
Reengineering Redox Sensitive GFP to Measure Mycothiol Redox Potential of during Infection
Mycobacterium tuberculosis (Mtb) survives under oxidatively hostile environments encountered inside host phagocytes. To protect itself from oxidative stress, Mtb produces millimolar concentrations of mycothiol (MSH), which functions as a major cytoplasmic redox buffer. Here, we introduce a novel system for real-time imaging of mycothiol redox potential (EMSH) within Mtb cells during infection. We demonstrate that coupling of Mtb MSH-dependent oxidoreductase (mycoredoxin-1; Mrx1) to redox-sensitive GFP (roGFP2; Mrx1-roGFP2) allowed measurement of dynamic changes in intramycobacterial EMSH with unprecedented sensitivity and specificity. Using Mrx1-roGFP2, we report the first quantitative measurements of EMSH in diverse mycobacterial species, genetic mutants, and drug-resistant patient isolates. These cellular studies reveal, for the first time, that the environment inside macrophages and sub-vacuolar compartments induces heterogeneity in EMSH of the Mtb population. Further application of this new biosensor demonstrates that treatment of Mtb infected macrophage with anti-tuberculosis (TB) drugs induces oxidative shift in EMSH, suggesting that the intramacrophage milieu and antibiotics cooperatively disrupt the MSH homeostasis to exert efficient Mtb killing. Lastly, we analyze the membrane integrity of Mtb cells with varied EMSH during infection and show that subpopulation with higher EMSH are susceptible to clinically relevant antibiotics, whereas lower EMSH promotes antibiotic tolerance. Together, these data suggest the importance of MSH redox signaling in modulating mycobacterial survival following treatment with anti-TB drugs. We anticipate that Mrx1-roGFP2 will be a major contributor to our understanding of redox biology of Mtb and will lead to novel strategies to target redox metabolism for controlling Mtb persistence.
Published in the journal:
. PLoS Pathog 10(1): e32767. doi:10.1371/journal.ppat.1003902
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1003902
Summary
Mycobacterium tuberculosis (Mtb) survives under oxidatively hostile environments encountered inside host phagocytes. To protect itself from oxidative stress, Mtb produces millimolar concentrations of mycothiol (MSH), which functions as a major cytoplasmic redox buffer. Here, we introduce a novel system for real-time imaging of mycothiol redox potential (EMSH) within Mtb cells during infection. We demonstrate that coupling of Mtb MSH-dependent oxidoreductase (mycoredoxin-1; Mrx1) to redox-sensitive GFP (roGFP2; Mrx1-roGFP2) allowed measurement of dynamic changes in intramycobacterial EMSH with unprecedented sensitivity and specificity. Using Mrx1-roGFP2, we report the first quantitative measurements of EMSH in diverse mycobacterial species, genetic mutants, and drug-resistant patient isolates. These cellular studies reveal, for the first time, that the environment inside macrophages and sub-vacuolar compartments induces heterogeneity in EMSH of the Mtb population. Further application of this new biosensor demonstrates that treatment of Mtb infected macrophage with anti-tuberculosis (TB) drugs induces oxidative shift in EMSH, suggesting that the intramacrophage milieu and antibiotics cooperatively disrupt the MSH homeostasis to exert efficient Mtb killing. Lastly, we analyze the membrane integrity of Mtb cells with varied EMSH during infection and show that subpopulation with higher EMSH are susceptible to clinically relevant antibiotics, whereas lower EMSH promotes antibiotic tolerance. Together, these data suggest the importance of MSH redox signaling in modulating mycobacterial survival following treatment with anti-TB drugs. We anticipate that Mrx1-roGFP2 will be a major contributor to our understanding of redox biology of Mtb and will lead to novel strategies to target redox metabolism for controlling Mtb persistence.
Introduction
It is estimated that nearly 2 billion people currently suffer from latent Mycobacterium tuberculosis (Mtb) infection and ∼1.4 million people succumb to tuberculosis (TB) annually [1] and [www.who.int/tb]. The ability of Mtb to adapt and resist killing by the immune system facilitates its survival, replication, and persistence. Following aerosol exposure, Mtb is engulfed by macrophages, and exposed to antimicrobial redox stresses including reactive oxygen and nitrogen species (ROS and RNS) [2]. Mycobacterial killing by ROS and RNS is important for host resistance as demonstrated by the increased susceptibility of NADPH oxidase (NOX2) and nitric oxide synthase (iNOS) deficient mice following Mtb challenge [3], [4]. Moreover, children with defective NOX2 suffer from chronic granulomatous disease, are susceptible to TB and even develop severe complications after vaccination with BCG [5]. Consistent with these observations, a recent study elegantly demonstrated the requirement of NADPH oxidase in exerting neutrophil-mediated killing of mycobacteria during infection [6].
In response to oxido-reductive stress, Mtb upregulates multiple redox sensing pathways, including SigH/RshA, DosR/S/T, MosR, and the WhiB family, to maintain redox homeostasis [7]–[12]. Furthermore, modified cell wall lipids [13], antioxidant enzymes, and cellular redox buffers, such as MSH, thioredoxins (TRXs), and ergothionine (ERG), assist Mtb in mitigating redox stress [14]. Collectively, these studies indicate that dynamic reprogramming of intrabacterial redox metabolism in response to host environment is vital for the persistence of Mtb. Despite its recognized importance, tools for monitoring changes in redox state of Mtb during infection do not exist. Customary approaches involving NAD+/NADH and MSH/MSSM measurements require cell disruption, which precludes real-time analyses, and are plagued by oxidation artifacts. Alternatively, redox-sensitive dyes which are commonly used to detect ROS generation in cells, suffer from non-specificity, irreversibility, and these dyes cannot deliver information regarding the redox potential of a specific redox couple [15], [16]. Therefore, development of a specific, sensitive, and non-invasive technology to study defined redox changes in Mtb would contribute significantly to delineating novel redox pathways involved in persistence, drug tolerance, as well as pathogenesis of Mtb, and may also have utility in high throughput screens to identify small-molecule modulators of intrabacterial redox homeostasis.
Genetically encoded reduction-oxidation sensitive GFP indicators (roGFPs) have been developed to measure the intracellular glutathione (GSH) redox potential (EGSH) through interaction with glutaredoxins (Grxs) in many organisms [17]. However, the preference of roGFPs for GSH limits their use in non-GSH producers like gram positive bacteria (e.g. Bacillus species, Staphylococcus aureus, Deinococcus radiodurans) and actinomycetes (e.g. Mycobacteria, Corynebacteria, Streptomyces, Nocardia), which respectively contain bacillithiol (BSH) and MSH as their major redox buffers [18], [19].
Because MSH is functionally analogous to GSH and plays a prominent role in maintaining the reduced state of the mycobacterial cytoplasm [20], [21], we engineered roGFP2 to generate a MSH-specific intracellular probe, Mrx1-roGFP2. Importantly, Mrx1-roGFP2 allows imaging of EMSH in diverse mycobacterial species and strains, including drug-resistant clinical isolates during infection. Lastly, we examine the potential of antibiotics in inducing intramycobacterial oxidative stress in the physiological context of infection and demonstrate the functional importance of MSH redox signaling in intramacrophage survival and sensitivity to anti-TB drugs. Our study provides an elegant tool to probe redox biology of Mtb under diverse environmental conditions including in vivo experimental models of TB.
Results
Design of the mycothiol-specific fluorescent biosensor
We selected roGFP2 as a fluorescent partner in the biosensor construct because it exhibits the largest dynamic range, it is brighter, pH insensitive, and is resistant to photoswitching [22]. The oxidation of two cysteines on either side of roGFP2 chromophore (S147C and Q204C) [22] generates a disulfide bond and increases the fluorescence intensity at ∼400 nm with concomitant decrease at ∼490 nm, while reduction reverses the spectrum. The 400/490 nm ratio thus reports the redox state of the cell or compartment in which it is expressed [22]. Because the sensor is ratiometric it eliminates errors due to variations in roGFP2 concentrations during different growth phases of an organism.
Conventional roGFP2 predominantly equilibrates with the cytosolic glutathione redox buffer through interaction with endogenous glutaredoxins [23]. However, the response kinetics of roGFP2 was slow and absolute specificity towards GSH/GSSG redox couple cannot be guaranteed [17], [24]. To resolve this, roGFP2 bioprobe was recently fused to human glutaredoxin 1 (Grx1; Grx1-roGFP2), which ensured complete specificity and rapid equilibration with intracellular GSH/GSSG couple [24]. On this basis, we explored the concept of covalently coupling roGFP2 to a mycothiol-specific oxidoreductase such as mycoredoxin (Mrx1) to generate a biosensor (Mrx1-roGFP2) that exclusively responds to perturbations in mycothiol redox potential (EMSH). The mechanistic basis of coupling Mrx1 to roGFP2 for measuring EMSH is depicted in Figure 1. Recently, a glutaredoxin (Grx1) homologue (mycoredoxin-1; Mrx1) which exclusively interacts with the mycothiol redox system has been reported in a non-pathogenic saprophytic mycobacteria, Mycobacterium smegmatis (Msm) [25]. We performed homology based analysis and identified three putative Mrx1 like proteins in Mtb H37Rv. Out of the three proteins (Rv3053c, Rv0508, and Rv3198A), Rv3198A demonstrate highest similarity with Mrx1 of Msm (72% identity). Based on this, we selected Rv3198A ORF as a putative mycoredoxin-encoding gene and named its product as Mtb Mrx1. The Mtb Mrx1 contains an active site (CGYC) similar to Msm Mrx1, supposedly required for thiol-disulfide exchange activity [25]. We independently replaced the two cysteine (Cys) residues present in the catalytic site of Mrx1 by alanine to generate Mrx1(CGYA) and Mrx1(AGYC). The wt Mrx1 along with its Cys variants were then separately fused to the N-terminus of roGFP2 via a 30-amino acid linker, (Gly-Gly-Ser-Gly-Gly)6. The resulting chimeras were affinity purified as His-tagged proteins and analyzed by spectrofluorometry. We observed that Mrx1-roGFP2 exhibits two distinct excitation peaks (390 nm and 490 nm) at a fixed emission wavelength of 510 nm. Therefore, in all subsequent experiments using spectrofluorometer the excitation ratio from these two wavelengths (390 and 490 nm) was measured to determine the extent of biosensor oxidation. Our analysis demonstrated that the intrinsic ratiometric changes exhibited by roGFP2 upon oxidation or reduction were recapitulated in Mrx1-roGFP2 (Figure S1A). In addition, we monitored the response of Mrx1-roGFP2 over a physiologically relevant pH range (pH 5.5–8.5) and found that the fluorescence excitation ratio exhibited by Mrx1-roGFP2 was insensitive to pH variations (Figure S1B). We also verified that the reported midpoint potential (−280 mV) and dynamic range (−320 mV to −240 mV) of roGFP2 were not influenced by the fusion (Figure S1C).

Since redox stress leads to an increase in oxidized mycothiol (MSSM) concentrations [26], we anticipated that the Mrx1 fusion would enable roGFP2 to selectively monitor this transformation. To test this proposal, we reduced uncoupled roGFP2, Mrx1-roGFP2, Mrx1(CGYA)-roGFP2, and Mrx1(AGYC)-roGFP2 and examined their oxidation by MSSM. Only Mrx1-roGFP2 (Lane 2, Figure 2A) and Mrx1(CGYA)-roGFP2 (Lane 4, Figure 2A) ratios increased upon MSSM addition, whereas uncoupled roGFP2 (Lane 1, Figure 2A) and Mrx1(AGYC)-roGFP2 (Lane 3, Figure 2A) remained non-responsive. This suggests that the Mrx1-roGFP2 reaction mechanism is similar to monothiol mechanism of glutaredoxins, wherein nucleophilic N-terminus cysteine interacts with GSSG to form mixed protein-glutathione disulfide intermediate followed by protein oxidation [24]. Importantly, Mrx1-roGFP2 did not respond to other disulfide-based compounds such as cystine (Cys2), GSSG, or 2-hydroxyethyl disulfide (HED), thus confirming the specificity of this biosensor towards MSSM (Figure 2B). Next, we examined the response of Mrx1-roGFP2 to reduced mycothiol (MSH). To continuously maintain a reduced state of MSH in our assays, we used MSH disulfide reductase (Mtr) enzyme. Mtr is known to catalyze the NADPH-dependent reduction of MSSM to MSH in the mycobacterial cells [27]. We first confirmed the activity of purified Mtr by monitoring NADPH oxidation in the presence of MSSM. A time-dependent decrease in 340 nm absorption due to NADPH consumption confirmed cycling of electrons from NADPH to MSSM by Mtr (Figure S1D). Next, oxidized Mrx1-roGFP2 was added as substrate for the MSH/Mtr/NADPH electron transfer assay as depicted in Figure 2C. By monitoring the decrease in the 390/490 excitation ratio, we examined the real-time response of oxidized Mrx1-roGFP2 to MSH generated via NADPH-dependent reduction of MSSM by Mtr. As shown in Figure 2D, a time-dependent decrease in 390/490 ratio confirms reduction of Mrx1-roGFP2 by MSH. The slow response of this biosensor towards MSH is consistent with the low catalytic turnover rate of Mtr [27]. No response was observed if either MSH was omitted from the Mtr/NADPH/Mrx1-roGFP2 mixture (Figure 2D) or catalytically inactive Mrx1(AGYC)-roGFP2 (Lane 3, Figure 2E) or uncoupled roGFP2 (Lane 1, Figure 2E) were used as substrates, whereas a response was readily detected in the case of Mrx1(CGYA)-roGFP2 (Lane 4, Figure 2E). Finally, to measure the sensitivity of the biosensor, we incubated pre-reduced Mrx1-roGFP2 with various ratios of MSH/MSSM at a physiological concentration (1 mM total) under anaerobic conditions and the ratiometric response was monitored. We found that a small increase in MSSM led to a significant increase in biosensor oxidation. For example, an increase in the amount of mycothiol oxidation (OxDMSH, see Materials and Methods for a mathematical explanation) from 0.00001 to 0.0001 (i. e an ∼100 nM increase in absolute MSSM) led to a larger increase in the biosensor oxidation (i. e from ∼40% to 90%) (Figure 2F). These results confirm that Mrx1-roGFP2 is capable of rapidly sensing nanomolar changes in MSSM against the backdrop of a highly reduced MSH pool (1 mM). Lastly, uncoupled roGFP2 remained completely non-responsive to changes in MSH/MSSM ratios (Figure 2F). Together, our results show that Mrx1-roGFP2 is exceptionally sensitive to measure physiological and dynamic changes in MSH/MSSM redox state.
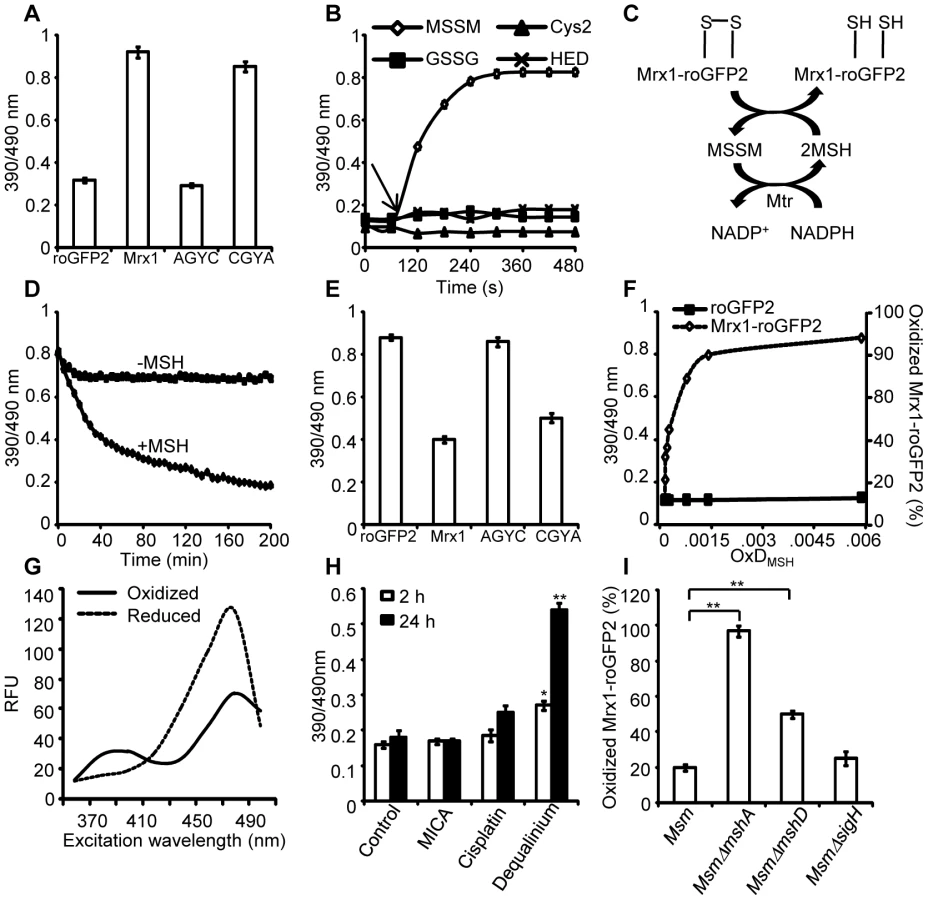
Mrx1-roGFP2 senses mycothiol redox state in mycobacteria
To investigate the redox responsiveness of Mrx1-roGFP2 in mycobacteria, we stably expressed Mrx1-roGFP2 in Msm. Next, we confirmed that the biosensor responds ratiometrically upon exposure of Msm to diamide or dithiothreitol (DTT) (Figure 2G). To examine if Mrx1-roGFP2 senses EMSH in vivo, we pharmacologically and genetically perturbed MSH levels in Msm. We first analyzed the response of Mrx1-roGFP2 upon depletion of the cytosolic MSH pool in Msm using dequalinium [an established small-molecule inhibitor of MSH ligase (MshC) [28]]. For comparison, we also tested inhibitors of TRX reductase (cisplatin) [29] and dihydrolipoamide dehydrogenase (5-methoxyindole-2-carboxylic acid) [30], both of which do not affect the cellular MSH pool. As expected, only dequalinium treatment led to a substantial increase in the fluorescence excitation ratio of Mrx1-roGFP2 (Figure 2H).
We further validated the MSH-specific response of Mrx1-roGFP2 by expressing it in MSH-negative (MsmΔmshA) and MSH-depleted (MsmΔmshD) strains of Msm [31], [32]. In addition to MSH, mycobacteria express the NADPH-dependent TRX system to efficiently counter oxidative stress [33]. Importantly, multiple components of the TRX system were down-regulated in mycobacterial strains lacking extracytoplasmic sigma factor, SigH [33], [34]. Therefore, to rule out the interaction of Mrx1-roGFP2 with the TRX system inside mycobacteria, we analyzed the biosensor response in SigH-deleted strain of Msm (MsmΔsigH). Oxidation of the biosensor was nearly quantitative (∼95%±3 oxidized) in MsmΔmshA as compared to ∼20%±2 in both wt Msm and MsmΔsigH, and ∼50%±4 in MsmΔmshD (Figure 2I). Since MsmΔmshD contains only 1% to 3% of total cellular MSH but produces two related thiols (Suc-mycothiol and formyl-mycothiol) [35], our data suggest that Mrx-1 can facilitate roGFP2 reduction with Suc-MSH and/or formyl-MSH, albeit suboptimally. A previous study reported a marked accumulation of ERG in MsmΔmshA [36]. Therefore, we also examined if Mrx1-roGFP2 can function as a sensor of ERG in mycobacteria. However, Mrx1-roGFP2 did not respond to ERG in vitro, further supporting mycothiol-specific response of Mrx1-roGFP2 (Figure S2A). Taken together, data generated from several independent techniques demonstrate that Mrx1-roGFP2 responds to the MSH redox buffer in mycobacteria.
Specific equilibration of Mrx1-roGFP2 with the MSH redox buffer enables precise measurement of the EMSH in various strains of Msm using the Nernst equation as described in SI Materials and Methods. Our studies reveal EMSH in wt Msm, MsmΔmshA, MsmΔmshD, and MsmΔsigH to be −300±2 mV, −239±7 mV, −275±7 mV, and −300±3 mV, respectively. Notably, the oxidizing EMSH values observed for MsmΔmshA and MsmΔmshD are consistent with our earlier findings indicating that the thiol-disulfide redox switch in Mrx1-roGFP2 is a specific substrate for the MSH reductive pathway.
Mrx1 enhances sensitivity of roGFP2
We next investigated whether coupling of roGFP2 with Mrx1 enhanced the sensitivity of the new biosensor towards transient changes in intracellular EMSH. Initial studies confirmed that H2O2 alone (0.5–5 mM) does not directly oxidize the Mrx1-roGFP2 protein in vitro (Figure S2B). By contrast, application of H2O2 to cells led to rapid (∼2 min) and substantial oxidation of the biosensor within Msm (Figure 3A). These results suggest that H2O2–mediated oxidation of MSH to MSSM is necessary for biosensor oxidation in Msm. Also, Mrx1-roGFP2 ratio rapidly increased upon exposure of Msm to diverse oxidants such as menadione, aldrithiol, and diamide (Figure S2C).
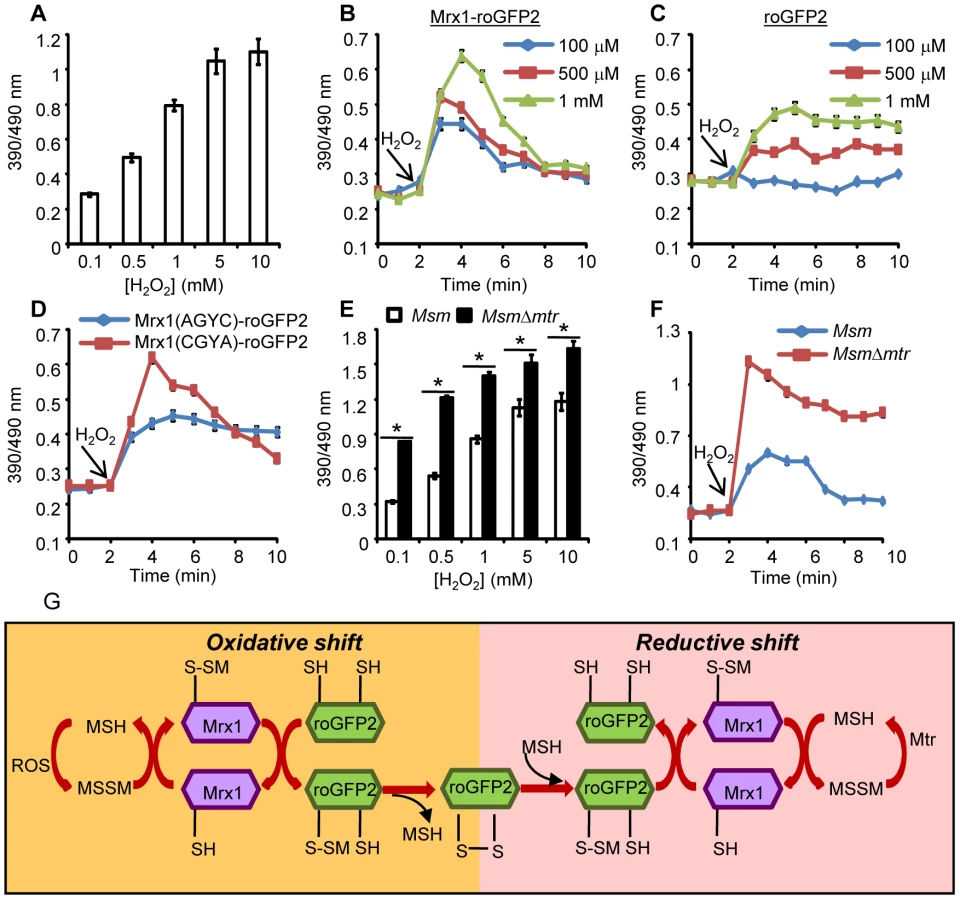
The sensitivity of Mrx1-roGFP2 in Msm was further investigated upon exposure to lower concentrations of H2O2. Addition of 100 µM, 500 μM and 1 mM of H2O2 resulted in rapid, but short-lived (∼5 min) increases in Mrx1-roGFP2 ratio, suggesting efficient mobilization of anti-oxidant response mechanisms in Msm (Figure 3B). In contrast, Msm expressing either uncoupled roGFP2 (Figure 3C) or Mrx1(AGYC)-roGFP2 (Figure 3D) responded slowly to lower concentrations of H2O2 and did not display an anti-oxidative response. The poor response shown by uncoupled roGFP2 and Mrx1(AGYC)-roGFP2 could either be due to their non-specific interaction with other mycobacterial redox systems (e.g. TRX, ERG etc) or suboptimal equilibration with the MSH/MSSM couple through mediation by endogenous Msm Mrx1. Finally, Mrx1(CGYA)-roGFP2 retained transient responses similar to Mrx1-roGFP2 (Figure 3D), further substantiating the crucial role of N-terminal Cys residue of Mrx1 in promoting a rapid and reversible equilibration of biosensor with the intracellular MSH/MSSM redox buffer. To decisively show that Mrx1-roGFP2 is capable of detecting dynamic changes in EMSH, we exploited another Msm mutant lacking MSH disulfide reductase (Mtr) activity (MsmΔmtr). The Mtr enzyme maintains intramycobacterial MSSM/MSH ratios by reducing MSSM to MSH upon oxidative stress, consequently its absence results in the depletion of MSH [37]. Exposure to H2O2 led to a ∼2-fold increase in the ratio of oxidized Mrx1-roGFP2 in MsmΔmtr as compared to wt Msm (Figure 3E). Importantly, the short-lived oxidative deflections of EMSH in response to limiting amounts of H2O2 were significantly extended in the MsmΔmtr mutant, thereby implicating Mtr in orchestrating an efficient anti-oxidative response in Msm (Figure 3F). On this basis, we propose a biochemical mechanism of sensing mycobacterial EMSH using Mrx1-roGFP2 bioprobe (Figure 3G).
It can be argued that the overexpression of a redox-based enzyme in our biosensor can influence cytoplasmic MSH/MSSM redox state to compromise redox measurements. However, we found that the absolute concentration of Mrx1-roGFP2 inside Msm cells is 1000–10000 fold lower (1 µM/cell; Figure S2D) as compared to the high millimolar concentrations of mycothiol (1–10 mM) present in mycobacteria. This shows that reducing equivalents (thiols) introduced by the expression of biosensor are significantly less than the combined pool of MSH and other thiols present in mycobacteria. Furthermore, ambient EMSH of Msm cells overexpressing either Mrx1-roGFP2 or its catalytically inactive derivative [Mrx1(AGYC)-roGFP2] was found to be comparable i. e −300±2 mV and −298±3 mV, respectively, indicating that Mrx1 activity does not perturb steady state EMSH. Lastly, Mrx1-roGFP2 harboring Msm, MsmΔmshA, and MsmΔmtr strains showed similar survival profile upon exposure to H2O2 as compared to control strains, suggesting no adverse influence of intracellular levels of biosensor on resistance to oxidative stress (Figure S2E). Taken together, we demonstrate that by integrating Mrx1 with roGFP2, the biosensor becomes catalytically self-sufficient in establishing a rapid and specific equilibration with the MSH redox buffer.
Measuring EMSH of slow growing Mtb strains
To obtain information about the basal redox potential differences between various species and strains of mycobacteria, we expressed Mrx1-roGFP2 in vaccine strain (M. bovis BCG), virulent laboratory strain (Mtb H37Rv), and several Indian clinical isolates of Mtb including single-drug resistant (BND 320), multi-drug resistant (MDR - Jal 2261, 1934, Jal 2287), and extensively-drug resistant (XDR - MYC 431). First, we confirmed that overexpression of Mrx1-roGFP2 does not affect metabolic activity and growth of Mtb using metabolic indicator dye, Alamar blue and by measuring culture absorbance (Figure S3A and S3B). Any downstream imaging analysis of the BSL3 class pathogens requires their chemical fixation by paraformaldehyde (PFA), which we found to oxidize the biosensor (Figure S3C). To circumvent PFA-mediated oxidation artifacts, we alkylated the thiols of Mrx1-roGFP2 using the cell permeable fast-acting thiol-modifier, N-ethyl maleimide (NEM). Control experiments clearly show that NEM treatment efficiently prevents oxidation during PFA-fixation of Mtb cells (Figure S3C). A similar chemical-fixation strategy was successfully exploited to measure the redox potential of glutathione (EGSH) in HeLa cells [24], and in the sub-cellular compartments and tissues of Drosophila using Grx1-roGFP2 [38]. With this system in hand, we confirmed that Mrx1-roGFP2 responds ratiometrically to oxidant (cumene hydroperoxide; CHP) and reductant (DTT) in Mtb H37Rv (Figure S4A). A concentration and time-dependent oxidation of Mrx1-roGFP2 upon H2O2 exposure was also detected in Mtb H37Rv (Figure S4B and S4C). Of note, induction of anti-oxidative response upon exposure to H2O2 was significantly delayed in Mtb (∼120 min) as compared to rapid response observed earlier in Msm (∼5 min) (Figure S4C), suggesting important variations in sensing and responding to redox stress between the two species.
Next, we evaluated the redox potential of various slow growing lab-adapted and clinical mycobacterial strains. The resulting data indicates that there is relatively little variation in the redox state within and between drug-resistant clinical (MDR/XDR) and drug-sensitive lab (Mtb H37Rv, M bovis BCG) strains, as exemplified by EMSH values around −273 mV to −280 mV (Table S1). This finding suggests that the steady-state EMSH is relatively unaffected by either genotypic or phenotypic variations within Mtb strains under laboratory growth conditions. However, EMSH in slow growing mycobacterial strains is notably oxidizing compared to Msm (−300±2 mV), which is consistent with an earlier report showing higher MSH/MSSM ratio in Msm (200∶1) as compared to BCG (50∶1) [39]. Lastly, to rule out any contribution of the chemical fixation procedure to the observed variation in EMSH between Mtb and Msm, we treated Msm expressing Mrx1-roGFP2 with NEM-PFA and confirmed that Msm maintains EMSH of −300±2 mV.
Macrophage induces redox heterogeneity within Mtb populations
We next determined whether Mrx1-roGFP2 could be used to quantify redox changes that occur in the natural context of infection. To investigate this issue, we infected THP-1 macrophages with Mtb H37Rv expressing Mrx1-roGFP2 at a multiplicity of infection (moi) of 10 and monitored intramycobacterial EMSH. To do this, we performed NEM-PFA based fixation technique followed by ratiometric fluorescence analysis by flow cytometry (see SI Materials and Methods). Since the flow cytometric based measurements are dependent on fixed wavelength lasers, we excited Mrx1-roGFP2 biosensor with the canonical 405 and 488 nm laser wavelengths at a fixed emission wavelength of 510 nm (see SI Materials and Methods). We first confirmed that intramycobacterial Mrx1-roGFP2 responds ratiometrically to oxidant; CHP and reductant; DTT inside macrophages (Figure 4A, 4B, 4C, and 4D). To measure changes in EMSH during infection, an in vitro redox calibration curve was generated by treating Mtb H37Rv with buffers of known redox potentials. By fitting Mrx1-roGFP2 ratio to the redox calibration curve, we precisely calculated the EMSH of Mtb inside macrophages (Figure S5A, see SI Materials and Methods). Intriguingly, flow cytometric analyses of ∼30,000 infected macrophages demonstrated the presence of cells with a gradient of intramycobacterial EMSH. For the purpose of measurements, infected macrophages were gated into three subpopulations on the basis of their corresponding intramycobacterial EMSH (Figure 4E and 4F). An EMSH-basal population with an intermediate EMSH of −275±5 mV, and two deflected populations were observed (Figure 4E and 4F). Deflected cells with a mean EMSH of −240±3 mV represent an EMSH-oxidized subpopulation, based on the observation that CHP treatment of infected macrophages results in a significant fraction of these gated cells (∼98%) (Figure 4E and 4F). The population with an average EMSH of −300±6 mV represents an EMSH-reduced subpopulation, as treatment of infected macrophages with the DTT results in ∼96% of the cells gating into this subpopulation (Figure 4E and 4F). Mtb cells present in media alone and analyzed in parallel did not show redox heterogeneity (Figure 4G and 4H), suggesting that the intramacrophage environment perturbs redox homeostasis to induce redox variability in Mtb. Furthermore, we infected macrophages with BCG, and measured intramycobacterial EMSH with and without NEM-PFA treatment. Both conditions induce similar degree of heterogeneity in intramycobacterial EMSH (Figure 4I, 4J, and 4K), demonstrating that redox heterogeneity has a biological basis and is not due to aberrant quenching of fluorescent signals during NEM-PFA treatment.
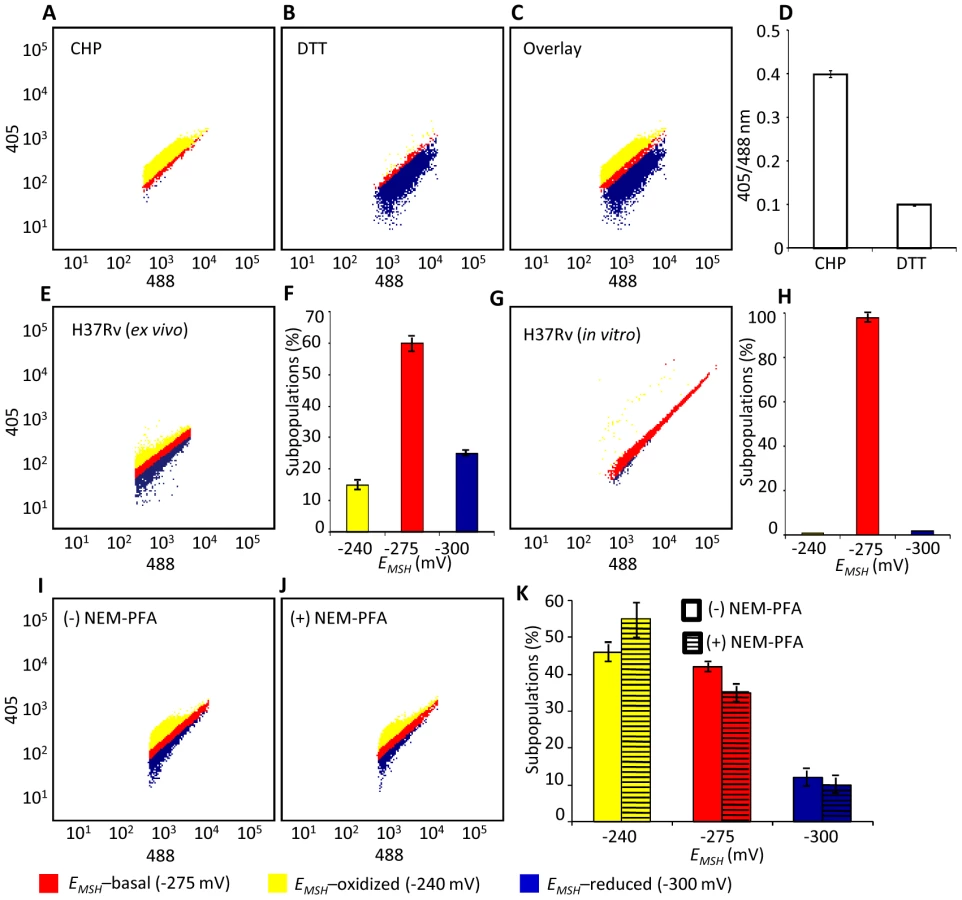
With the flow cytometry workflow in hand, we measured time-resolved changes in intramycobacterial EMSH during infection of THP-1 macrophages. Our results indicated that the initial period (0–24 h post-infection [p.i.]) of infection was associated with a gradual increase in cells with reduced EMSH (60±7%) followed by an oxidative shift (25±5%) at 48 h p.i. and then a significant recovery from oxidative stress, as revealed by a decrease in the population with oxidized EMSH (7±3%) at 72 h p.i. (Figure 5A). The observed redox heterogeneity and oscillatory patterns were confirmed by repeating experiments at least six times in quadruplicate and data from biologically independent experiments were combined and presented as mean ± standard deviation. We also verified the presence of both time-dependent heterogeneity and oscillations in intramycobacterial EMSH upon infection of THP-1 macrophages at a low moi of 1 (Figure S5B). However, we noticed that infection with lower moi induced higher proportion of bacteria with oxidized EMSH at each time point examined as compared to cells infected at a moi of 10 (Figure S5B and S5C).

Next, we investigated if the macrophage environment induces strain-specific variations in redox heterogeneity among various slow growing mycobacteria including BCG and Indian clinical MDR/XDR isolates. For this, we infected THP-1 macrophages with various strains of Mtb at a moi of 10 and time-resolved changes in intramycobacterial EMSH were measured as described in the earlier section. Interestingly, while heterogeneity in EMSH for BND 320 largely followed the reductive-oxidative-reductive oscillatory pattern of H37Rv (Figure 5B), distinct redox deviations were displayed by other strains. For example, Jal 2287, Jal 2261 and 1934 displayed overrepresentation of the EMSH-oxidized subpopulation at 48 h p.i. followed by a poor recovery at 48–72 h p.i., as compared to H37Rv (Figure 5C–5E). Noticeably, intramacrophage growth of MYC 431 displayed a loss of redox oscillatory pattern and showed a steady increase in EMSH-oxidized subpopulation over 48 h p.i. (Figure 5F). Intriguingly, intramacrophage profile of BCG displayed temporal changes in EMSH comparable to Jal 2261, 1934, and MYC 431. As shown in Figure 5G, infection with BCG showed a continuous decrease in EMSH-reduced subpopulation with a concomitant increase in EMSH-oxidized subpopulation over time (Figure 5G). Together, these findings for the first time revealed that macrophage environment triggers heterogeneity in EMSH of Mtb and uncovered redox variance among clinical field isolates.
Immune activation of macrophages induces oxidative shift in EMSH of Mtb
In order to examine the biosensor response upon stimulation of oxidant-mediated antimycobacterial stresses, we performed additional experiments in immunologically activated murine macrophages (RAW 264.7). Activated murine macrophages are known to control mycobacterial proliferation by producing ROS and RNS [2]. RAW 264.7 were activated with IFN-γ and LPS prior to infection with Mtb H37Rv [11]. Ratiometric flow cytomteric analysis showed a significant and sustained oxidative shift in EMSH of Mtb H37Rv inside activated macrophages at each time point investigated, whereas intramycobacterial EMSH inside naïve macrophages showed redox oscillations similar to THP-1 cells (Figure 5H and 5I). These results indicate that Mtb cells were able to recover from mild oxidative stress conditions inside naïve macrophages, whereas recovery was compromised in IFN-γ/LPS primed macrophages. Since nitric oxide (NO) generated via iNOS is considered to be one of the major contributors of redox stress in Mtb inside immune-activated murine macrophages [40], we treated IFN-γ/LPS activated RAW 264.7 macrophages with a well established iNOS inhibitor NG-methyl-L-arginine (NMLA;[41]) and monitored intramycobacterial EMSH. Strikingly, a substantial reduction in subpopulation with oxidized EMSH was observed upon addition of NMLA (Figure S6A). These results indicate that Mtb responds to host derived environmental cues by modulating EMSH, and further illustrates the utility of Mrx1-roGFP2 in dissecting redox signaling during infection.
Sub-vacuolar compartments are the source of redox heterogeneity
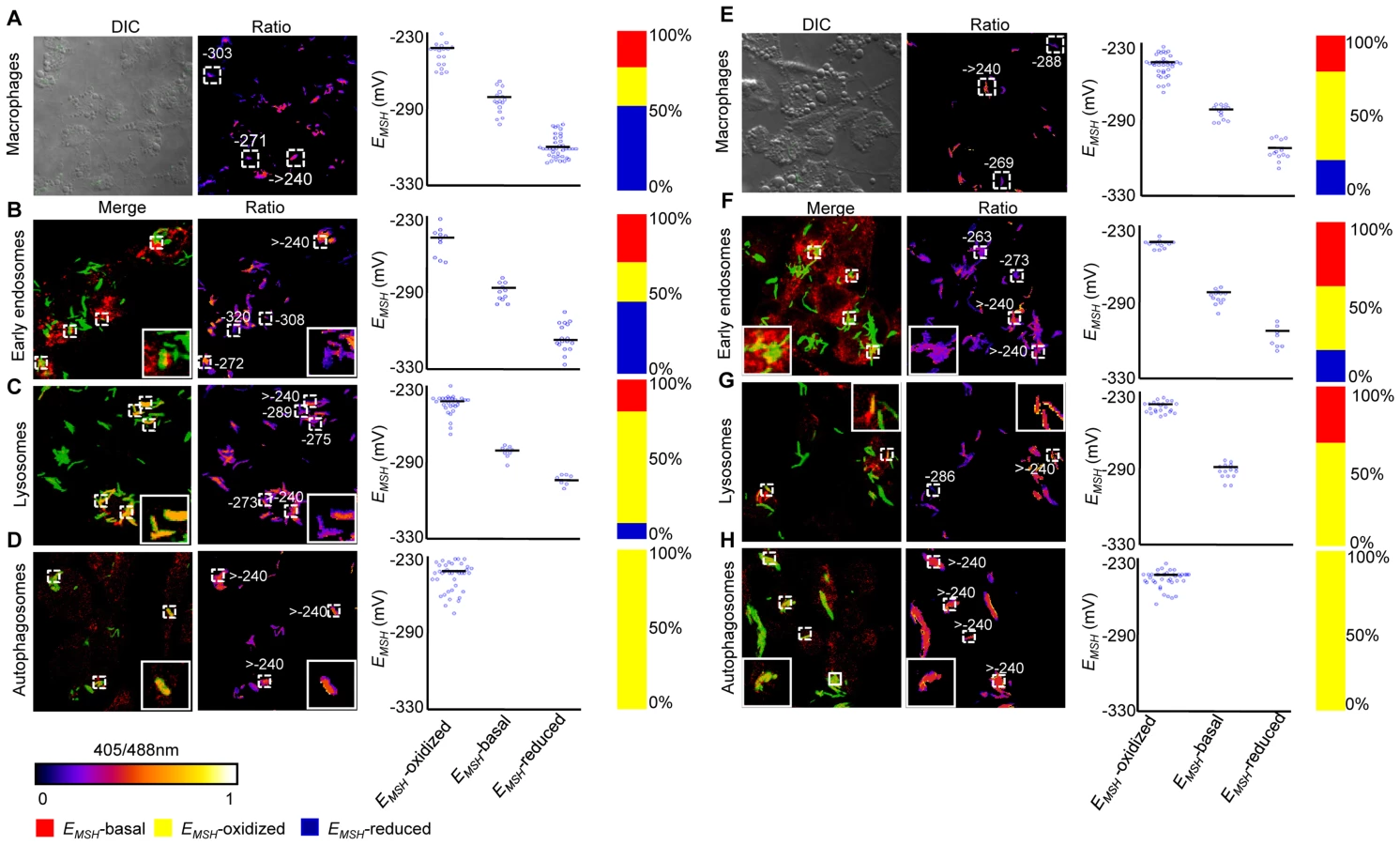
Intracellular Mtb exists in different vacuolar compartments, which may contribute to significant heterogeneity in mycobacterial gene expression, metabolic state and survival [42]. On this basis, we next asked whether trafficking into distinct vacuolar compartments could promote redox heterogeneity in the Mtb population using ratiometric confocal microscopy. First, we performed confocal imaging of Mtb cells inside THP-1 macrophages at 24 h p.i. Conforming to our flow cytometric findings, confocal analyses revealed that the intrabacterial EMSH varied markedly at a single-cell level. Similar to flow cytometry, this gradient in redox heterogeneity can be classified into EMSH-basal (−277±5 mV, 26%), EMSH-oxidized (−242±6 mV, 23%), and EMSH-reduced (−304±10 mV, 51%) sub-populations (Figure 6A and 6B). Mtb grown in media indicated an overrepresentation of the cells with uniform EMSH (Figure S6B), validating that both flow cytometry and confocal imaging revealed very similar ratiometric changes upon infection.
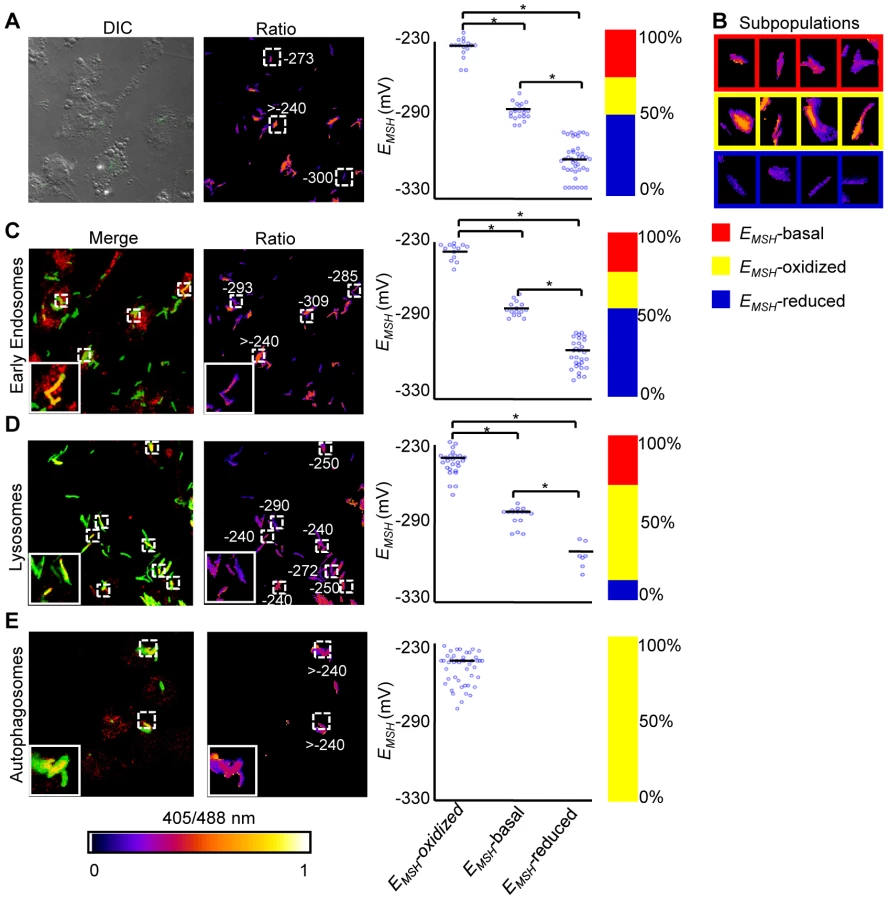
Next, we measured intrabacterial EMSH within early endosomes, lysosomes, and autophagosomes by visualizing the co-localization of Mtb H37Rv expressing Mrx1-roGFP2 with compartment specific fluorescent markers at 24 h p.i. using confocal microscopy (see SI Materials and Methods). For marking early endosomes, cells were stained with anti - early endosome autoantigen (EEA1) and anti-Rab5 antibodies. To study lysosomes, we used acidotropic dye Lysotracker and anti-cathepsin D antibody, whereas autophagosomes were labeled with anti-LC3 antibody (see SI Materials and Methods). The resulting data show that the majority of bacilli within early endosomes were likely to exhibit reduced (∼54%) as compared to oxidized (∼22%) or basal (∼24%) EMSH (Figure 6C and Figure S7A). Interestingly, in lysosomes, deflected subpopulations with EMSH-oxidized were clearly higher in proportion (58%), whereas EMSH-reduced (12%) and EMSH-basal (30%) subpopulations were underrepresented (Figure 6D and Figure S7B). The percent distribution of subpopulations with EMSH-reduced and EMSH-oxidized were significantly different between early endosomes and lysosomes (p<0.001), while EMSH-basal subpopulation remained comparable within these compartments. Furthermore, 100% of the Mtb population inside autophagosomes displayed a maximal oxidative shift in EMSH (Figure 6E). We also measured changes in EMSH during intramacrophage residence of drug-resistant strains Jal 2287 and MYC 431. While Jal 2287 displayed redox deviations similar to Mtb H37Rv (Figure 7A–7D), MYC 431 showed over-representation of subpopulations with EMSH-oxidized within the macrophage and sub-vacuolar compartments at 24 h p.i. (Figure 7E–7H). Taken together, our results suggest that distinct sub-vacuolar environments lead to the generation of Mtb subpopulations with a gradient of redox potentials.
Anti-TB drugs induce oxidative stress in Mtb during infection
Several studies have shown that MSH-deficient mycobacteria and other actinomycetes are sensitive to antibiotics [21], [43], [44]. On the other hand, MSH also contributes to susceptibility to INH and ETH [45], and mutations in mycothiol biosynthesis genes were identified in drug-resistant clinical isolates of Mtb [46]. While these constitute an important foundation linking antibiotic action with mycothiol redox homeostasis in vitro, they provide little insight into how antibiotics modulate intramycobacterial EMSH in the physiological setting of infection. To examine this, we characterized the effect of anti-TB drugs on redox heterogeneity in Mtb cells during intramacrophage residence. To this end, infected macrophages were exposed to anti-TB drugs (5-fold the in vitro MIC) with different modes of action (e.g. isoniazid [INH; mycolic acid inhibition], ethambutol [EMB; arabinogalactan inhibition], rifampicin [RIF; inhibition of transcription], and clofazimine [CFZ; redox cycling and ROS production; [47]) and the redox response was measured by flow cytometry. Treatment with all antibiotics induced variable levels of oxidative shift in EMSH of Mtb subpopulations at 12, 24, and 48 h p.i. (Figure 8A). The skew towards oxidizing EMSH was activated at an early time point (12 h p.i.) and increased significantly at 48 h p.i. (Figure 8A). Next, we examined if enhanced oxidizing EMSH correlated with the killing potential of anti-TB drugs during infection. At 12 h post-antibiotic treatment, the Mtb survival rate was comparable to the untreated control, as determined by colony forming unit (CFU) assay (Figure 8B). However, a modest (∼1.5-fold) to a significant reduction (∼5-fold) in intramacrophage bacillary load as compared to untreated control was detected at 24 and 48 h p.i., respectively (Figure 8B). These findings show that bactericidal antibiotics with different mechanisms of action induce oxidative changes in intramycobacterial EMSH during infection. To further validate these results, we infected macrophages with INH resistant clinical strains (BND 320 and Jal 2287) and measured intramycobacterial EMSH in response to INH at 48 h p.i. Since these strains are sensitive to CFZ, we used CFZ as a positive control in this experiment. Figure 8C clearly shows that the EMSH of strains genetically resistant to INH remained uninfluenced in response to INH. On the other hand, CFZ exposure generated considerable oxidative shift in EMSH of these strains (Figure 8C).

Lastly, to investigate if anti-TB drugs directly induce oxidative stress in vitro, we exposed Mtb cells grown in 7H9 medium supplemented with albumin, dextrose and sodium chloride (7H9-ADS) to INH, CFZ, RIF, and ETH (5× MIC) and tracked EMSH at 1, 2, 6, and 24 h post-treatment. This study revealed that only CFZ induces significant oxidative shift in EMSH of Mtb (Figure 8D). Other anti-TB drugs such as INH induce low levels of EMSH-oxidized at 24 h, post-treatment, while RIF and ETH do not influence the EMSH of Mtb (Figure 8D). Our results suggest that antibiotics do not perturb EMSH of Mtb per se, but co-opt host cellular responses to stimulate excessive oxidative stress during infection. These findings underscore the importance of studying redox-based mechanisms of drugs action under physiologically relevant microenvironmental conditions such as those encountered during TB infection in macrophage.
Redox heterogeneity induces differential susceptibility to anti-TB drugs
It has been suggested that long term anti-TB therapy is required because the mycobacterial population is functionally heterogeneous and harbors cells that are differentially sensitive to antibiotics [48]. However, the physiological determinants of phenotypic heterogeneity in Mtb population and its relation with antibiotic tolerance remains poorly characterized. Because macrophage environment quickly creates variability in mycobacterial cells to generate drug tolerant subpopulations [49], we hypothesize that heterogeneity in intrabacterial EMSH may be one of the factors that underlies emergence of Mtb populations with differential antibiotic susceptibility. We therefore sought to determine the susceptibility of Mtb cells with basal, oxidized, and reduced EMSH to antibiotics during infection of THP-1 cells. To do this, we analyzed the membrane integrity of Mtb cells expressing biosensor by assessing their capacity to exclude fluorescent nucleic-acid binding dye, propidium iodide (Pi), upon treatment with antibiotics during infection. The state of the bacterial membrane is a crucial physiological indicator, as Pi+ cells are considered damaged or dying [50]. Importantly, we found that NEM treatment of infected macrophages fixed the redox state of intracellular Mtb such that bacterial cells released from macrophages retained redox variations comparable to bacteria within macrophages (Figure S8). This allowed us to quantify bacterial viability by Pi staining of Mtb cells released from infected macrophages at various time points post antibiotic exposure. Infected THP-1 cells were exposed to anti-TB drugs (5× MIC) and intracellular bacteria were fixed with NEM at 12, 24, and 48 h p.i. Infected macrophages were lysed; released bacteria were stained with Pi, and ∼30,000 bacilli were analyzed by multi-parameter flow cytometry to simultaneously profile EMSH and viability status. As shown earlier, all antibiotics induce significant oxidative shift in intramycobacterial EMSH during infection. Furthermore, bacilli with oxidized EMSH were more sensitive to killing as evident by a time-dependent increase in Pi staining across all antibiotic treatments within this subpopulation as compared to other two subpopulations (Figure 9A, p<0.05). At 48 h p.i., 35–40% of EMSH-oxidized bacilli were Pi+. The EMSH-basal subpopulation demonstrates a modest increase in Pi+ staining (∼5–10%) at 24 and 48 h p.i. (Figure 9A). Surprisingly, EMSH-reduced subpopulation remained completely unaffected by antibiotics as shown by the absence of Pi+ cells within this group (Figure 9A). These results show that bacteria with lower EMSH are capable of excluding Pi and therefore maintain membrane integrity post-antibiotic treatment.

Thus, we find that redox heterogeneous bacteria vary in their susceptibility to antibiotics, consistent with the model that macrophage induced heterogeneity in intrabacterial EMSH creates physiologically distinct subpopulations of cells. Since environment inside autophagosomes induces substantial oxidative shift in intrabacterial EMSH, we hypothesized that treatment of infected macrophages with a well established autophagy inducer (rapamycin) would increase the localization of Mtb to autophagosomes and shift redox heterogeneity towards EMSH-oxidized. This would allow us to examine the influence of host antibacterial mechanisms (i. e autophagy) on intrabacterial EMSH and drug tolerance during infection. To examine this, infected macrophages were treated with a low non-lethal concentration of rapamycin (200 nM) and antibiotic-mediated redox stress and killing was monitored by assessing EMSH and Pi status. As shown in figure 9B, treatment of infected macrophages with rapamycin significantly increases fraction of Mtb cells exhibiting oxidizing EMSH as compared to untreated control (P<0.05). Furthermore exposure of infected macrophages to both rapamycin and antibiotics (INH or CFZ) induces oxidative shift in EMSH which supersedes that produced by either INH or CFZ or rapamycin alone (Figure 9B, p<0.05). Consistent with this, treatment of infected macrophages with rapamycin-INH or rapamycin-CFZ substantially increases the fraction of Pi+ Mtb cells, indicating augmented bacterial death by these combinations (Figure 9C, p<0.05).
Lastly, to show that EMSH-reduced subpopulation contributes to drug tolerance, we measured the sensitivity of Mtb towards anti-TB drugs in the presence of reducing agent DTT in vitro. First, we confirmed that exogenous addition of 5 mM DTT maintained a reductive EMSH (−320±2.9 mV) equivalent to the EMSH of reducing subpopulation and is non-deleterious for Mtb. In the absence of DTT treatment, antibiotics exposure led to a significant reduction (6–12-fold) in the survival of Mtb (Figure 9D). By contrast, ∼80% of Mtb survived an exposure to INH or ETH and ∼50% in the case of CFZ in the presence of DTT (Figure 9D). Collectively, our data suggest that oxidized EMSH potentiates antibiotic action, whereas EMSH-reduced promotes tolerance to anti-TB drugs and suggest that host-induced cell-to-cell variation in EMSH may be a novel mechanism by which Mtb resists antimicrobial treatment during infection.
Discussion
Basic research on persistence and drug-tolerance in Mtb is hampered by the lack of tools to study bacterial physiology during infection. Here, we developed a new biosensor, Mrx1-roGFP2, to image dynamic changes in the EMSH of Mtb during infection. Using confocal and flow cytometry, we quantified EMSH and unraveled mycothiol-linked redox heterogeneity in Mtb at the single-cell level during infection and highlighted the utility of bioprobes in exploring new mechanisms of drug action in Mtb bacilli. In Mrx1-roGFP2, the genetic coupling of roGFP2 with the mycothiol-specific oxidoreductase (Mrx1) ensures that roGFP2 functions as a physiological substrate for Mrx1 and therefore dynamically oxidizes and reduces in response to EMSH. Since cytoplasmic levels of chromosomally encoded Mrx1 may differ between mycobacterial species/strains or under diverse environmental conditions, one of the main advantages of coupling Mrx1 with roGFP2 is to facilitate rapid and continuous equilibration of biosensor with MSH/MSSM redox couple independent of endogenous Mrx1 pool. Our results agree with a recent study which showed that the homologue of Mtb Mrx1 in Msm specifically interacts with the mycothiol redox system in vitro [25]. Related non-disruptive approaches have been employed to develop GSH-specific and H2O2-specific in vivo bioprobes [24], [51].
The utility of Mrx1-roGFP2 in investigating mycobacterial physiology during infection comes from our flow cytometric and confocal data showing that the environment inside macrophage induces redox heterogeneity and oscillations in intrabacterial EMSH. The reductive-oxidative-reductive oscillations in EMSH during intramacrophage growth corresponds to the early induction of genes linked to reductive stress (e.g., whiB3, whiB7, and dosR) [10], [42], followed by extensive bacterial killing during intermediate phase of relative increase in subpopulation with oxidized EMSH at 48 h p.i as compared to 24 h p.i., and progressive increase in replication upon anti-oxidative shifts in EMSH at later stages of infection [42]. Interestingly, we observed some variations between confocal microscopy and flow cytometry based measurements of intrabacterial EMSH. For example, a higher percentage of subpopulation with oxidized EMSH was detected using confocal microscopy as compared to flow cytometry at 24 h p.i. (Figure 5A and 6A). However, although flow cytometry allows monitoring of large number of cells at multiple time points with high statistical power, the read out is derived from averaging Mtb cells inside infected macrophages. Therefore, confocal microscopy is a much more accurate indicator of intrabacterial EMSH at the level of individual bacteria inside macrophages. In agreement to this, the moi dependent variations in the distribution of subpopulations with different EMSH could also be due to averaging relatively higher number of bacteria present inside macrophages infected at a moi of 10 than 1 by flow cytometry. Nonetheless, both techniques were in reasonable agreement and complement one another to generate a highly resolved view of intrabacterial EMSH during infection. We also discovered that sub-vacuolar compartments such as endosomes, lysosomes and autophagosomes are the source of redox variability in Mtb populations. While lysosomes and autophagosomes enrich EMSH-oxidized bacteria, early endosomes induce a reductive shift in EMSH of Mtb. Similarly, a greater fraction of bacteria displayed oxidized EMSH at a moi of 1 than 10, which is consistent with the reported increase in phagosomal maturation and intracellular trafficking of Mtb to lysosomes at low moi [52]. Finally, our results showing a substantial oxidative shift in EMSH inside activated macrophages and its reversal upon treatment with iNOS inhibitor (NMLA) are in agreement with studies implicating the role of vacuolar NOX2 and iNOS systems in creating overwhelming oxidative stress within lysosomes and autolysosomes [2]. It is well known that Mtb actively remodels endosomal/phagosomal pathways during infection [53] to induce variability among phagosomes during infection [54]. This suggests that Mtb-induced alterations within sub-vacuolar compartments can generate a range of environmental conditions for the evolution of redox deviations in Mtb population.
Intriguingly, we found that redox heterogeneity is uniformly present in H37Rv and MDR/XDR strains inside macrophages. However, distinct strain-dependent variations in redox heterogeneity were also evident. Because macrophage compartments distinctly manipulate EMSH of Mtb, varied redox oscillatory behavior could simply be a consequence of differences in co-localization kinetics of Mtb strains within sub-vacuolar compartments over time. Alternatively, phenotypic variations such as uptake rate and intramacrophage growth kinetics may influence the EMSH of these strains. While the molecular mechanisms behind these redox variations and their influence on evolution of drug-resistance and fitness await further investigation, several studies have documented strain-specific differences in bacterial and host gene expression during infection [55], [56], and resistance to redox stress [57], [58].
Numerous studies using oxidant-sensitive dyes have demonstrated that bactericidal antibiotics, regardless of their target, exert toxicity by stimulating Fenton-catalyzed ROS production [59]–[61]. However, two recent studies demonstrate that measurements using dyes may be inconclusive owing to their non-specificity and that the bactericidal potential of antibiotics does not correlate with ROS generation in vitro [62], [63]. Furthermore, to the best of our knowledge, the contribution of antibiotics-stimulated oxidative stress in the physiological context of infection has not been evaluated. In this context, Mrx1-roGFP2 allowed us to investigate the redox-basis of drug action in Mtb. We show that Mtb maintains EMSH in response to antibiotic exposure (except in case of CFZ) during in vitro growth, whereas a significant oxidative shift was induced by all drugs during intramacrophage growth. This data along with the antibiotic sensitivity displayed by the MSH-deficient strains [21] suggest that MSH biosynthesis or recycling efficiently buffers toxicity associated with drugs during normal growing conditions. In this regard, mycothiol-S-conjugate detoxification system (Mca) maintains cytoplasmic MSH levels upon antibiotic exposure by rapidly converting MSH-antibiotic adducts to mercapturic acids and excreting them into the culture media [44]. Another Mtb antioxidant, ERG, was found to be over-expressed in MSH-mutants, however, it does not provide protection against antibiotics [64].
Importantly, we show that the antibiotic-mediated increase in EMSH-oxidized precedes bacterial death inside infected macrophages. Oxidative shift in EMSH was substantial at time points earlier than those at which drug antimycobacterial activities were achieved. Moreover, INH-resistant clinical strains remained uninfluenced by INH-mediated oxidative changes in EMSH during infection, thus supporting our conclusions. Our results suggest that an active cooperation between host factors and antibiotics could disrupt intramycobacterial EMSH during chemotherapy. This is in agreement with a recent study demonstrating the role of anti-TB drugs in inducing host ROS production and potentiating mycobactericidal activity by delivering Mtb into lysosomal and autophagosomal compartments [65]. Since lysosomes and autophagosomes predominantly contain Mtb cells with an oxidized EMSH, our findings present a novel insight into the mechanism of antibiotic action during infection. Together, our data suggest that direct effects of antibiotics on Mtb physiology and on host innate immune mechanisms such as autophagy and phagosomal maturation, jointly eliminate Mtb bacilli by stimulating overwhelming intramycobacterial oxidative stress in vivo. In line with this hypothesis, we show that treatment with autophagy inducer (rapamycin) significantly augments mycobactericidal activities of antibiotics during infection. While MSH has been shown to be dispensable for growth of Mtb in mice [66], future experiments should target MSH levels during antibiotic treatment to understand its function in modulating bacterial killing during antimicrobial therapy in vivo, where the heterogeneity in EMSH may be one of the critical determinants of persistence and drug tolerance. These exciting hypotheses can now be investigated by integrating Mrx1-roGFP2 technology with high-resolution live cell profiling of intramycobacterial EMSH at the single-cell level during infection.
How do these findings relate to human TB? Within human host, Mtb persists in a state of drug unresponsiveness in oxygen-depleted and lipid-rich granulomas [67]. The center of TB granuloma is hypoxic (3 mM Hg) and contains lipid-laden foamy macrophages and free fatty acids released from necrotic macrophages [67], [68]. Recent studies suggest that β-oxidation of host fatty acids by Mtb as the primary carbon source in an O2-deficient environment leads to massive accumulation of NADH/NADPH, which generates intrabacterial reductive stress in persisting cells during infection [10], [69]. Work presented here demonstrates that drug tolerance observed during persistence may be mediated by increased reductive capacity. Further strengthening this connection are recent findings demonstrating elimination of Mtb persisters by drugs which become active under reductive stress (e.g., metranidazole) or generate overwhelming ROS (e.g., CFZ) [70], [71]. Another common clinical observation is that Mtb genetically resistant to only a subset of anti-TB drugs resists clearance from all other antibiotics and thus, survive combination drug therapy [72]. One interesting possibility revealed by this work is that redox heterogeneity within genetically drug-resistant clinical isolates (MDR/XDR) provides a subpopulation that tolerates antibiotics against which bacteria are genetically susceptible. These understandings shed new light on drug resistant mechanisms and suggest that novel approaches that disrupt redox metabolism in Mtb may significantly impact eradication of both genetic and phenotypic drug resistant populations.
In summary, we have developed a genetically encoded, fluorescent reporter capable of monitoring intrabacterial EMSH within macrophages. We anticipate that Mrx1-roGFP2 will play an important role in high content screening of small-molecule inhibitors of intrabacterial redox homeostasis. Based on this work, one can readily imagine development of new biosensors to measure the redox state of specific and unusual redox thiols, such as BSH, trypanothione, ovothiol A found in a variety of pathogenic organisms. Finally, macrophage-induced redox heterogeneity and its connection to drug sensitivity may be relevant to other intracellular pathogens. For example, the macrophage environment induces expression of genes responsible for antioxidant production and drug-tolerance in Legionella pneumophilla [73]. Thus, our findings may have relevance to several intracellular pathogens causing chronic and relapsing infections where persistence and tolerance pose challenges for treatment.
Materials and Methods
Cloning, purification, and biochemical characterization of the Mrx1-roGFP2
The roGFP2 containing vector was obtained from Tobias P. Dick [24]. The roGFP2 open reading frame (ORF) was released using NcoI and HindIII and was cloned downstream of hsp60 promoter into similarly digested E. coli-mycobacterial shuttle vector, pMV762 [74] to generate pMV762-roGFP2. Mrx1-roGFP2 biosensor construct was generated by fusing the coding sequence of Mtb Mrx1 (Rv3198A) with roGFP2 having a 30-amino acid linker (GGSGG)6 between the two genes. Mrx1 coding sequence was amplified from Mtb genome (Forward primer: 5′ ATGCCCATGGTGATCACCGCTGCG 3′. Reverse primer: 5′ ATGCACTAGTACCCGCGATCTTTAC 3′). Cysteine mutations in Mrx1 coding sequence were introduced by site-directed mutagenesis as described [11]. Primers used were: Mrx1 (AGYC) forward primer: 5′ CTATACGACATCATGGGCTGGCTATTGCCTTCGAC 3′, reverse primer: 5′ GTCGAAGGCAATAGCCAGCCCATGATGTCGTATAG 3′, Mrx1 (CGYA) forward primer: 5′ CATCATGGTGTGGCTATGCCCTTCGACTCAAAACAG 3′, reverse primer: 5′ CTGTTTTGAGTCGAAGGGCATAGCCACACCATGATG 3′. All the fusion constructs were sub cloned into the expression vector pET28b (Novagen), expressed in the E. coli strain BL21 DE3 (Stratagene), and fusion proteins were purified via hexahistidine affinity chromatography as described [11]. Aerobically purified Mrx1-roGFP2 and ro-GFP2 were found to be in the fully oxidized state. To study the effect of various oxidants in vitro, the roGFP2 fusion proteins (1 µM) were first reduced with 10 mM DTT for 30 min on ice and desalted with Zeba Desalt spin columns (Pierce Biotechnology). In vitro measurements using various roGFP2 variants were performed on SpectraMax M3 microplate reader.
Mtr electron transfer assay
Mtb mtr ORF (Rv3198A) was cloned into pET28b (Novagen) and expressed in the E. coli strain BL21 DE3. Purification of Mtr protein was performed as described in the earlier section. Mycothiol is purchased from JEMA Biosciences, San Diego, CA, USA. To perform Mtr electron transfer assay, a mixture of 2.5 µM purified Mtr, 250 µM MSSM and 500 µM NADPH was prepared in 50 mM HEPES pH 8.0 in a 96-well plate. In the control reaction, Mtr was absent. The mixture was incubated at 37°C and consumption of NADPH was monitored at 340 nm for 60 min. To check the specificity of roGFP2 fusion proteins towards MSH, Mtr assay mixture was prepared as described above. After incubation at 37°C for 30 min, oxidized roGFP2 fusion proteins (1 µM) were added to the mix. Ratiometric sensor response was monitored for 200 min. A control reaction without MSSM was included.
Sensitivity of Mrx1-roGFP2 towards changes in OxDMSH
Pre-reduced uncoupled roGFP2 and Mrx1-roGFP2 (1 µM) were incubated with mycothiol solutions (1 mM total) containing increasing fractions of MSSM. The total concentration of MSH (MSH total) refers to MSH equivalents i. e MSHtotal = [MSH]+2[MSSM]. OxDMSH is the fraction of MSH total that exists as [MSSM] and can be conveniently calculated using the following formula:

Miscellaneous procedures
Detailed Materials and Methods are provided in the supporting information.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. DyeC, WilliamsBG (2010) The population dynamics and control of tuberculosis. Science 328 : 856–861.
2. EhrtS, SchnappingerD (2009) Mycobacterial survival strategies in the phagosome: defence against host stresses. Cell Microbiol 11 : 1170–1178.
3. MacMickingJD, NorthRJ, LaCourseR, MudgettJS, ShahSK, et al. (1997) Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci U S A 94 : 5243–5248.
4. CooperAM, SegalBH, FrankAA, HollandSM, OrmeIM (2000) Transient loss of resistance to pulmonary tuberculosis in p47(phox−/−) mice. Infect Immun 68 : 1231–1234.
5. LeePP, ChanKW, JiangL, ChenT, LiC, et al. (2008) Susceptibility to mycobacterial infections in children with X-linked chronic granulomatous disease: a review of 17 patients living in a region endemic for tuberculosis. Pediatr Infect Dis J 27 : 224–230.
6. YangCT, CambierCJ, DavisJM, HallCJ, CrosierPS, et al. (2012) Neutrophils exert protection in the early tuberculous granuloma by oxidative killing of mycobacteria phagocytosed from infected macrophages. Cell Host Microbe 12 : 301–312.
7. den HengstCD, ButtnerMJ (2008) Redox control in actinobacteria. Biochim Biophys Acta 1780 : 1201–1216.
8. BrugarolasP, MovahedzadehF, WangY, ZhangN, BartekIL, et al. (2012) The oxidation-sensing regulator (MosR) is a new redox-dependent transcription factor in Mycobacterium tuberculosis. J Biol Chem 287 : 37703–37712.
9. SinghA, GuidryL, NarasimhuluKV, MaiD, TrombleyJ, et al. (2007) Mycobacterium tuberculosis WhiB3 responds to O2 and nitric oxide via its [4Fe-4S] cluster and is essential for nutrient starvation survival. Proc Natl Acad Sci U S A 104 : 11562–11567.
10. SinghA, CrossmanDK, MaiD, GuidryL, VoskuilMI, et al. (2009) Mycobacterium tuberculosis WhiB3 maintains redox homeostasis by regulating virulence lipid anabolism to modulate macrophage response. PLoS Pathog 5: e1000545.
11. ChawlaM, ParikhP, SaxenaA, MunshiM, MehtaM, et al. (2012) Mycobacterium tuberculosis WhiB4 regulates oxidative stress response to modulate survival and dissemination in vivo. Mol Microbiol 85 : 1148–1165.
12. SongT, DoveSL, LeeKH, HussonRN (2003) RshA, an anti-sigma factor that regulates the activity of the mycobacterial stress response sigma factor SigH. Mol Microbiol 50 : 949–959.
13. FlynnJL, ChanJ (2001) Tuberculosis: latency and reactivation. Infect Immun 69 : 4195–4201.
14. KumarA, FarhanaA, GuidryL, SainiV, HondalusM, et al. (2011) Redox homeostasis in mycobacteria: the key to tuberculosis control? Expert Rev Mol Med 13: e39.
15. RotaC, ChignellCF, MasonRP (1999) Evidence for free radical formation during the oxidation of 2′-7′-dichlorofluorescin to the fluorescent dye 2′-7′-dichlorofluorescein by horseradish peroxidase: possible implications for oxidative stress measurements. Free Radic Biol Med 27 : 873–881.
16. TarpeyMM, WinkDA, GrishamMB (2004) Methods for detection of reactive metabolites of oxygen and nitrogen: in vitro and in vivo considerations. Am J Physiol Regul Integr Comp Physiol 286: R431–444.
17. MeyerAJ, DickTP (2010) Fluorescent protein-based redox probes. Antioxid Redox Signal 13 : 621–650.
18. NewtonGL, RawatM, La ClairJJ, JothivasanVK, BudiartoT, et al. (2009) Bacillithiol is an antioxidant thiol produced in Bacilli. Nat Chem Biol 5 : 625–627.
19. JothivasanVK, HamiltonCJ (2008) Mycothiol: synthesis, biosynthesis and biological functions of the major low molecular weight thiol in actinomycetes. Nat Prod Rep 25 : 1091–1117.
20. SareenD, NewtonGL, FaheyRC, BuchmeierNA (2003) Mycothiol is essential for growth of Mycobacterium tuberculosis Erdman. J Bacteriol 185 : 6736–6740.
21. BuchmeierNA, NewtonGL, KoledinT, FaheyRC (2003) Association of mycothiol with protection of Mycobacterium tuberculosis from toxic oxidants and antibiotics. Mol Microbiol 47 : 1723–1732.
22. HansonGT, AggelerR, OglesbeeD, CannonM, CapaldiRA, et al. (2004) Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J Biol Chem 279 : 13044–13053.
23. MeyerAJ, BrachT, MartyL, KreyeS, RouhierN, et al. (2007) Redox-sensitive GFP in Arabidopsis thaliana is a quantitative biosensor for the redox potential of the cellular glutathione redox buffer. Plant J 52 : 973–986.
24. GutscherM, PauleauAL, MartyL, BrachT, WabnitzGH, et al. (2008) Real-time imaging of the intracellular glutathione redox potential. Nat Methods 5 : 553–559.
25. Van LaerK, ButsL, FoloppeN, VertommenD, Van BelleK, et al. (2012) Mycoredoxin-1 is one of the missing links in the oxidative stress defence mechanism of Mycobacteria. Mol Microbiol 86 : 787–804.
26. RawatM, NewtonGL, KoM, MartinezGJ, FaheyRC, et al. (2002) Mycothiol-deficient Mycobacterium smegmatis mutants are hypersensitive to alkylating agents, free radicals, and antibiotics. Antimicrob Agents Chemother 46 : 3348–3355.
27. PatelMP, BlanchardJS (1999) Expression, purification, and characterization of Mycobacterium tuberculosis mycothione reductase. Biochemistry 38 : 11827–11833.
28. Gutierrez-LugoMT, BakerH, ShiloachJ, BoshoffH, BewleyCA (2009) Dequalinium, a new inhibitor of Mycobacterium tuberculosis mycothiol ligase identified by high-throughput screening. J Biomol Screen 14 : 643–652.
29. ArnerES, NakamuraH, SasadaT, YodoiJ, HolmgrenA, et al. (2001) Analysis of the inhibition of mammalian thioredoxin, thioredoxin reductase, and glutaredoxin by cis-diamminedichloroplatinum (II) and its major metabolite, the glutathione-platinum complex. Free Radic Biol Med 31 : 1170–1178.
30. HaramakiN, HanD, HandelmanGJ, TritschlerHJ, PackerL (1997) Cytosolic and mitochondrial systems for NADH - and NADPH-dependent reduction of alpha-lipoic acid. Free Radic Biol Med 22 : 535–542.
31. RawatM, JohnsonC, CadizV, Av-GayY (2007) Comparative analysis of mutants in the mycothiol biosynthesis pathway in Mycobacterium smegmatis. Biochem Biophys Res Commun 363 : 71–76.
32. FernandesND, WuQL, KongD, PuyangX, GargS, et al. (1999) A mycobacterial extracytoplasmic sigma factor involved in survival following heat shock and oxidative stress. J Bacteriol 181 : 4266–4274.
33. RamanS, SongT, PuyangX, BardarovS, JacobsWRJr, et al. (2001) The alternative sigma factor SigH regulates major components of oxidative and heat stress responses in Mycobacterium tuberculosis. J Bacteriol 183 : 6119–6125.
34. KaushalD, SchroederBG, TyagiS, YoshimatsuT, ScottC, et al. (2002) Reduced immunopathology and mortality despite tissue persistence in a Mycobacterium tuberculosis mutant lacking alternative sigma factor, SigH. Proc Natl Acad Sci U S A 99 : 8330–8335.
35. NewtonGL, TaP, FaheyRC (2005) A mycothiol synthase mutant of Mycobacterium smegmatis produces novel thiols and has an altered thiol redox status. J Bacteriol 187 : 7309–7316.
36. TaP, BuchmeierN, NewtonGL, RawatM, FaheyRC (2011) Organic hydroperoxide resistance protein and ergothioneine compensate for loss of mycothiol in Mycobacterium smegmatis mutants. J Bacteriol 193 : 1981–1990.
37. HolsclawCM, MuseWB3rd, CarrollKS, LearyJA (2011) Mass Spectrometric Analysis of Mycothiol levels in Wild-Type and Mycothiol Disulfide Reductase Mutant Mycobacterium smegmatis. Int J Mass Spectrom 305 : 151–156.
38. AlbrechtSC, BarataAG, GrosshansJ, TelemanAA, DickTP (2011) In vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell Metab 14 : 819–829.
39. UngKS, Av-GayY (2006) Mycothiol-dependent mycobacterial response to oxidative stress. FEBS Lett 580 : 2712–2716.
40. ChanJ, XingY, MagliozzoRS, BloomBR (1992) Killing of virulent Mycobacterium tuberculosis by reactive nitrogen intermediates produced by activated murine macrophages. J Exp Med 175 : 1111–1122.
41. ChanJ, TanakaK, CarrollD, FlynnJ, BloomBR (1995) Effects of nitric oxide synthase inhibitors on murine infection with Mycobacterium tuberculosis. Infect Immun 63 : 736–740.
42. RohdeKH, VeigaDF, CaldwellS, BalazsiG, RussellDG (2012) Linking the transcriptional profiles and the physiological states of Mycobacterium tuberculosis during an extended intracellular infection. PLoS Pathog 8: e1002769.
43. LiuYB, LongMX, YinYJ, SiMR, ZhangL, et al. (2013) Physiological roles of mycothiol in detoxification and tolerance to multiple poisonous chemicals in Corynebacterium glutamicum. Arch Microbiol 195 : 419–29.
44. RawatM, UppalM, NewtonG, SteffekM, FaheyRC, et al. (2004) Targeted mutagenesis of the Mycobacterium smegmatis mca gene, encoding a mycothiol-dependent detoxification protein. J Bacteriol 186 : 6050–6058.
45. XuX, VilchezeC, Av-GayY, Gomez-VelascoA, JacobsWRJr (2011) Precise null deletion mutations of the mycothiol synthesis genes reveal their role in isoniazid and ethionamide resistance in Mycobacterium smegmatis. Antimicrob Agents Chemother 55 : 3133–3139.
46. BrossierF, VezirisN, Truffot-PernotC, JarlierV, SougakoffW (2011) Molecular investigation of resistance to the antituberculous drug ethionamide in multidrug-resistant clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother 55 : 355–360.
47. YanoT, Kassovska-BratinovaS, TehJS, WinklerJ, SullivanK, et al. (2011) Reduction of clofazimine by mycobacterial type 2 NADH:quinone oxidoreductase: a pathway for the generation of bactericidal levels of reactive oxygen species. J Biol Chem 286 : 10276–10287.
48. ConnollyLE, EdelsteinPH, RamakrishnanL (2007) Why is long-term therapy required to cure tuberculosis? PLoS Med 4: e120.
49. AdamsKN, TakakiK, ConnollyLE, WiedenhoftH, WingleeK, et al. (2011) Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145 : 39–53.
50. HewittCJ, Nebe-Von-CaronG (2001) An industrial application of multiparameter flow cytometry: assessment of cell physiological state and its application to the study of microbial fermentations. Cytometry 44 : 179–187.
51. GutscherM, SobottaMC, WabnitzGH, BallikayaS, MeyerAJ, et al. (2009) Proximity-based protein thiol oxidation by H2O2-scavenging peroxidases. J Biol Chem 284 : 31532–31540.
52. WelinA, RaffetsederJ, EklundD, StendahlO, LermM (2011) Importance of phagosomal functionality for growth restriction of Mycobacterium tuberculosis in primary human macrophages. J Innate Immun 3 : 508–518.
53. ClemensDL, HorwitzMA (1996) The Mycobacterium tuberculosis phagosome interacts with early endosomes and is accessible to exogenously administered transferrin. J Exp Med 184 : 1349–1355.
54. RussellDG, MwandumbaHC, RhoadesEE (2002) Mycobacterium and the coat of many lipids. J Cell Biol 158 : 421–426.
55. HomolkaS, NiemannS, RussellDG, RohdeKH (2010) Functional genetic diversity among Mycobacterium tuberculosis complex clinical isolates: delineation of conserved core and lineage-specific transcriptomes during intracellular survival. PLoS Pathog 6: e1000988.
56. KooMS, SubbianS, KaplanG (2012) Strain specific transcriptional response in Mycobacterium tuberculosis infected macrophages. Cell Commun Signal 10 : 2.
57. MancaC, PaulS, BarryCE3rd, FreedmanVH, KaplanG (1999) Mycobacterium tuberculosis catalase and peroxidase activities and resistance to oxidative killing in human monocytes in vitro. Infect Immun 67 : 74–79.
58. IdhJ, MekonnenM, AbateE, WedajoW, WerngrenJ, et al. (2012) Resistance to first-line anti-TB drugs is associated with reduced nitric oxide susceptibility in Mycobacterium tuberculosis. PLoS One 7: e39891.
59. KohanskiMA, DwyerDJ, HayeteB, LawrenceCA, CollinsJJ (2007) A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130 : 797–810.
60. DwyerDJ, KohanskiMA, CollinsJJ (2009) Role of reactive oxygen species in antibiotic action and resistance. Curr Opin Microbiol 12 : 482–489.
61. BrynildsenMP, WinklerJA, SpinaCS, MacdonaldIC, CollinsJJ (2013) Potentiating antibacterial activity by predictably enhancing endogenous microbial ROS production. Nat Biotechnol 31 : 160–165.
62. KerenI, WuY, InocencioJ, MulcahyLR, LewisK (2013) Killing by bactericidal antibiotics does not depend on reactive oxygen species. Science 339 : 1213–1216.
63. LiuY, ImlayJA (2013) Cell death from antibiotics without the involvement of reactive oxygen species. Science 339 : 1210–1213.
64. Sao EmaniC, WilliamsMJ, WiidIJ, HitenNF, ViljoenAJ, et al. (2013) Ergothioneine is a secreted antioxidant in Mycobacterium smegmatis. Antimicrob Agents Chemother 57L: 3202–7.
65. KimJJ, LeeHM, ShinDM, KimW, YukJM, et al. (2012) Host cell autophagy activated by antibiotics is required for their effective antimycobacterial drug action. Cell Host Microbe 11 : 457–468.
66. VilchezeC, Av-GayY, AttarianR, LiuZ, HazbonMH, et al. (2008) Mycothiol biosynthesis is essential for ethionamide susceptibility in Mycobacterium tuberculosis. Mol Microbiol 69 : 1316–1329.
67. RussellDG, VanderVenBC, LeeW, AbramovitchRB, KimMJ, et al. (2010) Mycobacterium tuberculosis wears what it eats. Cell Host Microbe 8 : 68–76.
68. ViaLE, LinPL, RaySM, CarrilloJ, AllenSS, et al. (2008) Tuberculous granulomas are hypoxic in guinea pigs, rabbits, and nonhuman primates. Infect Immun 76 : 2333–2340.
69. BoshoffHI, XuX, TahlanK, DowdCS, PetheK, et al. (2008) Biosynthesis and recycling of nicotinamide cofactors in Mycobacterium tuberculosis. An essential role for NAD in nonreplicating bacilli. J Biol Chem 283 : 19329–19341.
70. LinPL, DartoisV, JohnstonPJ, JanssenC, ViaL, et al. (2012) Metronidazole prevents reactivation of latent Mycobacterium tuberculosis infection in macaques. Proc Natl Acad Sci U S A 109 : 14188–14193.
71. GrantSS, KaufmannBB, ChandNS, HaseleyN, HungDT (2012) Eradication of bacterial persisters with antibiotic-generated hydroxyl radicals. Proc Natl Acad Sci U S A 109 : 12147–12152.
72. FortuneSM (2012) The Surprising Diversity of Mycobacterium tuberculosis: Change You Can Believe In. J Infect Dis 206 : 1642–1644.
73. RankinS, LiZ, IsbergRR (2002) Macrophage-induced genes of Legionella pneumophila: protection from reactive intermediates and solute imbalance during intracellular growth. Infect Immun 70 : 3637–3648.
74. SinghA, MaiD, KumarA, SteynAJ (2006) Dissecting virulence pathways of Mycobacterium tuberculosis through protein-protein association. Proc Natl Acad Sci U S A 103 : 11346–11351.
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2014 Číslo 1
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Lyme Disease: Call for a “Manhattan Project” to Combat the Epidemic
- Origin, Migration Routes and Worldwide Population Genetic Structure of the Wheat Yellow Rust Pathogen f.sp.
- IFNγ/IL-10 Co-producing Cells Dominate the CD4 Response to Malaria in Highly Exposed Children
- Human and Plant Fungal Pathogens: The Role of Secondary Metabolites
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy