
The N-Terminus of the RNA Polymerase from Infectious Pancreatic Necrosis Virus Is the Determinant of Genome Attachment
The RNA-dependent RNA polymerase VP1 of infectious pancreatic necrosis virus (IPNV) is a single polypeptide responsible for both viral RNA transcription and genome replication. Sequence analysis identifies IPNV VP1 as having an unusual active site topology. We have purified, crystallized and solved the structure of IPNV VP1 to 2.3 Å resolution in its apo form and at 2.2 Å resolution bound to the catalytically-activating metal magnesium. We find that recombinantly-expressed VP1 is highly active for RNA transcription and replication, yielding both free and polymerase-attached RNA products. IPNV VP1 also possesses terminal (deoxy)nucleotide transferase, RNA-dependent DNA polymerase (reverse transcriptase) and template-independent self-guanylylation activity. The N-terminus of VP1 interacts with the active-site cleft and we show that the N-terminal serine residue is required for formation of covalent RNA∶polymerase complexes, providing a mechanism for the genesis of viral genome∶polymerase complexes observed in vivo.
Published in the journal:
. PLoS Pathog 7(6): e32767. doi:10.1371/journal.ppat.1002085
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002085
Summary
The RNA-dependent RNA polymerase VP1 of infectious pancreatic necrosis virus (IPNV) is a single polypeptide responsible for both viral RNA transcription and genome replication. Sequence analysis identifies IPNV VP1 as having an unusual active site topology. We have purified, crystallized and solved the structure of IPNV VP1 to 2.3 Å resolution in its apo form and at 2.2 Å resolution bound to the catalytically-activating metal magnesium. We find that recombinantly-expressed VP1 is highly active for RNA transcription and replication, yielding both free and polymerase-attached RNA products. IPNV VP1 also possesses terminal (deoxy)nucleotide transferase, RNA-dependent DNA polymerase (reverse transcriptase) and template-independent self-guanylylation activity. The N-terminus of VP1 interacts with the active-site cleft and we show that the N-terminal serine residue is required for formation of covalent RNA∶polymerase complexes, providing a mechanism for the genesis of viral genome∶polymerase complexes observed in vivo.
Introduction
Virus-encoded RNA-dependent RNA polymerases (RdRPs) are vital components in the life cycle of RNA viruses, being responsible for both genome replication and transcription. RdRPs from viruses that are considered unrelated [e.g. hepatitis C virus (family Flaviviridae), Foot-and-Mouth disease virus (family Picornaviridae) and bacteriophage Φ6 (family Cystoviridae)] share a similar structure and mechanism of catalysis and are therefore potential targets for generic antiviral drugs [1]. They all have the canonical “right-hand” polymerase fold with palm, fingers and thumb domains and contain a number of conserved sequence motifs (A–G), the conserved residues GDD within motif C being essential for catalytic activity [2], [3].
Members of the family Birnaviridae possess bi-segmented double-stranded RNA (dsRNA) genomes and form virions with non-enveloped single-shelled icosahedral capsids [4]. Infectious pancreatic necrosis virus (IPNV), which infects salmonids, and infectious bursal disease virus (IBDV), a poultry virus, are pathogens of significant economic importance and are the best characterized members of this virus family [5], [6]. From sequence analysis it was proposed that the RdRPs of Birnaviridae possess a non-canonical active site where the polymerase sequence motifs are re-ordered C-A-B, motif C containing ADN in place of the conserved GDD catalytic residues [3]. The structure of the RdRP VP1 from IBDV confirmed this hypothesis [7], revealing that the polymerase catalytic site was spatially similar to canonical viral RdRPs despite the different connectivity of the catalytic motifs. However, it remained unclear how substrate and metal ions bound at the active site during catalysis.
An additional unusual feature of birnavirus RdRPs is their ability to self-guanylylate: auto-catalyzing the covalent addition of a GMP moiety to a serine residue of the polymerase in a template-independent manner to form VP1pG [8], [9], [10]. This self-guanylylation has been presumed to be involved in priming the initiation of replication [11]. VP1 is observed in the virion as two forms, free polymerase and covalently attached to the 5′ ends of the segments of the viral RNA genome [12], and recent studies have shown that recombinant IBDV VP1 produces both free and protein-attached RNA products [9]. The exact site of self-guanylylation is unclear. Biochemical characterization mapped the self-guanylylation to residue S163 of IPNV VP1 [10], but no evidence for guanylylation was observed at an equivalent site in structures of the IBDV enzyme [7], [13]. Recent studies of IBDV VP1 suggest that the self-guanylylation reaction occurs in the first 175 residues of the enzyme at a locus distinct from the polymerase catalytic site [9].
We have cloned, expressed and solved the structure of the RdRP VP1 from IPNV in both its apo form and bound to a catalytic metal (magnesium). We show that IPNV VP1 possesses significant terminal (deoxy)nucleotide transferase and reverse transcriptase activity in addition to its RNA replication/transcription activity. Unexpectedly our structures reveal that the first 30 residues of VP1, not observed in the structure of IBDV VP1, bind at the active site of the enzyme. Mutation of the N-terminal serine residue of the mature protein to alanine abolishes its ability to form covalent RNA∶polymerase complexes, suggesting a mechanism of VP1∶genome attachment in vivo.
Results
The structure of C-terminally truncated (ΔC55) IPNV VP1
Recombinantly expressed IPNV VP1 carrying a C-terminal hexahistidine affinity tag is predominantly monomeric in solution as determined by analytical gel filtration and multi-angle light scattering (data not shown). Crystals of full-length VP1 with the C-terminal hexahistidine tag removed diffracted to 3.8 Å resolution but proved refractory to further optimization. A second construct lacking the C-terminal 55 residues (ΔC55) was therefore cloned, expressed, purified and crystallised. These crystals diffracted well and the structure was solved by molecular replacement using the structure of IBDV VP1 [7] as a search model. The final structure contains 5 molecules in the asymmetric unit and has been refined to 2.3 Å resolution with residuals R = 0.166, Rfree = 0.187 (Table S1). The stereochemical quality of the structure is excellent, with 3756 of 3835 residues (98%) occupying favored regions of the Ramachandran plot (Table S1).
The structure of ΔC55 IPNV VP1 comprises the polymerase domain (residues 31–790), an ordered helix of the N-terminus (residues 11–19; see below) and two residues of the C-terminal affinity tag. The IPNV polymerase closely resembles IBDV VP1, with an r.m.s. deviation in Cα positions of 1.2 Å over 678 residues (Figure 1). The structure of the core polymerase, comprising the fingers, palm and thumb domains, is especially well conserved (1.0 Å Cα r.m.s. deviation over 463 residues), with significant structural rearrangements occurring only at the tips or back of the fingers and thumb, distal to the catalytic palm domain (Figure 1). The structure of IPNV VP1 confirms the previously-identified rearrangement of the catalytic palm domain [3], [7], with the non-canonical ADN catalytic motif (residues 387–389) lying at the apex of a β-hairpin in the active site (Figure 1). As in IBDV VP1, extensive N - and C-terminal extensions wrap around the core fingers, palm and thumb domains (Figure 1). The sequence and structure of the N-terminal extension is largely conserved, differences occurring only in the conformation of the loop between residues 82–91. The general conformation of the C-terminal extension is similar to IBDV, passing from the inner-base of the thumb to the back of the fingers via the outer edge of the palm. However, many of the surface-exposed loops adopt different conformations and the helix-turn-helix between residues 736–779 is rotated by approximately 50° relative to the equivalent region of IBDV VP1.

Strong electron density was observed near the outer edge of the palm domain, between the side chains of N184, N409 and N514 and the backbone carbonyl oxygen atoms of V177, N182 and G512 (Figure 2). This feature, some 15 Å from the putative active site, was modeled as a K+ ion based on the strength of the electron density, the ligand-to-K+ distances of 2.6–2.8 Å [14] and the hard nature of the six ligands.

The structure of Mg2+-bound ΔC55 VP1
To characterize binding of catalytic metals at the active site of the enzyme, crystals of ΔC55 VP1 were soaked in reservoir solution supplemented with 50 mM MgCl2 and 10 µM GTP and lacking citrate (which chelated metals, frustrating soaking experiments). The Mg-bound ΔC55 VP1 structure was refined to 2.2 Å resolution with residuals R = 0.161, Rfree = 0.181 (Table S1). A single Mg2+ ion was observed bound at the active site, coordinated by four solvent molecules and the side chain oxygen atoms of N389 and D402 in an octahedral geometry (Figure 2).
The coordination geometry of the bound K+ ion does not change upon addition of Mg2+, suggesting that Mg2+ does not replace K+ in this position. As in the structure of IBDV VP1 soaked with Mg2+ and GTP [13], no significant structural rearrangements were observed between the structures of apo and Mg-bound ΔC55 VP1 and no electron density for a bound GTP molecule was observed.
The N-terminus of VP1 interacts with the active site
Additional electron density resembling a protein helix was observed in the active site cleft of both apo and Mg-bound ΔC55 VP1. Based on the presence of strong density suggesting a sulfur-containing methionine side chain, this helix was tentatively assigned as residues 11–19 from the otherwise-disordered N-terminal tail of the protein. As the 48 Å separating the final residue of the helix (M19) and the first residue of the polymerase domain (I31) is too great to be spanned by the intervening 10 residues, the N-terminal helix presumably derives from an adjacent polymerase domain in the crystal.
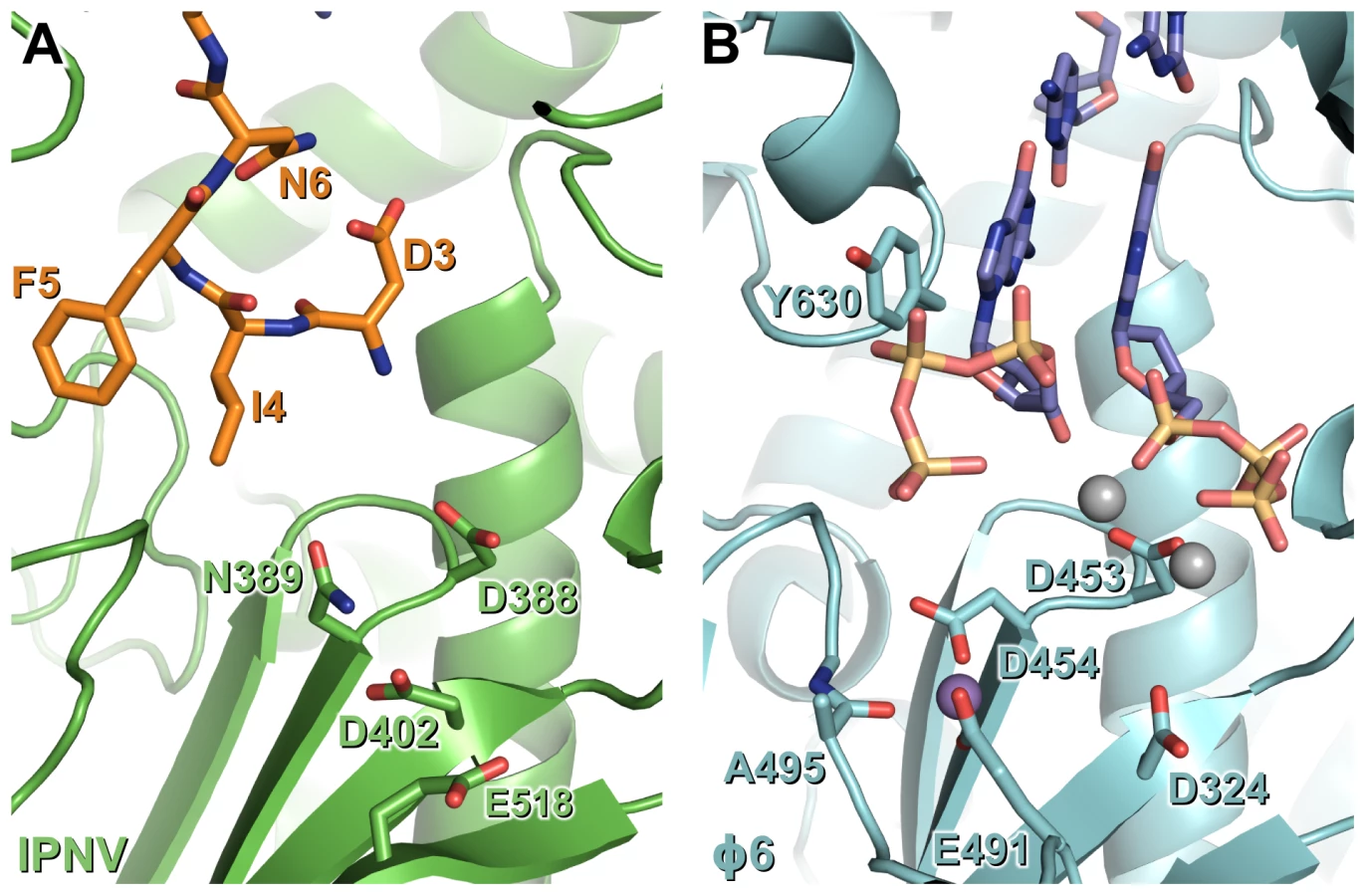
The identity of the N-terminal helix was confirmed by solving the structure of ΔC55 VP1 in an alternate crystal form at 3.0 Å resolution with residuals R = 0.188, Rfree = 0.216 (‘large unit cell’, Table S1). Clear electron density connects the helix with residue 31 of an adjacent molecule in the crystal in 7 of the 8 molecules present in the asymmetric unit (Figure 3, Figure S1). In addition, residues 3–10 are seen in this structure extending toward the catalytic active site (Figure 3, Figure S1). As in the high-resolution structures, the helix formed by residues 13–19 lies in a hydrophobic pocket within the active site cleft formed by loops of the fingers domain and the N-terminal extension (Figure 3, Figure S1). Residues 11–12 form a short isolated β strand which interacts with residue 241 of the fingers domain, while residue 10 spans the two sides of the active site cleft. Residues 7–10 interact with a loop between residues 594–598 of the thumb, this loop being partly mobile and modeled as having two conformations in the refined structure. Residues 3–6 interact with the thumb, the palm and the C-terminal extension, the side chain of F5 being buried in a hydrophobic pocket between the side chains of residues W563, L578, R582 and F662 (Figure 3). While the conformation of S2 (the first residue of mature VP1 assuming removal of the initiator methionine by methionine aminopeptidase) could not be assigned unambiguously, it will clearly lie extremely close to the catalytic residues of the active site (Figure 3).

The structure of full-length VP1
The structure of full-length IPNV VP1 was solved using ΔC55 VP1 as a search model and refined to 3.8 Å resolution with residuals R = 0.186, Rfree = 0.213 (Table S1). As for ΔC55 VP1 in the large unit cell, electron density for residues 3–19 was present in the active site. Some residual electron density could be observed linking residue 19 to the first residue (E27) of an adjacent polymerase molecule, but it was not sufficiently well-resolved to allow modeling of the intervening residues. Despite being crystallized in the presence of Mn2+ and ATP, no metal ions or nucleotides were observed at the active site. While additional electron density was seen connected to the C-terminus of the molecular replacement search model in three of the four copies of VP1 present in the asymmetric unit only eight additional residues (791–798) could be placed, suggesting that residues 799–845 are not ordered. Surprisingly the C-terminus of the fourth molecule in the asymmetric unit (from residue 687 onwards) is significantly refolded compared to the other IPNV VP1 molecules (Figure S2) and IBDV VP1. The strand that runs along the side of the palm domain is shifted ‘upward’ to lie at the outer rim of the active site cleft. Neither the helix that lay between the base of the palm and fingers (residues 706–716), the three-helix bundle (residues 724–781) nor the intervening loop region are evident; instead the polypeptide has flipped up and projects away from the back of the fingers. A continuous stretch of density returning from this projection covers the back of the fingers and extends to cover the back base of the fingers and the N-terminal extension, but given the low resolution of the data we were unable to include this region in the final refined model.
Biochemical characterization of IPNV VP1
Full-length VP1, the ΔC55 construct used to generate well-diffracting crystals, and an N - and C-terminally truncated construct (ΔN27C55) that represents the well-folded VP1 polymerase domain (residues 28–790) were biochemically characterized. All recombinant VP1 constructs are capable of initiating de novo synthesis with non-specific single-stranded (ss) and double-stranded (ds) RNA templates, but not with ssDNA or dsDNA molecules (Figure 4, Table 1 and data not shown). VP1-catalysed polymerization proceeds readily in the presence of divalent cations and nucleotides (NTPs or dNTPs) and produces full-length double-stranded products (dsRNA or RNA/DNA hybrids, see below) of all tested ssRNA templates. Full-length and truncated VP1 are less productive than the highly-active RdRP from bacteriophage Φ6, which was used for comparison (Figure 4, Table 1). VP1 can perform in vitro semi-conservative transcription with dsRNA templates, albeit less efficiently than replication (Table 1).


VP1 is devoid of activity in the absence of divalent cations, whereas addition of Mg2+ or Mn2+ at concentrations exceeding 2 mM stimulates RNA synthesis (Figure S3). Adding Ca2+ as the sole divalent cation to the reaction mixture does not induce catalysis (data not shown) and supplementing the Mg2+ - or Mn2+-containing reaction mixtures with Ca2+ completely inhibits polymerization (data not shown). The addition of Zn2+ yields low levels of RNA synthesis at a concentration of 2 mM or less, whereas higher concentrations efficiently inhibit catalysis (data not shown). Supplementing reaction mixtures with K+ does not stimulate RNA synthesis, and K+ on its own is unable to induce catalysis (data not shown). Interestingly, adding both Mg2+ and Mn2+ to the reaction mixture at their respective optimal concentration has an adverse effect on RNA synthesis and RNA polymerization is most efficient when using half the optimal concentration of both ions (5 mM Mg2+ and 1 mM Mn2+; data not shown).
VP1 is not able to initiate primer-dependent RNA synthesis but displaces various complementary RNA oligonucleotides annealed to ssRNA templates (Figure S4) despite poor in vitro transcription activity (Table 1). Synthesis of a significant proportion of RNA strands is initiated by means of back-priming (Table 1), as observed previously for IBDV VP1 [15]. IPNV VP1 shows only a slight preference for cytidine as the terminal template nucleotide (Table 1). The elongation rate of IPNV VP1 is slow when compared with the Φ6 RdRP (Table 1; Figure 4) and VP1 reaches a higher product yield with short (<500 nucleotide) ssRNA templates (Figure 4).
Based on structural data, we designed a set of mutant polymerases to verify the identity of the catalytic center. Single point mutations were introduced at the putative active site (D388N, D402N and S400A) and at the K+ binding site (N184S and N514H). The replication activity of all these mutant polymerases was below the detection limit of our assays (<5% of wild-type VP1 activity, data not shown).
Terminal (deoxy)nucleotide transferase and reverse transcriptase activity
IPNV VP1 is capable of utilizing free NTPs or dNTPs for the template-independent addition of one or more nucleotides to the 3′ terminus of single - or double-stranded RNA, so-called terminal (deoxy)nucleotide transferase (TNTase and TdNTase) activity (Figure 4). The TNTase activity of VP1 is very weak compared to that of Φ6 RdRP, although this activity is significantly enhanced in the truncated VP1 constructs (Figure 4, Table 1). Somewhat surprisingly, ΔC55 VP1 has a 5-fold higher TdNTase activity than the full-length enzyme, surpassing the equivalent activity of the Φ6 RdRP by more than two-fold, while the TdNTase activity of ΔN27C55 VP1 is severely compromised (Table 1).
In addition to the TdNTase activity of IPNV VP1, the polymerase is capable of utilizing a ssRNA template and dNTPs for de novo RNA-directed DNA polymerization resulting in a RNA/DNA hybrid (Figure 4, Figure S5). This reverse transcriptase activity is less efficient than in vitro RNA replication when utilizing the same ssRNA template. Both full-length and ΔC55 VP1 yield RNA/DNA hybrids of various heterologous ssRNA templates, ΔC55 being roughly 5-fold more active than the full-length enzyme, while ΔN27C55 VP1 reverse transcriptase activity is extremely low (Table 1, Figure 4).
Self-guanylylation and covalent RNA∶polymerase complex formation
Upon incubation with [α32P]-GTP both full-length and ΔC55 VP1 acquire radio-labels that do not dissociate during denaturing gel electrophoresis (Figure 5), presumably because the proteins have covalently bound [α32P]-GMP to form VP1pG (so-called self-guanylylation activity) [8]. A mutant form of VP1 where the N-terminal serine residue of the mature protein has been mutated to alanine (S2A) maintains the ability to form VP1pG, while ΔN27C55 VP1 is not radio-labeled under denaturing conditions and therefore lacks self-guanylylation activity (Figure 5).

Replication of ssRNA by IPNV VP1 yields high molecular weight products in addition to the expected dsRNA, manifesting as ladders and smears of radioactivity on native agarose gel electrophoresis [8]. The formation of ladders is most marked when using short RNA templates and is consistent with the formation of ‘concatenated’ RNA products containing integral repeats of the template RNA (Figure 4A and data not shown). RNA concatenation activity is strongest for ΔN27C55 VP1, while formation of the larger heterogeneous smears is more pronounced for full-length and ΔC55 VP1 (Figure 4A). Following denaturing sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS PAGE; Figure 5) two major products are observed: free RNA product and RNA∶polymerase complexes that represent covalent association of VP1 with the nascent RNA [9]. However, while ΔN27C55 and S2A VP1 are capable of producing RNA product, they lack the ability to form covalent RNA∶polymerase complexes.
Discussion
We have expressed and purified the IPNV RNA-dependent RNA polymerase VP1 and determined its structure to 2.2 Å resolution. The structure closely resembles that of IBDV VP1 (Figure 1), confirming that birnavirus polymerases constitute a family of RNA virus polymerases with a permuted motif C relative to other (canonical) viral single-polypeptide RdRPs [3], [7]. Recombinant IPNV VP1 is highly active, utilizing NTPs to catalyze the replication/transcription of ssRNA/dsRNA templates in a reaction that does not rely on added RNA primers (primer-independent or de novo initiation; Figure 4) in order to produce both free RNA product and RNA that remains covalently bound to the polymerase (Figure 5). VP1 is also capable of adding NTPs and dNTPs to the 3′ terminus of reaction products in a template-independent fashion (TNTase/TdNTase activity; Figure 4). Furthermore, it can utilize dNTPs and ssRNA templates in the absence of DNA primers to produce an RNA/DNA hybrid (reverse transcriptase activity; Figure 4, Figure S5). Mutations of residues D388, S400 and D402 abolish replication, transcription and TNTase/TdNTase activity, confirming that the catalytic active site lies within the palm domain. The spatial organization of the active site is similar to that of IBDV VP1 (Figure 1) and of canonical viral RdRPs such as the Φ6 RdRP [7], [9], [16]. Removal of the C-terminal 55 residues (residues 791–845) of VP1 enhances the replication, transcription and TNTase/TdNTase activities of the enzyme; however, as this region was not ordered in the structure of full-length VP1, its role in modulating activity remains unclear. Further removal of the N-terminal 27 amino acids yields an enzyme that is competent to replicate/transcribe RNA and has enhanced TNTase activity, but has a reduced ability to utilize dNTPs (Table 1) and lacks both self-guanylylation activity and the ability to covalently associate with RNA product (Figure 5). A VP1 mutant where the N-terminal serine of the mature enzyme is replaced with alanine (S2A) maintains RNA replication and self-guanylylation activity but does not form covalent RNA∶polymerase complexes (Figure 5).
Metal binding at the VP1 catalytic site
IPNV VP1 requires metal ions for catalysis, optimal activity being obtained in the presence of 5 mM Mg2+ and 1 mM Mn2+ (Figure S3 and data not shown). However, when soaked with an excess of Mg2+ ions we observe only one Mg2+ ion at the active site, coordinated by the side chains of N389 and D402 (Figure 2). It was reported that three Mg2+ ions were bound in the 3.15 Å structure of Mg-bound IBDV VP1 [13], however none of these occupy the Mg2+ site we observe in IPNV VP1. We performed additional experiments, soaking ΔC55 IPNV VP1 crystals with up to 200 mM Mg2+ and 40 mM Mn2+, but these failed to reveal any additional bound Mg2+ (data not shown). In the light of these results and inspection of the IBDV electron density maps, we ascribe differences in the observed metal binding to difficulties in interpreting the low resolution electron density of the earlier IBDV VP1 structure.
In initiation or elongation complexes of canonical viral polymerases two catalytic metal ions (plus one structural metal in the case of Φ6 RdRP) are observed at the active site [16], [17], [18], [19]. In such complexes the catalytic metal ions bridge the conserved Asp residues of catalytic motifs A and C, lying above the tip of the β hairpin (formed by motif C), and the triphosphate group of the substrate nucleotide. In the structure of reovirus RdRP solved in the absence of substrate nucleotides a single Mn2+ ion is observed in the active site in a position corresponding to that we observe for the single Mg2+ of IPNV VP1 [18], being coordinated by the side chains of D735, D585 and E780 (equivalent to VP1 residues N389, D402 and half-way between D559 and S400, respectively; Figure 2). Upon formation of an initiation complex two Mn2+ ions bind the reovirus RdRP active site (Figure 2). It is therefore likely that in the presence of template and nucleotide substrates a second metal ion will bind at the active site of IPNV VP1 and the resulting initiation complex will contain two metal ions coordinated by D388, D402 and the nucleotide triphosphate moiety at the tip of the catalytic motif C β hairpin.
Birnavirus polymerases possess a structural K+ ion
In addition to the Mg2+ ion bound at the catalytic active site, in all structures of IPNV VP1 we observe a K+ ion bound at the junction of the palm and fingers domains (Figure 2). We find that density consistent with a K+ ion is also present in previously published structures of IBDV VP1, although it was not identified as such at the time. The residues that interact with the K+ ion (N184, N409 and N514) are conserved across the Birnaviridae (Figure S6) and we propose that binding of this K+ ion is a general feature of birnavirus polymerases. The region at which the K+ ion binds is not structurally conserved in canonical RdRPs and the K+ ion is not analogous to ‘structural’ metal ions such as the divalent cation bound on top of the palm near the catalytic residues in the Φ6 RdRP [20]. Mutations N184S or N514H (both predicted to prevent K+ binding) abolish polymerase activity, consistent with the K+ ion performing a role in structuring the palm and maintaining its orientation relative to the fingers.
VP1 reverse transcriptase activity
IPNV VP1 has significant reverse transcriptase activity, utilizing dNTPs and RNA to form DNA/RNA hybrids (Figure 4, Figure S5). VP1 RNA-dependent RNA polymerization and reverse transcriptase activities share a requirement for RNA (not DNA) template, are optimally active under similar conditions, and are both abolished by mutations to the active site residues D388, S400 and D402, consistent with the two activities sharing a common catalytic mechanism.
The molecular features of VP1 that allow it to utilize dNTPs in addition to NTPs are not obvious. Mutation of F155 to valine in Moloney murine lukemia virus reverse transcriptase removes its specificity for dNTPs [17], [21]. Birnavirus RdRPs have a conserved glutamic acid at the equivalent position (E407 in IPNV, Figure S6), while in other viral RdRPs this residue is aspartic acid [7]. It had been suggested that this aspartate residue might recognize substrate NTPs by forming a hydrogen bond with the NTP O2′ atom [22], but structures of Φ6 and Norwalk virus RdRP initiation complexes contradict this hypothesis [16], [19]. The presence of asparagine in catalytic motif B near the tip of the motif C hairpin is unable to explain the preference of viral RdRPs for NTPs over dNTPs [23], [24] since this residue is asparagine in IPNV VP1 (N480, Figure 1). It is also unlikely that serine in the loop preceding the motif B helix (IPNV S471) is crucial for (d)NTP selectivity [7], as serine or threonine at this position is generally conserved across RdRPs not competent for reverse transcriptase activity.
Removal of the N-terminal 27 residues of IPNV VP1 greatly diminishes the reverse transcriptase activity of the enzyme and ΔN27C55 VP1 has enhanced TNTase activity but severely attenuated TdNTase activity. Based on a structural alignment with the Φ6 RdRP we observe that the N-terminus of the polymerase domain is adjacent to the (d)NTP entry tunnel (Figure S7), suggesting that the N-terminal segment of VP1 plays a role in recruiting dNTPs for catalysis. Further work is required to understand the molecular features that define NTP versus dNTP selectivity in viral RdRPs.
Flexibility of the VP1 C-terminus
Residues 688 onwards are completely refolded in one of the four molecules present in the full-length VP1 structure (Figure S2). The functional implications of this reorganization are not obvious. Models of a VP1 elongation complex based on superposition of the reovirus RdRP elongation complex [18] or of HIV reverse transcriptase in complex with a DNA template [17] require refolding of a small segment of the VP1 C-terminal extension, residues 654–670, in order to accommodate the nascent RNA duplex (not shown). It is possible that the purpose of the C-terminal extension is to rigidify the polymerase during initiation, but that this extension must peel away to allow the growing nucleotide chain to exit the active site. The structure of VP1 where residues 688 onwards are refolded might thus represent an intermediate unfolding step rather than a conformation of biological significance in its own right.
IPNV VP1 self-guanylylation
Birnavirus RdRPs hydrolyze GTP to form a covalent VP1pG complex in a template-independent manner, so-called self-guanylylation [8]. We see no evidence of covalent GMP modification at S163, the IPNV VP1 residue previously identified as being guanylylated [10], nor at any other position in the structures presented here or in crystals of VP1 co-crystallized with GTP (data not shown). Removal of the N-terminal 27 residues abolishes VP1 self-guanylylation activity (ΔN27C55 VP1, Figure 5), consistent with recent studies of IBDV VP1 that identified the site of self-guanylylation as residing within the first 175 residues of the polymerase [9]. We observe that ΔN27C55 VP1 maintains significant replication and transcription activity, demonstrating that self-guanylylation is not required for general “protein priming” of RNA polymerase activity. Further, as S2A VP1 self-guanylylates but lacks the ability to form covalent RNA∶polymerase complexes, the self-guanylylation activity is not sufficient for covalent attachment of RNA product to the enzyme (see below). While it is therefore clear that the N-terminal 27 residues of VP1 are required for self-guanylylation, the precise site and mechanism of this activity remains enigmatic.
Role of the N-terminus in RNA∶polymerase complex formation
In 16 of the 17 independent molecular views of the IPNV VP1 structure presented here the flexible N-terminal tail of an adjacent VP1 molecule in the crystalline lattice lies anchored in the active site cleft (Figure 3). The affinity of this self-interaction is obviously not high as VP1 behaves predominantly as a monomer in solution (data not shown). However, the high concentration of VP1 in crystal structures may well reflect the environment of the viral capsid more closely than biochemical assays performed in dilute solution.
There are two main sites of interaction between the N-terminal tail and the active site cleft of IPNV VP1: F5 binds in a pocket formed by residues from the thumb and palm, and a helix (S13–M19) plus the two residues that precede it (K11–A12) bind a hydrophobic pocket formed by the fingers and N-terminal extension. F5 and the residues with which it interacts (W563, L578, R582 and F662) are highly conserved across birnavirus RdRPs (Figure S6), the conformations of these residues being identical in IBDV and IPNV VP1. A hydrophobic helix-binding pocket is also evident in IBDV VP1. The edge of the pocket proximal to the active site (IPNV residues 238–242) is similarly positioned in IPNV and IBDV VP1. The loops which form the edges of the pocket distal from the active site (IPNV residues 86–91 and 227–234) are very poorly ordered in IBDV VP1 structures, indicating significant flexibility, and they would require only modest rearrangement to accommodate a bound N-terminal helix. In crystal structures of IBDV VP1 there is no evidence for the N-terminal tail binding the active site [13]; however, the molecular packing does not bring N-termini of adjacent molecules into close enough proximity to interact with the active site, precluding the interaction.
The observed self-interaction brings the N-terminal residue of VP1 (S2, assuming removal of the initiator methionine) within approximately 5 Å of the catalytic site (Figure 3), and removal of the N-terminal tail or mutation of S2 to alanine abolishes the ability of VP1 to form covalent RNA∶polymerase complexes (Figure 5). Superposition of VP1 onto the initiation complex of the Φ6 RdRP shows the VP1 N-terminal tail to be located in approximately the same position as the 5′ nucleotide of a nascent RNA daughter strand (Figure 6). The N-terminal interaction is not required for “protein priming” since ΔN27C55 VP1 maintains significant replication activity (Figure 5). Instead, we propose that residue S2 at the N-terminus of VP1 represents the site of covalent attachment of RNA product to the polymerase. As there is no strong preference for templates with 3′ cytidine bases it is unlikely that the covalently-attached RNA is formed by extension of a self-guanylylation guanosine that Watson-Crick base pairs with template. Further, S2A VP1 is incapable of forming covalent RNA∶polymerase complexes despite maintaining self-guanylylation activity. This suggests that nascent RNA is directly ligated to S2, independently of initiation or of any self-guanylylation activity. Such a hypothesis, based on the observed interaction of the VP1 N-terminus with the active site and requirement for an N-terminal serine to generate covalent RNA∶polymerase complexes, provides an elegant molecular mechanism for the birnavirus VP1∶genome association observed in vivo.
Materials and Methods
Cloning, expression and purification of IPNV VP1
A pET-21a(+)–derived plasmid encoding full-length VP1 from IPNV strain Jasper (UniProt ID P22173) plus a C-terminal VEH6 tag [10] was the generous gift of Dr P. Dobos (University of Guelph). VP1 constructs lacking the C-terminal 55 residues (ΔC55) and N-terminal 27 plus C-terminal 55 residues (ΔN27C55 VP1) were cloned as described in Text S1. Site-directed mutagenesis of full-length IPNV VP1 was performed using the QuikChange site-directed mutagenesis kit (Stratagene). Full-length, ΔC55, ΔN27C55 and mutant VP1 were expressed and purified by Ni-immobilization affinity and gel-filtration chromatography using standard protocols (see Text S1). Pure VP1 was used immediately for crystallization or stored at −20°C in 50% v/v glycerol for kinetic analysis (conditions under which the enzyme remained stable and active for several months).
Crystallization and data collection
Crystals of ΔC55 VP1 were grown in sitting drops containing 200 nL protein (5.8–6.8 mg/mL) and 100 nL reservoir solution (20–18% w/v PEG3350, 0.10–0.09 M bis-Tris propane pH 7.5, 0.2–0.18 M sodium citrate) equilibrated against 95 µL reservoirs at 20.5°C. Crystals were cryoprotected by soaking in mother-liquor supplemented with 20–25% glycerol for 1–10 min. Crystals of Mg-soaked ΔC55 VP1 were prepared by diluting the mother-liquor with ∼0.5 µL 20% w/v PEG3350, 0.1 M bis-Tris propane pH 8.25 and 20% v/v glycerol, transferring the crystals to a fresh drop containing 20% w/v PEG3350, 0.1 M bis-Tris propane pH 8.25, 20% v/v glycerol, 50 mM MgCl2 and 10 µM GTP, and incubating for 10 min. Crystals of full-length VP1 were grown in sitting drops containing 100 nL protein (5.3 mg/mL) plus 5 mM MnCl2 and 100 nL reservoir solution (20% w/v PEG3350, 0.1 M bis-Tris propane pH 7.5, 0.2 M sodium citrate, 20 mM ATP, 5% v/v MPD, 10 mM NaOH) equilibrated against 95 µL reservoirs at 20.5°C. All crystals were dipped into perfluoropolyether oil (PFO-X125/03, Lancester Synthesis) prior to cryocooling in a 100 K stream of N2 gas. Initial diffraction data collected on Diamond beam line I24 guided crystal optimization, from which diffraction data were recorded at Diamond beam line I02 (ΔC55 VP1) or ESRF beam line ID14-2 (Full-length VP1).
Structure solution and refinement
Diffraction data were processed using MOSFLM [25] and SCALA [26] as implemented by xia2 [27]. Data processing statistics are shown in Table S1. The structure of Mg-soaked ΔC55 VP1 was solved by molecular replacement with PHASER [28] using the structure of IBDV VP1, PDB ID 2PGG [7], as a starting model. The structures of apo ΔC55 VP1, apo ΔC55 VP1 in an alternate (large) unit cell and of full-length VP1 were solved by molecular replacement using the polymerase domain of Mg-soaked ΔC55 VP1 (residues 31–792) as a starting model.
Manual building was performed using COOT [29] and structures were refined using BUSTER-TNT [30] in consultation with the MolProbity web server [31]. Non-crystallographic local structure similarity restraints [32] were used during the refinement of all structures. For full-length VP1 and apo ΔC55 VP1 in the large unit cell local structure similarity restraints [32] were also used to deter deviation of the models from the high-resolution apo ΔC55 VP1 structure unless compelling evidence for a difference was present in the electron density. As the test sets for all structures were chosen randomly (apo and Mg-bound ΔC55 VP1 sharing the same random set) the presence of non-crystallographic symmetry may artificially lower the value of Rfree by a small amount but will not render the metric invalid [33]. Structural superpositions were performed using SSM [34] and Theseus [35]. Molecular graphics were prepared using PyMOL (DeLano Scientific) and sequence alignments with ALINE [36]. Structure factors and final refined coordinates have been deposited in the PDB with accession codes 2yi8 (apo ΔC55 VP1), 2yi9 (Mg-bound ΔC55 VP1), 2yia (apo ΔC55 VP1, large unit cell) and 2yib (full-length VP1).
Biochemical analyses
The RdRP from bacteriophage Φ6 was expressed and purified as described previously [37] and RNA templates were generated as described in Text S1. Please refer to Table S2 for an overview of the RNA templates. The reaction conditions for RNA polymerization by IPNV VP1 were optimized using the 723 nucleotide (nt) ssRNA sΔ+13 template (Figure S3) and found to be similar to the ‘standard’ Φ6 RdRP reaction conditions (50 mM HEPES-KOH pH 7.5, 20 mM NH4Ac, 6% w/v PEG 4000, 5 mM MgCl2, 1 mM MnCl2, 0.1 mM EDTA, 0.1% v/v Triton X-100) [38]; these conditions were hence used for all subsequent experiments unless otherwise noted. Under optimized reaction conditions all VP1-catalysed reactions except reverse transcription were readily observable by ethidium bromide staining of agarose gel electrophoresis, indicating a high level of polymerase activity. Replication (ssRNA template) and transcription (dsRNA template) assays were carried out in the presence of 1 mM ATP and GTP and 0.2 mM CTP and UTP supplemented with labeled [α32P]-UTP (replication, transcription) or [α32P]-GTP (replication). TNTase, TdNTase and self-guanylylation assays contained only labeled nucleotides of [α32P]-UTP (TNTase), [α32P]-dTTP (TdNTase) or [α32P]-GTP (self-guanylylation). Reverse transcription was performed with 1 mM dATP and dGTP and 0.2 mM dCTP supplemented with 0.3 µM (0.1 mCi/mL) labeled [α32P]-dTTP. After 2 h incubation, reactions were stopped by the addition of 2× or 10× loading buffer [39], [40] and analysis of the reaction products was performed using 0.8% w/v agarose gel (TBE) electrophoresis [40]. [α32P]-GTP labeled self-guanylylation and replication assays were terminated with 7× SB loading buffer [40], boiled for 3 min and analyzed in 8% SDS polyacrylamide gels. Oligonucleotide displacement was assayed as previously described [41]. Signals were collected by autoradiography on BAS1500 image plates (Fujifilm), which were scanned using a Fuji BAS-1500 phosphorimager (Fujifilm). Digital image analysis (densitometry) was performed using AIDA Image Analyzer software (version 3.44; Raytest Isotopenmeβgeräte), measuring the band intensities in 1D Evaluation mode using Lane and Peak Determination.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. BruennJA 2003 A structural and primary sequence comparison of the viral RNA-dependent RNA polymerases. Nucleic Acids Res 31 1821 1829
2. DelarueMPochOTordoNMorasDArgosP 1990 An attempt to unify the structure of polymerases. Protein Eng 3 461 467
3. GorbalenyaAEPringleFMZeddamJLLukeBTCameronCE 2002 The palm subdomain-based active site is internally permuted in viral RNA-dependent RNA polymerases of an ancient lineage. J Mol Biol 324 47 62
4. CoulibalyFChevalierCGutscheIPousJNavazaJ 2005 The birnavirus crystal structure reveals structural relationships among icosahedral viruses. Cell 120 761 772
5. MullerHIslamMRRaueR 2003 Research on infectious bursal disease–the past, the present and the future. Vet Microbiol 97 153 165
6. Rodriguez Saint-JeanSBorregoJJPerez-PrietoSI 2003 Infectious pancreatic necrosis virus: biology, pathogenesis, and diagnostic methods. Adv Virus Res 62 113 165
7. PanJVakhariaVNTaoYJ 2007 The structure of a birnavirus polymerase reveals a distinct active site topology. Proc Natl Acad Sci U S A 104 7385 7390
8. DobosP 1993 In vitro guanylylation of infectious pancreatic necrosis virus polypeptide VP1. Virology 193 403 413
9. PanJLinLTaoYJ 2009 Self-guanylylation of birnavirus VP1 does not require an intact polymerase activity site. Virology 395 87 96
10. XuHTSiWDDobosP 2004 Mapping the site of guanylylation on VP1, the protein primer for infectious pancreatic necrosis virus RNA synthesis. Virology 322 199 210
11. DobosP 1995 Protein-primed RNA synthesis in vitro by the virion-associated RNA polymerase of infectious pancreatic necrosis virus. Virology 208 19 25
12. CalvertJGNagyESolerMDobosP 1991 Characterization of the VPg-dsRNA linkage of infectious pancreatic necrosis virus. J Gen Virol 72 2563 2567
13. GarrigaDNavarroAQuerol-AudiJAbaituaFRodriguezJF 2007 Activation mechanism of a noncanonical RNA-dependent RNA polymerase. Proc Natl Acad Sci U S A 104 20540 20545
14. HardingMM 2002 Metal-ligand geometry relevant to proteins and in proteins: sodium and potassium. Acta Crystallogr D Biol Crystallogr 58 872 874
15. von EinemUIGorbalenyaAESchirrmeierHBehrensSELetzelT 2004 VP1 of infectious bursal disease virus is an RNA-dependent RNA polymerase. J Gen Virol 85 2221 2229
16. ButcherSJGrimesJMMakeyevEVBamfordDHStuartDI 2001 A mechanism for initiating RNA-dependent RNA polymerization. Nature 410 235 240
17. HuangHChopraRVerdineGLHarrisonSC 1998 Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 282 1669 1675
18. TaoYFarsettaDLNibertMLHarrisonSC 2002 RNA synthesis in a cage–structural studies of reovirus polymerase λ3. Cell 111 733 745
19. ZamyatkinDFParraFAlonsoJMHarkiDAPetersonBR 2008 Structural insights into mechanisms of catalysis and inhibition in Norwalk virus polymerase. J Biol Chem 283 7705 7712
20. PoranenMMSalgadoPSKoivunenMRWrightSBamfordDH 2008 Structural explanation for the role of Mn2+ in the activity of Φ6 RNA-dependent RNA polymerase. Nucleic Acids Res 36 6633 6644
21. GaoGOrlovaMGeorgiadisMMHendricksonWAGoffSP 1997 Conferring RNA polymerase activity to a DNA polymerase: a single residue in reverse transcriptase controls substrate selection. Proc Natl Acad Sci U S A 94 407 411
22. BressanelliSTomeiLRousselAIncittiIVitaleRL 1999 Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc Natl Acad Sci U S A 96 13034 13039
23. GoharaDWCrottySArnoldJJYoderJDAndinoR 2000 Poliovirus RNA-dependent RNA polymerase (3Dpol): structural, biochemical, and biological analysis of conserved structural motifs A and B. J Biol Chem 275 25523 25532
24. KorneevaVSCameronCE 2007 Structure-function relationships of the viral RNA-dependent RNA polymerase: fidelity, replication speed, and initiation mechanism determined by a residue in the ribose-binding pocket. J Biol Chem 282 16135 16145
25. LeslieAGW 1992 Mosflm - a programme for processing X-ray diffraction data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography 26
26. EvansP 2006 Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62 72 82
27. WinterG 2010 xia2: an expert system for macromolecular crystallography data reduction. Journal of Applied Crystallography 43 186 190
28. McCoyAJGrosse-KunstleveRWAdamsPDWinnMDStoroniLC 2007 Phaser crystallographic software. J Appl Crystallogr 40 658 674
29. EmsleyPCowtanK 2004 Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60 2126 2132
30. BlancERoversiPVonrheinCFlensburgCLeaSM 2004 Refinement of severely incomplete structures with maximum likelihood in BUSTER-TNT. Acta Crystallogr D Biol Crystallogr 60 2210 2221
31. DavisIWLeaver-FayAChenVBBlockJNKapralGJ 2007 MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35 W375 383
32. SmartOSBrandlMFlensburgCKellerPPaciorekW 2008 Annual Meeting of the American Crystallographic Association, Knoxville, TN. Abstract TP139
33. KleywegtGJBrungerAT 1996 Checking your imagination: applications of the free R value. Structure 4 897 904
34. KrissinelEHenrickK 2004 Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr D Biol Crystallogr 60 2256 2268
35. TheobaldDLWuttkeDS 2006 THESEUS: maximum likelihood superpositioning and analysis of macromolecular structures. Bioinformatics 22 2171 2172
36. BondCSSchuttelkopfAW 2009 ALINE: a WYSIWYG protein-sequence alignment editor for publication-quality alignments. Acta Crystallogr D Biol Crystallogr 65 510 512
37. MakeyevEVBamfordDH 2000 The polymerase subunit of a dsRNA virus plays a central role in the regulation of viral RNA metabolism. EMBO J 19 6275 6284
38. LaurilaMRMakeyevEVBamfordDH 2002 Bacteriophage Φ6 RNA-dependent RNA polymerase: molecular details of initiating nucleic acid synthesis without primer. J Biol Chem 277 17117 17124
39. PagratisNRevelHR 1990 Minus-strand RNA synthesis by the segmented double-stranded RNA bacteriophage Φ6 requires continuous protein synthesis. Virology 177 281 288
40. SambrookJRussellD 2001 Molecular cloning: a laboratory manual (3rd edition) Cold Spring Harbor, New York Cold Spring Harbor Laboratory Press
41. SarinLPPoranenMMLehtiNMRavanttiJJKoivunenMRL 2009 Insights into the pre-initiation events of bacteriophage Φ6 RNA-dependent RNA polymerase: towards the assembly of a productive binary complex. Nucleic Acids Res 37 1182 1192
42. ShwedPSDobosPCameronLAVakhariaVNDuncanR 2002 Birnavirus VP1 proteins form a distinct subgroup of RNA-dependent RNA polymerases lacking a GDD motif. Virology 296 241 250
43. PetkovDLinnemannEKapczynskiDRSellersHS 2007 Full-length sequence analysis of four IBDV strains with different pathogenicities. Virus Genes 34 315 326
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- High Affinity Nanobodies against the VSG Are Potent Trypanolytic Agents that Block Endocytosis
- Structural and Mechanistic Studies of Measles Virus Illuminate Paramyxovirus Entry
- Sporangiospore Size Dimorphism Is Linked to Virulence of
- The Binding of Triclosan to SmeT, the Repressor of the Multidrug Efflux Pump SmeDEF, Induces Antibiotic Resistance in
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy