
Compensatory T Cell Responses in IRG-Deficient Mice Prevent Sustained Infections
The obligate intracellular pathogen Chlamydia trachomatis is the most common cause of bacterial sexually transmitted diseases in the United States. In women C. trachomatis can establish persistent genital infections that lead to pelvic inflammatory disease and sterility. In contrast to natural infections in humans, experimentally induced infections with C. trachomatis in mice are rapidly cleared. The cytokine interferon-γ (IFNγ) plays a critical role in the clearance of C. trachomatis infections in mice. Because IFNγ induces an antimicrobial defense system in mice but not in humans that is composed of a large family of Immunity Related GTPases (IRGs), we questioned whether mice deficient in IRG immunity would develop persistent infections with C. trachomatis as observed in human patients. We found that IRG-deficient Irgm1/m3(-/-) mice transiently develop high bacterial burden post intrauterine infection, but subsequently clear the infection more efficiently than wildtype mice. We show that the delayed but highly effective clearance of intrauterine C. trachomatis infections in Irgm1/m3(-/-) mice is dependent on an exacerbated CD4+ T cell response. These findings indicate that the absence of the predominant murine innate effector mechanism restricting C. trachomatis growth inside epithelial cells results in a compensatory adaptive immune response, which is at least in part driven by CD4+ T cells and prevents the establishment of a persistent infection in mice.
Published in the journal:
. PLoS Pathog 7(6): e32767. doi:10.1371/journal.ppat.1001346
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1001346
Summary
The obligate intracellular pathogen Chlamydia trachomatis is the most common cause of bacterial sexually transmitted diseases in the United States. In women C. trachomatis can establish persistent genital infections that lead to pelvic inflammatory disease and sterility. In contrast to natural infections in humans, experimentally induced infections with C. trachomatis in mice are rapidly cleared. The cytokine interferon-γ (IFNγ) plays a critical role in the clearance of C. trachomatis infections in mice. Because IFNγ induces an antimicrobial defense system in mice but not in humans that is composed of a large family of Immunity Related GTPases (IRGs), we questioned whether mice deficient in IRG immunity would develop persistent infections with C. trachomatis as observed in human patients. We found that IRG-deficient Irgm1/m3(-/-) mice transiently develop high bacterial burden post intrauterine infection, but subsequently clear the infection more efficiently than wildtype mice. We show that the delayed but highly effective clearance of intrauterine C. trachomatis infections in Irgm1/m3(-/-) mice is dependent on an exacerbated CD4+ T cell response. These findings indicate that the absence of the predominant murine innate effector mechanism restricting C. trachomatis growth inside epithelial cells results in a compensatory adaptive immune response, which is at least in part driven by CD4+ T cells and prevents the establishment of a persistent infection in mice.
Introduction
Chlamydia trachomatis is an obligate intracellular bacterial pathogen that causes frequent infections in humans and significant morbidity throughout the world [1]. Ocular infection with C. trachomatis is the leading cause of preventable blindness worldwide and genital infection with C. trachomatis is the most common bacterial sexually transmitted infection (STI) in the United States [2], [3]. The major complications of C. trachomatis genital tract infections arise primarily in women. Acute genitourinary infections with C. trachomatis remain asymptomatic in a high proportion of infected individuals, and therefore often go untreated. In a substantial number of infected untreated women C. trachomatis can establish persistent infections, which over time result in pelvic inflammatory disease and tubal scarring and can ultimately cause infertility [4], [5].
C. trachomatis is a highly specialized, human-adapted pathogen with a narrow host range. Like many other pathogens with a very restricted host range, C. trachomatis has evolved to cause persistent infections in its preferred host enabling C. trachomatis to establish reservoirs for new infections and assure its survival as a pathogen within the human population [6]. Generally speaking, if a highly specialized pathogen enters a non-typical or “accidental” host, the non-typical host will either succumb to the infection and die or, more commonly, will rapidly clear the infection [7]. However, it is extremely rare for chronic infection to develop in a non-typical, immune-competent host. This basic principle holds true for experimental infections of laboratory mice with C. trachomatis. In contrast to human infections, C. trachomatis is rapidly cleared from mice when the organisms are instilled in the vagina or directly into the uterus [8], [9]. If it were possible to model elements of human C. trachomatis pathogenesis in mice – with a mouse model of chronic human infection - it would accelerate the study of this disease and therapies to combat it. A first step toward this goal is to understand the underlying mechanisms that promote persistent C. trachomatis infections in the human host and prevent the establishment of chronic C. trachomatis infections in the murine host.
A milestone in dissecting the basis for host tropisms of C. trachomatis was the discovery that IFNγ-induced cell-autonomous resistance in epithelial and other non-hematopoietic cell types like fibroblasts fundamentally differs between mice and humans. In human epithelial cells, IFNγ exerts its antimicrobial effect on C. trachomatis predominantly through the induction of indole-2,3-dioxygenase (IDO). The enzyme IDO degrades intracellular tryptophan stores, thus starving C. trachomatis, a natural auxotroph for tryptophan, of this essential nutrient [10], [11]. In contrast to human cells, most IFNγ-activated murine epithelial cells express insufficient amounts of IDO to restrict bacterial growth, with the notable exception of alveolar epithelial cells [10], [12], [13], [14]. Accordingly, IDO is not required for the clearance of vaginal C. trachomatis infections in mice [10], [15]. Instead, mice restrict Chlamydia species through a cell-autonomous resistance system that is executed by members of a large family of IFNγ-inducible GTPases called Immunity Related GTPases or IRGs [15], [16], [17], [18], [19], [20]. Remarkably, the divergent IFNγ responses of these two host species, mice and humans, are reflected in the counter-immune mechanisms that exist in two closely related Chlamydia species with distinct host tropism. Whereas genital strains of the human pathogen C. trachomatis can utilize exogenous indole to produce tryptophan to overcome IDO-mediated growth restriction, the rodent-adapted species Chlamydia muridarum has evolved a mechanism to evade IRG-driven immune responses [19], [21]. Given that the human pathogen C. trachomatis is highly susceptible to an IRG-driven immune response that is absent in its typical host, humans, but present in epithelial cell and fibroblasts of its non-typical host, mice, we investigated in this study whether the removal of the IRG resistance system would render mice permissive for persistent C. trachomatis genital infections.
Here, we report that mice deficient for the expression of two pivotal IRG regulatory proteins, Irgm1 (also called Lrg-47) and Irgm3 (also called Igtp), initially develop high bacterial burden after genital infection compared to wildtype mice. However, in spite of the initial delay in immune clearance, Irgm1/m3(-/-) mice are ultimately able to resolve a C. trachomatis infection as rapidly as wildtype mice. An exacerbated CD4+ T cell response is essential for the efficient clearance of genital C. trachomatis infections in Irgm1/m3(-/-) mice and the prevention of persistence. Our data show that the absence of early innate immune defenses and the resulting unrestricted expansion of C. trachomatis trigger an amplified T cell response that results in sterilizing immunity.
Results
Deletion of Irgm3 relieves the expansion defect observed in Irgm1-/- CD4+ T cells
We previously reported that Irgm1 and its paralog Irgm3 are required for resistance to C. trachomatis in an in vivo systemic infection model [19]. It is well established that IRG genes like Irgm1 and Irgm3 mediate a cell-autonomous antimicrobial response that directly targets vacuolar pathogens like C. trachomatis [22], [23], [24]. More recently it has been shown that at least one IRG family member, Irgm1, is also required for the proper expansion of CD4+ T cells [25]. To determine if Irgm1 and Irgm3 deficient mice were more susceptible to C. trachomatis infections due to an intrinsic T cell deficiency, we first tested whether expression of Irgm1 and/or Irgm3 in T cells was required for the activation and expansion of CD4+ T cells during an intrauterine infection with C. trachomatis. Towards this goal we crossed the C. trachomatis-specific, MHC class II restricted T cell receptor transgene NR1 onto the Irgm1-/-, Irgm3-/- and Irgm1/m3(-/-) genetic backgrounds. From these mice we derived naïve T cells and transferred these T cells into recipient wildtype mice. Although transfer of an antigen experienced or pre-activated population of NR1 cells can accelerate bacterial clearance, the naïve activation state and the low number of C. trachomatis-specific T cells transferred in these experiments did not result in accelerated immune clearance, as shown previously [26]. Therefore, the transfer of a relatively small number of IRG-deficient NR1 cells into a wildtype host allowed us to monitor pathogen specific immunity without altering the normal course of the immune response [26], [27]. One day after NR1 cell transfer, we directly instilled C. trachomatis into the uterus of these mice by a transcervical infection method. Six days post-infection we monitored the activation and expansion of the transferred NR1 cells in the uterus and the draining lymph nodes of the genital tract. The transferred NR1 cells expressed an allele of the congenic surface marker CD90 that was distinct from the allele expressed by T cells derived from the recipient mice allowing us to specifically detect transferred T cells. We observed that the IRG genotype of the NR1 cells had no apparent impact on the expression of surface activation marker CD62L and CD25 (Fig. 1A) or on the expression of the markers CD127, CD69 and CD44 (data not shown). Similarly, transferred NR1 cells of distinct IRG genotypes were indistinguishable in regards to the proportion of cells expressing the cytokines IFNγ, TNFα and IL-2 (data not shown). However, Irgm1-/- NR1 cells accumulated to significantly lower numbers in the uterus than wildtype NR1 cells did (Fig. 1B), and showed a trend towards lower cell numbers in the draining lymph nodes of the genital tract (Fig. 1B). These data are consistent with the previous observation that Irgm1-deficient CD4+ T cells fail to expand following an infection in mice [25]. Remarkably, we also found that the expansion defect of Irgm1-/- CD4+ cells was reversed by the concomitant removal of Irgm3 (Fig. 1B), suggesting that Irgm1/m3(-/-) T cells could be fully immune-competent. To directly determine whether Irgm1/m3(-/-) NR1 cells could convey protection to an intrauterine infection with C. trachomatis, we transferred Th1 skewed wildtype and Irgm1/m3(-/-) NR1 cells into Ifng-/- mice and subsequently instilled C. trachomatis directly into the uterus of these mice. In these experiments the early clearance of the infection is solely dependent on IFNγ secreted by the transferred NR1 cells, since the recipient mice are deficient for IFNγ production [26]. We found that NR1 cells doubly deficient in Irgm1 and Irgm3 conveyed protection against intrauterine C. trachomatis infection with an efficiency similar to wildtype NR1 cells (Fig. 2). In sum, these data indicated that the removal of Irgm3 salvaged the T cell intrinsic defect of Irgm1-/- cells and that Irgm1/3(-/-) CD4+ T cells were fully functional.


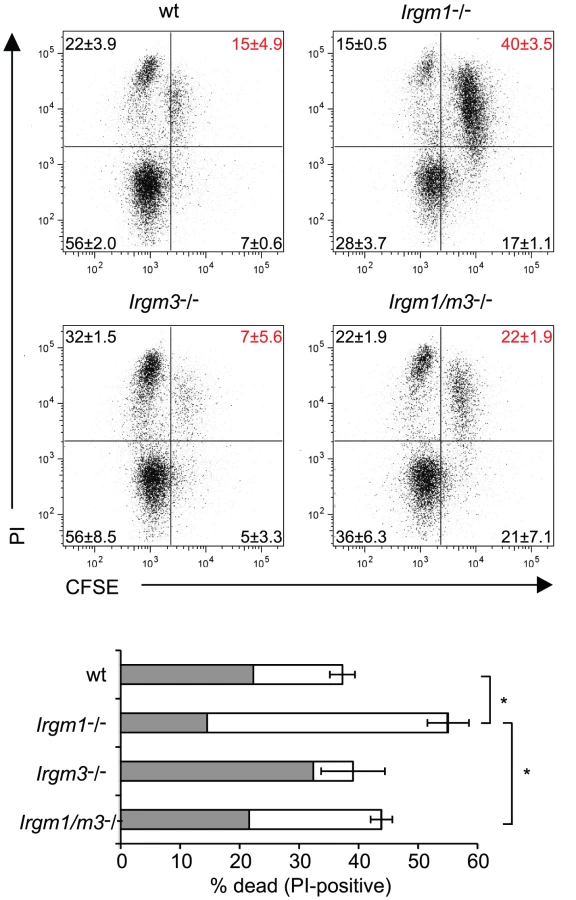
The inability of Irgm1-/- NR1 cells to expand to similar numbers as wildtype NR1 cells at the site of the infection could be explained by the propensity of Irgm1-deficient mature CD4+ T cells to prematurely die when activated for proliferation [25]. Considering that the concomitant removal of Irgm3 ‘rescues’ the T cell expansion defect of Irgm1-/- NR1 cells, we hypothesized that the simultaneous deletion of Irgm3 might serve to reverse the premature cell death phenotype of Irgm1-/- CD4+ T cells. To test this idea, we labeled wildtype, Irgm1-/-, Irgm3-/- and Irgm1/m3(-/-) CD4+ T cells with CFSE and then activated them for proliferation using plate-bound anti-CD3 and anti-CD28 antibodies for 72 hours. As reported previously [25], we observed increased total cell death of Irgm1-/- T cells compared to wildtype cells using propidium iodide incorporation as a measure for cell death (Fig. 3). The increase in cell death in Irgm1-/- T cells compared to wildtype cells was most pronounced in the population of CFSEhigh cells that had only undergone a few rounds of cell division. However, Irgm1/m3(-/-) T cells showed a significant decrease in cell death compared Irgm1-/- T cells and the total percentage of dead Irgm3-/- and Irgm1/m3(-/-) T cells was similar to wildtype T cells (Fig. 3). Taken together, these data showed that the deletion of Irgm3 reverses the T cell expansion and cell survival defect found in Irgm1-deficient T cells.
Irgm1/m3(-/-) fibroblasts are deficient for IFNγ-induced cell-autonomous resistance to C. trachomatis
IFNγ-activated cells lacking Irgm1 expression are impaired in their ability to contain the intracellular growth of various intracellular pathogens including Salmonella typhimurium, Toxoplasma gondii and C. trachomatis [22], [23], [24]. Recently, it has been shown that Irgm1-deficient cells can regain the ability to fully restrict growth of S. typhimurium when an additional genetic lesion in Irgm3 is introduced, suggesting that Irgm1 and Irgm3 are not essential to restrict S. typhimurium inside an infected cell [28]. In contrast, IFNγ-activated cells doubly deficient in Irgm1 and Irgm3 remain at least as susceptible to a T. gondii infection as cells harboring single Irgm gene deletions [28]. To determine whether Irgm1 and Irgm3 are essential for IFNγ-induced cell-autonomous resistance to C. trachomatis, we generated Irgm1/m3(-/-) mouse embryonic fibroblasts (MEFs). MEFs lacking both Irgm1 and Irgm3 displayed a near complete loss of IFNγ-induced resistance to C. trachomatis growth. Whereas only very few C. trachomatis inclusion were detectable in IFNγ-activated wildtype MEFs, the number of inclusions found in IFNγ-treated and untreated Irgm1/m3(-/-) MEFs was similar (Fig. 4A). To more accurately quantify C. trachomatis replication in these MEFs, we harvested DNA from infected cells at 4 and 30 hours post-infection (hpi) and measured the amount of Chlamydia DNA using qPCR. Neither IFNγ treatment nor IRG deficiency had any appreciable effect on C. trachomatis burden at 4 hpi, suggesting normal bacterial attachment and entry under all conditions. By 30 hpi IFNγ activation, C. trachomatis yields were reduced by nearly 2 logs in wildtype MEFs. In contrast, we only observed a 2-fold reduction in IFNγ-treated Irgm1/m3(-/-) MEFs (Fig. 4B). Because it was formally possible that C. trachomatis was able to replicate but unable to differentiate into infectious elementary bodies in IFNγ-activated Irgm1/m3(-/-) MEFs, we also assayed the release of infectious EBs by monitoring the production of inclusion forming units (IFUs) in these cells. Consistent with our findings by qPCR (Fig. 4B), we found that the ability of IFNγ-activated MEFs to restrict the production of infectious C. trachomatis progeny was severely compromised in Irgm1/m3(-/-) MEFs (Fig. 4C). Collectively, our results showed that Irgm1/m3(-/-) MEFs have lost their ability to efficiently restrict growth of C. trachomatis upon IFNγ activation.

Because these experiments were conducted in tryptophan-rich culture media, we could not dismiss a possible role for the tryptophan-degrading enzyme IDO in restricting C. trachomatis growth in IFNγ-activated MEFs. To test for such a function of IDO, we first measured induction of mouse IDO1 expression upon IFNγ activation. In contrast to the approximately 1000-fold induction of Irgm3 and Irgb10 mRNA expression, IDO1 mRNA was only moderately induced (5-fold), and more importantly, inhibition of C. trachomatis growth upon IFNγ treatment remained unchanged in the presence or absence of tryptophan in the culture media (Fig. S1A and B). Because ectopic expression of IDO resulted in significant restriction of C. trachomatis growth in MEFs (Fig. S1C), we conclude that endogenous IDO expression levels are too low in IFNγ-stimulated MEFs to restrict C. trachomatis growth.
Initially delayed but subsequently accelerated clearance of intrauterine C. trachomatis infections in Irgm1/m3(-/-) mice
Since MEFs isolated from Irgm1/3-/- mice failed to constrain C. trachomatis growth in the presence of IFNγ, we expected that these mice would also be unable to control C. trachomatis genital infections. To test this idea, we infected wild type, Ifngr1-/-, and Irgm1/m3(-/-) mice transcervically with C. trachomatis and measured the bacterial burden in the genital tract over 45 days. As expected, mice lacking the gene encoding the IFNγ receptor (Ifngr1-/-) showed greater bacterial burden than wildtype mice over the entire time course (Fig. 5). Initially Irgm1/m3(-/-) mice also developed high bacterial burden similar to the burden observed in Ifngr1-/- mice, yet by day 15 post-infection the number of organisms present in Irgm1/m3(-/-) mice was reduced to levels similar to the ones found in wildtype mice (Fig.5). By day 45 post-infection, C. trachomatis was still detectable in Ifngr1-/- mice but undetectable in wildtype and Irgm1/m3(-/-) mice. These data show that although Irgm1/m3(-/-) mice were initially defective in clearing C. trachomatis infections, they ultimately were able to control the infection. Therefore, we conclude that Irgm1/m3(-/-) mice do not exhibit an infection of extended duration as had been observed with the Ifngr1-/- mice.

An exacerbated CD4+ T cell response mediates the clearance of intrauterine C. trachomatis infection in Irgm1/m3(-/-) mice
We then began to investigate how Irgm1/m3(-/-) mice were able to clear C. trachomatis infections despite the initially high bacterial burden observed at day 5 and earlier. One possible explanation was that the increased antigen burden in the Irgm1/m3(-/-) mice stimulates the expansion and activation of C. trachomatis-specific effector T cells. To test the idea that host Irgm1/m3-deficiency could impact the development of wildtype T cells, we transferred naïve, CFSE-labeled, wildtype C. trachomatis-specific NR1 cells into wildtype, Ifngr1-/- or Irgm1/m3(-/-) host mice. Although we had already shown that Irgm1/m3(-/-) T cells appear to be indistinguishable from wildtype T cells (Fig. 1 and 2), we opted to use wildtype NR1 cells in these experiments in order to exclude any cell-autonomous effects on T cells that could be caused by the presence of the Ifngr1-/- or the Irgm1/m3(-/-) alleles. One day after the transfer of wildtype NR1 cells into mice of the indicated genotypes, mice were infected with C. trachomatis in the uterus. We found that C. trachomatis infection stimulated robust proliferation of transferred NR1 cells regardless of the genotype of the recipient mice (Fig. 6A, top panel). Though the genotype of the recipient mouse had no discernable impact on NR1 cell proliferation based on CSFE dye dilution, we did observe a significant increase in the total number of NR1 cells in the iliac lymph node (Fig. 6A and 6B) and the uterus (Fig. 6C) of Irgm1/m3(-/-) mice on day 6 post-infection compared to Ifngr1-/- and wildtype mice. The greater expansion of NR1 cells in Irgm1/m3(-/-) mice corresponded to an increase in the population of NR1 cells expressing the IL-2 high affinity receptor CD25 on NR1 cells resident in C. trachomatis-infected Irgm1/m3(-/-) mice compared to infected Ifngr1-/- and wildtype mice (Fig. 6A). Additionally, a significantly larger proportion of NR1 cells expressed IFNγ, the hallmark cytokine of Th1 activation, and the proinflammatory cytokine TNFα in combination with IFNγ, when transferred into Irgm1/m3(-/-) recipient mice compared to Ifngr1-/- and wildtype recipient mice (Fig. 6D). As expected for conventional Th1 cells, we did not detect significant IL-17 expression in NR1 cells transferred into either wildtype or Irgm1/m3(-/-) mice on day 6 post-infection (data not shown).

These results suggested that enhanced expansion of C. trachomatis-specific T cells and a boost in their activation status may compensate for the loss of innate immune restriction due to the Irgm1 and Irgm3 mutations. To prove that a compensatory amplification of the T cell response was responsible for clearance of C. trachomatis from the genital tract, we subjected groups of Irgm1/m3(-/-) and wildtype mice to multiple treatments with either anti-CD4 depleting or control antibodies and then determined bacterial burden in the uterus at 15 days post-infection. As expected, depleting CD4+ T cells resulted in greater bacterial burden in both wildtype as well as Irgm1/m3(-/-) mice relative to control mice of the same genotype, confirming that the adaptive immune response contributes to immune clearance at later times in the course of the infection (Fig. 7). Depletion of CD4+ T cells, however, had a greater effect on bacterial burden in Irgm1/m3(-/-) mice than it did in wildtype mice, increasing bacterial yield in the uterus by two logs instead of one log. These data confirmed that Irgm1/m3(-/-) mice compared to wildtype mice developed an exacerbated T cell response that was instrumental in the rapid clearance of genital infection with C. trachomatis.

Neutrophil infiltration to the uterus is enhanced in C. trachomatis infected Irgm1/m3(-/-) mice compared to wildtype mice
We then began to investigate the mechanism by which an exacerbated CD4+ T cell response clears C. trachomatis infections in mice lacking cell-autonomous IRG resistance in epithelial cells. Because of the recent demonstration that Chlamydia-specific T cells can control Chlamydia replication inside epithelial cells by a Nos2-dependent mechanism [29], we tested whether Nos2 was responsible for the elimination of C. trachomatis in Irgm1/m3(-/-) mice. To test this hypothesis, we generated Irgm1/m3(-/-)Nos2(-/-) triple knockout mice. Although C. trachomatis-infected Irgm1/m3(-/-)Nos2-/- mice trended towards higher bacterial burden compared to Irgm1/m3(-/-) or wildtype mice, this effect was not statistically significant in our experiments (Fig. S2).
While exploring alternative CD4-dependent immune mechanisms targeting C. trachomatis in Irgm1/m3(-/-) mice, we identified an increase in the uterine population of cells expressing the neutrophil surface marker GR1 relative to wildtype mice at day15 post-infection. An increase in the number of GR1+ cells was also observed in the iliac lymph nodes but not in the spleen of Irgm1/m3(-/-) mice (Fig. 8A). Similar to the depletion of CD4+ T cells, the depletion of the GR1+ cell population or the simultaneous depletion of CD4+ and GR1+ cells in Irgm1/m3(-/-) mice resulted in a dramatic increase in bacterial burden (Fig. 8B), suggesting an important role for neutrophils in the elimination of C. trachomatis in Irgm1/m3(-/-) mice.

Lastly, we explored the question as to why Irgm1/m3(-/-) mice cleared C. trachomatis infections more efficiently than Ifngr1-/- mice did (Fig. 5). Both Irgm1/m3(-/-) and Ifngr1-/- mice lack IRG-mediated cell-autonomous resistance, yet differ in the number and activation status of their Chlamydia-specific T cell population (Fig. 6). To determine whether T cells in conjunction with neutrophils are responsible for the relatively more efficient elimination of C. trachomatis in Irgm1/m3(-/-) mice compared to Ifngr1-/- mice, we treated Irgm1/m3(-/-), Ifngr1-/- and wildtype mice with anti-CD4 and anti-GR1 depleting antibodies, and determined bacterial burden at 15 days post infection. As expected, bacterial burden was dramatically elevated in all CD4/GR1-depeleted animals regardless of their genetic background. However, the most dramatic effect on bacterial burden was observed in Irgm1/m3(-/-) mice (Fig. S3) elevating the bacterial yield to the same levels observed in similarly treated Ifngr1-/- mice. These results suggest that IFNγ directs T cell - and neutrophil-dependent clearance of C. trachomatis infections.
Discussion
Genital infections with C. trachomatis are among the most common STI worldwide and constitute the most frequent bacterial STI in the United States. Asymptomatic and consequently unrecognized and untreated C. trachomatis infections can ascend from the cervix to the fallopian tube and establish persistence resulting in irreversible tissue damage and infertility [30], [31]. In contrast to humans, mice clear genitourinary infection with C. trachomatis rapidly and members of the IRG gene family play an important role in conveying resistance to C. trachomatis infections in the mouse. Whereas the importance for IRG proteins in resistance to C. trachomatis infections in mice is undisputed, the function of the constitutively expressed human ortholog IRGM in the pathogenesis of human C. trachomatis infections is less clear. Although IRGM induces antimicrobial xenophagy in human cells [32], [33], [34], [35], most human cells restrict growth of C. trachomatis by an IDO-dependent and apparently IRGM-independent mechanism ([10], [11], [15] and Fig. S1B). Although IRGM may still prove to be important in providing resistance to C. trachomatis infections in some cell types or tissues, IRGM is less likely to play a prominent role in the pathogenesis of human C. trachomatis infections. For these reasons, we sought to determine whether mice deficient in IRG-dependent resistance to C. trachomatis - and thus resembling humans in that regard - would develop persistent C. trachomatis infections.
We have previously shown that two members of the IRG gene family, Irgm1 and Irgm3, mediate resistance to C. trachomatis in an in vivo model of systemic infection [19]. Because the gene Irgm1 not only conveys cell-autonomous resistance to vacuolar pathogens inside an infected host cell but is also required to prevent the premature cell death of activated, proliferating CD4+ T cells, we first tested the hypothesis that the inability of IRG-deficient mice to efficiently restrict C. trachomatis infections could partly be due to a diminished CD4+ T cell response. We found that Irgm1-/- CD4+ T cells specific for a C. trachomatis epitope failed to efficiently expand in the uterus of genitally infected animals and prematurely died when activated for proliferation ex vivo. These results are consistent with the previously described expansion defect of Irgm1-/- CD4+ T cells [25]. However, we also found that the simultaneous removal of Irgm3 in Irgm1/m3(-/-) CD4+ T cells ‘rescued’ the phenotype of Irgm1-/- CD4+ T cells. Two alternative models may account for these observations: the first model is based on the published finding that IFNγ-induced IRG proteins form large protein aggregates in the absence of Irgm1 expression [36], [37]. These protein aggregates are not found in IFNγ-activated wildtype cells and are likely to have cytotoxic properties. The concomitant removal of Irgm3 has been shown to reduce the levels of IRG protein aggregates inside a cell, possibly below a threshold level of toxicity [28]. Because T cells have a relatively small cytoplasmic volume compared to most differentiated cells, they may be particularly susceptible to the cytotoxic effects of IRG protein aggregates. According to the “aggregate model” recently proposed by Hunn and Howard [38], the expansion defect of Irgm1-/- CD4+ T cells could be due to the cytotoxicity of IRG protein aggregates that form when the finely balanced network of IRG protein interactions is artificially disrupted by the Irgm1 gene deletion. In a second, alternative, model IFNγ-induced IRG mediated cell death in T cells is a regulated biological process that plays an important role in fine-tuning T cell homeostasis. Our observations can be reconciled with a model in which Irgm3 protein acts as an inducer of cell death whereas Irgm1promotes cell survival, functioning as an Irgm3 antagonist. According to this model, regulated changes in the expression of Irgm1 relative to Irgm3 or post-translational modifications of either Irgm protein could shift the balance towards either T cell survival or T cell death in wildtype T cells. Removal of the pro-survival factor Irgm1 in Irgm1-/- T cells would result in uncontrolled cell death due to the unrestricted action of Irgm3. Further removal of the pro-death factor Irgm3 in Irgm1/3 T cells would restore cell survival. In support of the latter model, we found that anti-CD3 stimulated Irgm3-/- CD4+ T cells showed improved cell viability compared to wildtype T cells at least during the first few rounds of anti-CD3 stimulated cell division (Fig. 2), suggesting that Irgm3 may indeed function as a pro-cell death molecule. Additionally, we observed that Irgm3-/- mice (but not Irgm1-/- or wildtype mice) expressing the NR1 transgene relatively frequently developed lymphoma with advanced age (JC and MNS, unpublished data), indicating a potential role for Irgm3 as a tumor suppressor gene. Beyond the uncertainty of the biological functional importance of IRG proteins in murine T cells, the additional unanswered question remains as to whether IRGM, the single human IRG ortholog constitutively and ubiquitously expressed in human cells, plays any role in regulating T cell homeostasis.
Because Irgm1/3 double deficiency does not intrinsically affect T cell expansion or function, we were able to investigate whether the absence of IRG-mediated resistance in epithelial cells in an otherwise immune-competent host would impact the course and duration of a genital C. trachomatis infection. We found that Irgm1/m3(-/-) mice initially failed to effectively clear C. trachomatis during the early course of an infection. This observation can be explained with a nearly complete defect in IFNγ-induced cell-autonomous resistance in cells doubly deficient in Irgm1 and Irgm3 (Fig. 3). At later time points, however, Irgm1/m3(-/-) mice cleared genital C. trachomatis infections rapidly due to an exacerbated C. trachomatis-specific CD4+ T cell response. The observation that Irgm1/m3(-/-) mice developed a more pronounced T cell response towards C. trachomatis than Ifngr1-/- mice did, can be explained by the important role IFNγ plays in stimulating the adaptive immune response, for example, through improved antigen presentation allowing for direct cytolysis by degranulating CD4 T cells [29] and/or enhanced neutrophil recruitment and survival [39], [40], [41], [42]. It has also been reported that IFNγ acts as a positive regulator of CD25 expression on CD4+ T cells in a mouse model of myocarditis, though it is unclear whether IFNγ directly or indirectly controls CD25 expression in CD4+ T cells [43]. The boost in CD25 surface expression on wildtype NR1 cells resident in C. trachomatis-infected Irgm1/m3(-/-) mice compared to C. trachomatis-infected Ifngr1-/- mice may therefore be the net result of high antigen burden and simultaneous IFNγ activation of antigen presenting cells. In addition to an increase in CD25 expression on NR1 cells, we also observed greater accumulation of NR1 cells in C. trachomatis infected Irgm1/m3(-/-) mice compared to C. trachomatis infected wildtype or Ifngr1-/- mice. It seems likely that these two phenotypes are linked: the surface protein CD25 is identical to the alpha chain of the high-affinity, trimeric IL-2 receptor and expression of CD25 has been shown to be upregulated on activated conventional T cells [44], [45]. Although signaling through the IL-2 receptor is not essential for T cell effector function in vivo, IL-2 is known to promote T cell survival and expansion [46], [47], [48], [49]. The increase in CD25 expression in NR1 cells and the resulting boost in IL-2 mediated cell survival may therefore be the underlying cause for the greater expansion of NR1 cells that we observed in C. trachomatis infected Irgm1/m3(-/-) mice. Collectively, these data indicate that the virtual absence of cell-autonomous host resistance during the early course of a C. trachomatis infection triggers a compensatory adaptive immune response in an otherwise fully immune-competent host and ultimately prevents the establishment of a sustained genital infection with C. trachomatis in mice.
These results prompt the question of how C. trachomatis can prevent clearance by the adaptive immune response in its natural host, humans, which lack a robust IRG-dependent immune response that targets C. trachomatis directly. We propose that the answer to this question lies in the adaption of C. trachomatis to a different cell-autonomous IFNγ response – one found in humans where the pathogen has evolved but absent from urogenital epithelial cells in mice [50]. A wealth of in vitro experimental data shows that IFNγ activation of human epithelial cells results in the expression of IDO, which leads to the depletion of intracellular tryptophan stores [10], [51], [52], [53], [54]. In response to tryptophan starvation, C. trachomatis transforms from an active, replicating, state into a quiescent form [55]. Replication of these organisms stops, yet they endure in a quiescent form until immune response wanes and tryptophan becomes available again [4], [5], [55], [56], [57]. The IDO-induced, non-replicating quiescent organisms are less likely to trigger a strong adaptive immune response due to reduced antigenic burden. Additionally, the quiescent bacterium may be more resistant to immune effector mechanisms than its replicating form. We therefore propose that in order to avoid clearance by the adaptive immune response, C. trachomatis has evolved to co-opt the IDO-driven cell-autonomous immune response as an inducer of bacterial quiescence. Accordingly, a humanized mouse model for chronic C. trachomatis infections would require both the removal of the murine IRG response and the recreation of IDO-mediated cell-autonomous immunity in urogenital epithelial cells.
Materials and Methods
Ethics statement
All experiments were approved by the Institutional Animal Care and Use Committee of Harvard Medical School. Harvard maintains an animal care and use program certified by The Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC) and all procedures are conducted in accordance with guidelines established by the American Veterinary Medical Association.
Mice
All mice were maintained and bred under specific pathogen-free conditions. Control C57BL/6J (wildtype) B6.Ifngr1-/- and B6.Ifng-/- mice were obtained from The Jackson Laboratory. The targeted gene deletions of Irgm1 and Irgm3, mice doubly deficient for Irgm1 and Irgm3 and NR1 mice expressing a TCR transgene specific for the C. trachomatis antigen Cta1 have been described previously [27], [28], [58], [59]. The NR1 T cell receptor transgene was crossed onto the Irgm1-/-, Irgm3-/- and Irgm1/m3(-/-) genetic backgrounds.
C. trachomatis strains and measurement of inclusion forming units and bacterial genomes
C. trachomatis serovar L2 434/Bu were propagated in McCoy cells and purified as described [17]. Cells were routinely cultured in high glucose DMEM (Gibco) supplemented with 10% fetal calf serum. For those experiments in which we monitored the antimicrobial effects of IDO-mediated tryptophan depletion, cells were transferred into DMEM F-12 culture medium lacking tryptophan (US Biological) supplemented with 3% fetal calf serum. Limiting the amount of calf serum was necessary to reduce the amount of serum-derived tryptophan to levels at which IFNγ-activated HeLa cells maximally restricted growth of C. trachomatis. Where indicated media was supplemented with tryptophan at a final concentration of 0.05 mg/ml. To quantify the bacterial load in Chlamydia-infected cells and in the uteri of infected animals, a previously described quantitative PCR assay was applied [17]. Briefly, total nucleic acid from infected cells or spleen homogenates was prepared using the QIAamp DNA mini kit from Qiagen. Chlamydia 16S DNA and mouse GAPDH DNA content of individual samples was then quantified by qPCR on an ABI 7000 sequence detection system using primer pairs and dual-labeled probes. Standard curves were generated from known amounts of Chlamydia and mouse DNA, and these curves were used to calculate the mass of Chlamydia DNA per unit mass of mouse DNA in the samples. Alternatively, infected cells in culture were lysed by sonication and combined with their culture supernatants. Serial dilutions of the lysate were applied to McCoy cell monolayers. Inclusions were counted by immunofluorescence microscopy 30 hours post-infection.
Cell culture and C. trachomatis infections ex vivo, and microscopy
Mouse embryonic fibroblasts were generated from the indicated mouse strains as previously described [17]. Cells were treated with 100 U/ml recombinant mouse IFNγ (Chemicon International) over night before infection or left untreated. Cells were infected with C. trachomatis at a multiplicity of infection (MOI) of 2 in SPG buffer (220 mM sucrose, 12.5 mM phosphate, and 4 mM L-glutamic acid (pH 7.5) by centrifugation at 1928 x g for 1 h at 37°C and then returned to standard medium. Chlamydia inclusions were detected in infected cells using mouse anti-Hsc70 antibody (Abcam), followed by anti-mouse secondary fluorescently labeled antibody. Cell nuclei were visualized with 4′,6-diamidino-3-phenylindole (DAPI) staining and epifluorescent images were acquired with a Nikon Eclipse TE2000-U microscope using a Nikon Plan Apo 20x /0.75 N.A. Phase 2 objective. Images were saved as TIFF files and imported into Adobe Illustrator for labeling.
Transfer of C. trachomatis-specific CD4+ T cells, anti-CD4 and anti-GR1 antibody depletion, and intrauterine infection
Before transfer, C. trachomatis-specific CD4+ T cells were isolated from peripheral lymphoid tissues of mice transgenic for the NR1 TCR (Vα2+, Vβ8.3+) and labeled with 5 µM carboxyfluorescein-diacetate-succinimidyl-ester (CFSE) in serum-free medium as described [26]. One day before an intrauterine infection with C. trachomatis recipient CD90.2+ mice were injected i.v. with 106 lymphocytes derived from NR1 transgenic animals of the indicated genetic backgrounds and congenically marked with the surface marker CD90.1+. To infect the genital tract, mice were treated with 2.5 mg of medroxyprogestrone acetate s.c. and one week later 2*106 inclusion-forming units of C. trachomatis L2 were instilled into the uterus using a commercially available non-surgical embryo transfer device (Paratechs). In order to deplete CD4+ T cells, mice were injected intraperitoneally with 0.5 mg anti-CD4+ mAb (GK 1.5) two days prior to infection and with 0.25 mg of the same antibody on days 0, 2, 5, 7, 9 and 12 post-infection. To deplete GR1+ cells, mice were injected with the anti-GR1+ depleting antibody RB6-8C5 and control mice were injected with the isogenic rat IgG2b antibody LTF-2 (BioXCell) following the same injection regiment.
Tissue preparation and flow cytometry
At the indicated times post-infection, lymph nodes and uteri were collected. Uteri were digested with 1 mg/ml type XI collagenase and 50, 000 units/ml DNase for 45 minutes at 37°C. Single-cell suspensions were prepared for staining via mechanical disaggregation. Tissues were mechanically disaggregated and immediately stained for activation markers or stimulated for 5 h with 50 ng/ml PMA and 500 ng/ml ionomycin in the presence of brefeldin A to determine intracellular cytokine staining. Cells were preincubated with anti-FcRγ (Bio X-Cell) before staining with anti-CD4 Pacific Blue (Biolegend), anti-CD90.1 peridinin chlorophyll-a protein (BD Bioscience), and Live/Dead Aqua (Invitrogen). For activation marker analysis, we examined anti-CD62L allophycocyanin-Alexa 750 (Ebioscience), and anti-CD25 allophycocyanin (BD Bioscience). For intracellular staining, the following antibodies were used: anti-IFNγ-PE or -Alexa 700; anti-IL2-PE or -allophycocyanin; and anti-TNFα-PE or -PE-CyChrome 7 (BD Biosciences). Cells were permeabilized with the Cytofix/Cytoperm Plus kit according to the manufacturer's instructions (BD Bioscience). In all samples, an unbiased total of 106 lymphocytes were collected based on forward and side scatter gating. Post-acquisition, lymphocytes were gated based on forward and side scatter, dead cells were excluded, and NR1+ cells were delineated by gating on CD4+, CD90.1+, Vα2+, Vβ8.3+ T cells events. Data were collected on a modified FACSCalibur (Cytek Development) or an LSRII (BD Bioscience) and analyzed using Flow Jo (Tree Star).
Proliferation and cell death assay
Spleens were mechanically disaggregated, red blood cells were lysed and cell suspension was enriched for CD4+ T cells using the Dynal Mouse CD4+ Negative Isolation Kit (Invitrogen) according to the manufacturer's instructions. Isolated cells were CFSE labeled and 105 cells were plated out in wells of a 96-well flat bottom plates coated with anti-CD3 (500 ng/ well) and anti-CD28 (50 ng/ well) antibodies. Three days later cell viability was assessed by flow cytometry with propidium iodide staining.
T cell protection assay
CD4+ T cells were purified from NR1 mice using a mouse CD4+ isolation kit (Dynal; Invitrogen) per the manufacturer's directions. The T cells were cultured in RPMI 1640 (Invitrogen) supplemented with 10% FCS, L-glutamine, HEPES, 50 µM 2-ME, 50 U/ml penicillin, and 50 µg/ml streptomycin. To stimulate the T cells, irradiated feeder splenocytes were pulsed with 5 µM Cta1133–152 peptide and co-cultured with the CD4+-enriched NR1 cells at a stimulator to T cell ratio of 4∶1. To polarize T cells towards Th1, T cells were incubated with 10 ng/ml IL-12 (Peprotech) and 10 µg/ml anti-IL-4 (Biolegend) for 5–7 days. 107 Th1-skewed C. trachomatis-specific CD4+ T cells were transferred into mice, and 24 h later mice were infected in the uterus as described above. Uteri were harvested 6 days after infection. To assess the protective capacity of the skewed cells, uteri from infected mice were homogenized, and DNA was prepared as described above and used for qPCR.
Statistical analysis
All groups were evaluated for statistical significance through the use of unpaired two-tailed t tests. Where it appeared necessary to highlight significant differences between data points, the level of significance is depicted as: *, p<0.05; **, p<0.01; and ***, p<0.005.
Supporting Information
{kind=link}
Zdroje
1. BellandR
OjciusDM
ByrneGI
2004 Chlamydia. Nat Rev Microbiol 2 530 531
2. ResnikoffS
PascoliniD
Etya'aleD
KocurI
PararajasegaramR
2004 Global data on visual impairment in the year 2002. Bull World Health Organ 82 844 851
3. World Health Organization 2003 World Health Report, Shaping the future. World Health Organization Geneva
4. BeattyWL
MorrisonRP
ByrneGI
1994 Persistent chlamydiae: from cell culture to a paradigm for chlamydial pathogenesis. Microbiol Rev 58 686 699
5. MpigaP
RavaoarinoroM
2006 Chlamydia trachomatis persistence: an update. Microbiol Res 161 9 19
6. MonackDM
MuellerA
FalkowS
2004 Persistent bacterial infections: the interface of the pathogen and the host immune system. Nat Rev Microbiol 2 747 765
7. BrownNF
WickhamME
CoombesBK
FinlayBB
2006 Crossing the line: selection and evolution of virulence traits. PLoS Pathog 2 e42
8. ItoJIJr
HarrisonHR
AlexanderER
BillingsLJ
1984 Establishment of genital tract infection in the CF-1 mouse by intravaginal inoculation of a human oculogenital isolate of Chlamydia trachomatis. J Infect Dis 150 577 582
9. PerryLL
SuH
FeilzerK
MesserR
HughesS
1999 Differential sensitivity of distinct Chlamydia trachomatis isolates to IFN-gamma-mediated inhibition. J Immunol 162 3541 3548
10. RoshickC
WoodH
CaldwellHD
McClartyG
2006 Comparison of gamma interferon-mediated antichlamydial defense mechanisms in human and mouse cells. Infect Immun 74 225 238
11. ThomasSM
GarrityLF
BrandtCR
SchobertCS
FengGS
1993 IFN-gamma-mediated antimicrobial response. Indoleamine 2,3-dioxygenase-deficient mutant host cells no longer inhibit intracellular Chlamydia spp. or Toxoplasma growth. J Immunol 150 5529 5534
12. BurianK
EndreszV
DeakJ
KormanyosZ
PalA
2010 Transcriptome analysis indicates an enhanced activation of adaptive and innate immunity by chlamydia-infected murine epithelial cells treated with interferon gamma. J Infect Dis 202 1405 1414
13. DesvignesL
ErnstJD
2009 Interferon-gamma-responsive nonhematopoietic cells regulate the immune response to Mycobacterium tuberculosis. Immunity 31 974 985
14. PengK
MonackDM
2010 Indoleamine 2,3-dioxygenase 1 is a lung-specific innate immune defense mechanism that inhibits growth of Francisella tularensis tryptophan auxotrophs. Infect Immun 78 2723 2733
15. NelsonDE
VirokDP
WoodH
RoshickC
JohnsonRM
2005 Chlamydial IFN-gamma immune evasion is linked to host infection tropism. Proc Natl Acad Sci U S A 102 10658 10663
16. Al-ZeerMA
Al-YounesHM
BraunPR
ZerrahnJ
MeyerTF
2009 IFN-gamma-inducible Irga6 mediates host resistance against Chlamydia trachomatis via autophagy. PLoS One 4 e4588
17. Bernstein-HanleyI
BalsaraZR
UlmerW
CoersJ
StarnbachMN
2006 Genetic analysis of susceptibility to Chlamydia trachomatis in mouse. Genes Immun 7 122 129
18. Bernstein-HanleyI
CoersJ
BalsaraZR
TaylorGA
StarnbachMN
2006 The p47 GTPases Igtp and Irgb10 map to the Chlamydia trachomatis susceptibility locus Ctrq-3 and mediate cellular resistance in mice. Proc Natl Acad Sci U S A 103 14092 14097
19. CoersJ
Bernstein-HanleyI
GrotskyD
ParvanovaI
HowardJC
2008 Chlamydia muridarum evades growth restriction by the IFN-gamma-inducible host resistance factor Irgb10. J Immunol 180 6237 6245
20. MiyairiI
TatireddigariVR
MahdiOS
RoseLA
BellandRJ
2007 The p47 GTPases Iigp2 and Irgb10 regulate innate immunity and inflammation to murine Chlamydia psittaci infection. J Immunol 179 1814 1824
21. CaldwellHD
WoodH
CraneD
BaileyR
JonesRB
2003 Polymorphisms in Chlamydia trachomatis tryptophan synthase genes differentiate between genital and ocular isolates. J Clin Invest 111 1757 1769
22. MacMickingJD
2005 Immune control of phagosomal bacteria by p47 GTPases. Curr Opin Microbiol 8 74 82
23. MartensS
HowardJ
2006 The interferon-inducible GTPases. Annu Rev Cell Dev Biol 22 559 589
24. TaylorGA
2007 IRG proteins: key mediators of interferon-regulated host resistance to intracellular pathogens. Cell Microbiol 9 1099 1107
25. FengCG
ZhengL
JankovicD
BaficaA
CannonsJL
2008 The immunity-related GTPase Irgm1 promotes the expansion of activated CD4+ T cell populations by preventing interferon-gamma-induced cell death. Nat Immunol 9 1279 1287
26. GondekDC
RoanNR
StarnbachMN
2009 T cell responses in the absence of IFN-gamma exacerbate uterine infection with Chlamydia trachomatis. J Immunol 183 1313 1319
27. RoanNR
GierahnTM
HigginsDE
StarnbachMN
2006 Monitoring the T cell response to genital tract infection. Proc Natl Acad Sci U S A 103 12069 12074
28. HenrySC
DaniellXG
BurroughsAR
IndaramM
HowellDN
2009 Balance of Irgm protein activities determines IFN-gamma-induced host defense. J Leukoc Biol 85 877 885
29. JayarapuK
KerrM
OfnerS
JohnsonRM
2010 Chlamydia-specific CD4 T cell clones control Chlamydia muridarum replication in epithelial cells by nitric oxide-dependent and -independent mechanisms. J Immunol 185 6911 6920
30. JoynerJL
DouglasJMJr
FosterM
JudsonFN
2002 Persistence of Chlamydia trachomatis infection detected by polymerase chain reaction in untreated patients. Sex Transm Dis 29 196 200
31. MardhPA
2004 Tubal factor infertility, with special regard to chlamydial salpingitis. Curr Opin Infect Dis 17 49 52
32. IntemannCD
ThyeT
NiemannS
BrowneEN
Amanua ChinbuahM
2009 Autophagy gene variant IRGM -261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog 5 e1000577
33. LapaquetteP
GlasserAL
HuettA
XavierRJ
Darfeuille-MichaudA
2010 Crohn's disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol 12 99 113
34. SinghSB
DavisAS
TaylorGA
DereticV
2006 Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 313 1438 1441
35. SinghSB
OrnatowskiW
VergneI
NaylorJ
DelgadoM
2010 Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat Cell Biol 12 1154 1165
36. HunnJP
Koenen-WaismanS
PapicN
SchroederN
PawlowskiN
2008 Regulatory interactions between IRG resistance GTPases in the cellular response to Toxoplasma gondii. EMBO J 27 2495 2509
37. PapicN
HunnJP
PawlowskiN
ZerrahnJ
HowardJC
2008 Inactive and active states of the interferon-inducible resistance GTPase, Irga6, in vivo. J Biol Chem 283 32143 32151
38. HunnJP
HowardJC
2010 The mouse resistance protein Irgm1 (LRG-47): a regulator or an effector of pathogen defense? PLoS Pathog 6 e1001008
39. BoehmU
KlampT
GrootM
HowardJC
1997 Cellular responses to interferon-gamma. Annu Rev Immunol 15 749 795
40. EllisTN
BeamanBL
2004 Interferon-gamma activation of polymorphonuclear neutrophil function. Immunology 112 2 12
41. MikhakZ
FarsidjaniA
LusterAD
2009 Endotoxin augmented antigen-induced Th1 cell trafficking amplifies airway neutrophilic inflammation. J Immunol 182 7946 7956
42. StrehlB
SeifertU
KrugerE
HeinkS
KuckelkornU
2005 Interferon-gamma, the functional plasticity of the ubiquitin-proteasome system, and MHC class I antigen processing. Immunol Rev 207 19 30
43. AfanasyevaM
GeorgakopoulosD
BelardiDF
BedjaD
FairweatherD
2005 Impaired up-regulation of CD25 on CD4+ T cells in IFN-gamma knockout mice is associated with progression of myocarditis to heart failure. Proc Natl Acad Sci U S A 102 180 185
44. FonteneauJF
LarssonM
BhardwajN
2002 Interactions between dead cells and dendritic cells in the induction of antiviral CTL responses. Curr Opin Immunol 14 471 477
45. LanRY
SelmiC
GershwinME
2008 The regulatory, inflammatory, and T cell programming roles of interleukin-2 (IL-2). J Autoimmun 31 7 12
46. GanusovVV
MilutinovicD
De BoerRJ
2007 IL-2 regulates expansion of CD4+ T cell populations by affecting cell death: insights from modeling CFSE data. J Immunol 179 950 957
47. MalekTR
BayerAL
2004 Tolerance, not immunity, crucially depends on IL-2. Nat Rev Immunol 4 665 674
48. PeiY
ZhuP
DangY
WuJ
YangX
2008 Nuclear export of NF90 to stabilize IL-2 mRNA is mediated by AKT-dependent phosphorylation at Ser647 in response to CD28 costimulation. J Immunol 180 222 229
49. WillerfordDM
ChenJ
FerryJA
DavidsonL
MaA
1995 Interleukin-2 receptor alpha chain regulates the size and content of the peripheral lymphoid compartment. Immunity 3 521 530
50. CoersJ
StarnbachMN
HowardJC
2009 Modeling infectious disease in mice: co-adaptation and the role of host-specific IFNgamma responses. PLoS Pathog 5 e1000333
51. DaubenerW
MacKenzieCR
1999 IFN-gamma activated indoleamine 2,3-dioxygenase activity in human cells is an antiparasitic and an antibacterial effector mechanism. Adv Exp Med Biol 467 517 524
52. DaubenerW
SporsB
HuckeC
AdamR
StinsM
2001 Restriction of Toxoplasma gondii growth in human brain microvascular endothelial cells by activation of indoleamine 2,3-dioxygenase. Infect Immun 69 6527 6531
53. MacKenzieCR
LangenR
TakikawaO
DaubenerW
1999 Inhibition of indoleamine 2,3-dioxygenase in human macrophages inhibits interferon-gamma-induced bacteriostasis but does not abrogate toxoplasmastasis. Eur J Immunol 29 3254 3261
54. SpekkerK
CzeslaM
InceV
HeselerK
SchmidtSK
2009 Indoleamine 2,3-dioxygenase is involved in defense against Neospora caninum in human and bovine cells. Infect Immun 77 4496 4501
55. BeattyWL
BelangerTA
DesaiAA
MorrisonRP
ByrneGI
1994 Tryptophan depletion as a mechanism of gamma interferon-mediated chlamydial persistence. Infect Immun 62 3705 3711
56. KaneCD
VenaRM
OuelletteSP
ByrneGI
1999 Intracellular tryptophan pool sizes may account for differences in gamma interferon-mediated inhibition and persistence of chlamydial growth in polarized and nonpolarized cells. Infect Immun 67 1666 1671
57. RottenbergME
Gigliotti-RothfuchsA
WigzellH
2002 The role of IFN-gamma in the outcome of chlamydial infection. Curr Opin Immunol 14 444 451
58. CollazoCM
YapGS
SempowskiGD
LusbyKC
TessarolloL
2001 Inactivation of LRG-47 and IRG-47 reveals a family of interferon gamma-inducible genes with essential, pathogen-specific roles in resistance to infection. J Exp Med 194 181 188
59. TaylorGA
CollazoCM
YapGS
NguyenK
GregorioTA
2000 Pathogen-specific loss of host resistance in mice lacking the IFN-gamma-inducible gene IGTP. Proc Natl Acad Sci U S A 97 751 755
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- High Affinity Nanobodies against the VSG Are Potent Trypanolytic Agents that Block Endocytosis
- Structural and Mechanistic Studies of Measles Virus Illuminate Paramyxovirus Entry
- Sporangiospore Size Dimorphism Is Linked to Virulence of
- The Binding of Triclosan to SmeT, the Repressor of the Multidrug Efflux Pump SmeDEF, Induces Antibiotic Resistance in
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy