS, et al. (2003) A role for triggering receptor expressed on myeloid cells-1 in host defense during the early-induced and adaptive phases of the immune response. J Immunol 170 : 3812–3818.
55. WangF, LiuS, WuS, ZhuQ, OuG, et al. (2012) Blocking TREM-1 signaling prolongs survival of mice with Pseudomonas aeruginosa induced sepsis. Cell Immunol 272 : 251–258.
56. HamermanJA, TchaoNK, LowellCA, LanierLL (2005) Enhanced Toll-like receptor responses in the absence of signaling adaptor DAP12. Nat Immunol 6 : 579–586.
57. TurnbullIR, McDunnJE, TakaiT, TownsendRR, CobbJP, et al. (2005) DAP12 (KARAP) amplifies inflammation and increases mortality from endotoxemia and septic peritonitis. J Exp Med 202 : 363–369.
58. VinceJE, WongWW, KhanN, FelthamR, ChauD, et al. (2007) IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell 131 : 682–693.
59. Dayer SchneiderJ, SeiboldI, Saxer-SekulicN, ParedesBE, SaurerL, et al. (2009) Lack of TNFR2 expression by CD4(+) T cells exacerbates experimental colitis. Eur J Immunol 39 : 1743–1753.
60. TitusRG, MarchandM, BoonT, LouisJA (1985) A limiting dilution assay for quantifying Leishmania major in tissues of infected mice. Parasite Immunol 7 : 545–555.
61. BachmannMF, EcabertB, KopfM (1999) Influenza virus: a novel method to assess viral and neutralizing antibody titers in vitro. J Immunol Methods 225 : 105–111.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 1
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Lyme Disease: Call for a “Manhattan Project” to Combat the Epidemic
- Origin, Migration Routes and Worldwide Population Genetic Structure of the Wheat Yellow Rust Pathogen f.sp.
- IFNγ/IL-10 Co-producing Cells Dominate the CD4 Response to Malaria in Highly Exposed Children
- Human and Plant Fungal Pathogens: The Role of Secondary Metabolites
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy