
A Dynamic Landscape for Antibody Binding Modulates Antibody-Mediated Neutralization of West Nile Virus
Neutralizing antibodies are a significant component of the host's protective response against flavivirus infection. Neutralization of flaviviruses occurs when individual virions are engaged by antibodies with a stoichiometry that exceeds a required threshold. From this “multiple-hit” perspective, the neutralizing activity of antibodies is governed by the affinity with which it binds its epitope and the number of times this determinant is displayed on the surface of the virion. In this study, we investigated time-dependent changes in the fate of West Nile virus (WNV) decorated with antibody in solution. Experiments with the well-characterized neutralizing monoclonal antibody (MAb) E16 revealed a significant increase in neutralization activity over time that could not be explained by the kinetics of antibody binding, virion aggregation, or the action of complement. Additional kinetic experiments using the fusion-loop specific MAb E53, which has limited neutralizing activity because it recognizes a relatively inaccessible epitope on mature virions, identified a role of virus “breathing” in regulating neutralization activity. Remarkably, MAb E53 neutralized mature WNV in a time - and temperature-dependent manner. This phenomenon was confirmed in studies with a large panel of MAbs specific for epitopes in each domain of the WNV envelope protein, with sera from recipients of a live attenuated WNV vaccine, and in experiments with dengue virus. Given enough time, significant inhibition of infection was observed even for antibodies with very limited, or no neutralizing activity in standard neutralization assays. Together, our data suggests that the structural dynamics of flaviviruses impacts antibody-mediated neutralization via exposure of otherwise inaccessible epitopes, allowing for antibodies to dock on the virion with a stoichiometry sufficient for neutralization.
Published in the journal:
. PLoS Pathog 7(6): e32767. doi:10.1371/journal.ppat.1002111
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002111
Summary
Neutralizing antibodies are a significant component of the host's protective response against flavivirus infection. Neutralization of flaviviruses occurs when individual virions are engaged by antibodies with a stoichiometry that exceeds a required threshold. From this “multiple-hit” perspective, the neutralizing activity of antibodies is governed by the affinity with which it binds its epitope and the number of times this determinant is displayed on the surface of the virion. In this study, we investigated time-dependent changes in the fate of West Nile virus (WNV) decorated with antibody in solution. Experiments with the well-characterized neutralizing monoclonal antibody (MAb) E16 revealed a significant increase in neutralization activity over time that could not be explained by the kinetics of antibody binding, virion aggregation, or the action of complement. Additional kinetic experiments using the fusion-loop specific MAb E53, which has limited neutralizing activity because it recognizes a relatively inaccessible epitope on mature virions, identified a role of virus “breathing” in regulating neutralization activity. Remarkably, MAb E53 neutralized mature WNV in a time - and temperature-dependent manner. This phenomenon was confirmed in studies with a large panel of MAbs specific for epitopes in each domain of the WNV envelope protein, with sera from recipients of a live attenuated WNV vaccine, and in experiments with dengue virus. Given enough time, significant inhibition of infection was observed even for antibodies with very limited, or no neutralizing activity in standard neutralization assays. Together, our data suggests that the structural dynamics of flaviviruses impacts antibody-mediated neutralization via exposure of otherwise inaccessible epitopes, allowing for antibodies to dock on the virion with a stoichiometry sufficient for neutralization.
Introduction
Flaviviruses are a group of ∼70 RNA viruses that cause morbidity and mortality on a global scale, with greater than 100 million human infections annually [1]. Viruses within this genus of medical concern include yellow fever virus, tick-borne encephalitis virus, Japanese encephalitis virus, dengue virus (DENV) and West Nile virus (WNV). WNV is a mosquito-borne flavivirus maintained in nature in an enzootic cycle with birds. WNV infections of humans result in a spectrum of clinical symptoms depending, in part, on the age and immune status of the individual. While most infections are sub-clinical, symptomatic cases range from self-limiting fever to acute flaccid paralysis and encephalitis [2]. Since its introduction into North America in 1999, as many as three million people have been infected by WNV [3], with ∼1000 severe infections occurring in the United States annually (www.cdc.gov). To date, there are no WNV-specific treatments or vaccines licensed for use in humans.
Flaviviruses are small spherical virions that encapsidate an ∼11 kb genomic RNA of positive-sense polarity [1]. This RNA is translated as a single polyprotein that is processed by viral and host cell proteases into ten functionally distinct proteins. Flaviviruses encode three structural proteins that comprise the virus particle and seven non-structural proteins that function to process the viral polyprotein, replicate the viral genome, and antagonize the host's protective response to infection [1], [4], [5], [6], [7], [8], [9], [10], [11]. Flaviviruses bud into the endoplasmic reticulum as immature viruses that incorporate 60 heterotrimeric spikes of the envelope (E) and precursor to membrane (prM) proteins [12], [13]. Maturation of virions during egress from the cell is associated with a pH-dependent change in the arrangement and oligomeric state of the E protein and cleavage of prM by a host cell furin-like serine protease [14], [15], [16]. While prM cleavage is a required step in the formation of an infectious virion [17], several lines of evidence suggest that a significant fraction of infectious virions retain some uncleaved prM [18], [19], [20], [21], [22]. The E protein is a class II fusion protein composed of three structurally distinct domains (domains I-III; E-DI-III) connected to the viral membrane by a helical stem region [23]. On mature virions, 180 copies of the E protein are arranged as anti-parallel dimers in an unusual herringbone T = 3 pseudo-icosahedral array [24], [25], [26]. In this configuration, E proteins are not quasi-equivalent, but exist in three distinct chemical environments defined by their proximity to the two-, three-, or five-fold symmetry axis. This arrangement adds complexity to the antigenic surface of virus particles as antibodies may not be able to bind all 180 E proteins due to steric constraints on antibody binding or occlusion of the epitope itself [27], [28].
Humoral immunity is a critical aspect of protection against flavivirus infection (reviewed in [29]). Eliciting a protective antibody response is a primary goal in the development of safe and effective flavivirus vaccines [30]. The majority of neutralizing antibodies against flaviviruses are directed against the E protein; neutralizing monoclonal antibodies (MAbs) have been identified that bind to all three E protein domains (reviewed in [29], [31]). While antibodies that bind the prM protein have been detected in humans, they possess limited neutralizing activity [32], [33], [34], [35], [36].
Antibody-mediated neutralization of WNV is a “multiple-hit” phenomenon that occurs when virions are engaged by antibody with a stoichiometry that exceeds a required threshold [29], [37]. Our estimate of this threshold is ∼30 antibody molecules per virion [38]. Two factors principally govern the neutralizing activity of an antibody. Antibody affinity (or avidity) controls the fraction of accessible epitopes on the intact virion occupied at a particular concentration of antibody. From a vaccination standpoint, eliciting antibodies that bind viral antigens with high affinity is desirable because they can reach the stoichiometric threshold required for neutralization at lower concentrations. However, high-affinity interactions do not always translate into significant functional potency. Because of steric constraints arising from the dense icosahedral arrangement of E proteins on the mature virion, not all epitopes are displayed on intact virions equivalently. Epitope accessibility is a second independent parameter that defines the circumstances that allow for neutralization, and differs markedly between antibodies that recognize structurally distinct epitopes [21], [28], [38], [39], [40]. In fact, many epitopes, including those recognized by antibodies commonly elicited in vivo, are not displayed on the surface of the mature virion enough times to allow for neutralization even when fully occupied [21], [38], [41].
An important concept of the “multiple-hit” model of neutralization is that infectious virions may be decorated with non-neutralizing quantities of antibody. Evidence in support of this includes increases in the neutralizing activity of antibodies observed in the presence of anti-IgG secondary antibodies or complement [37], [42], [43], and the phenomenon of antibody-dependent enhancement (ADE) of infection [44]. Because the rate of antibody-virion interactions is orders of magnitude faster than the rate of virus binding to cells, viruses are likely engaged by antibody for significant periods prior to productive interactions with target cells. Here, we investigated the fate of virions decorated by antibody with a stoichiometry that does not exceed the threshold required for neutralization. Our data revealed time - and temperature-dependent increases in neutralization of WNV that would not be predicted for static virions bound by antibodies under steady-state conditions. Because both viral proteins and intact virions are dynamic [45], [46], [47], [48], the increased neutralization can be explained by changes in epitope accessibility occurring as virions sample different states of a dynamic ensemble of conformations. That kinetic aspects of neutralization were observed for every WNV - and DENV-reactive MAb examined, as well as WNV-polyclonal immune sera, suggests a widespread impact of the dynamic motion of virions on epitope accessibility and antibody-mediated neutralization.
Results
Antibody modulates the infectious half-life of WNV in solution
To investigate whether prolonged exposure of virions to “non-neutralizing” quantities of antibody has functional consequences, we performed a series of kinetic experiments with the WNV E-DIII-lateral ridge specific MAb E16 at a concentration sufficient to neutralize the infectivity of half the virus particles (the EC50). At this concentration, half the virions in a population are bound by antibody with a stoichiometry that exceeds the neutralization threshold, whereas the other half are bound by fewer antibody molecules. WNV reporter virus particles (RVPs) encoding a GFP reporter gene were incubated with 10 ng/ml E16 for one hour at room temperature to allow for steady-state binding (Figure S1). After the room temperature incubation, RVP-antibody complexes were shifted to 37°C for the indicated periods prior to the addition of target Raji-DC-SIGNR cells. The infectivity of RVPs in the presence and absence of antibody was determined by flow cytometric analysis, and is expressed relative to the level of infectivity observed when cells were added to viral immune complexes immediately after the initial room temperature incubation (Figure 1a). Using this approach, we observed an antibody-independent decrease in RVP infectivity with prolonged incubation at 37°C (intrinsic decay), consistent with our previously published findings [49]. Incubation in the presence of E16 resulted in a 4.7-fold reduction in the apparent half-life of WNV RVPs relative to the intrinsic rate of decay observed in the absence of antibody (n = 23; p<0.0001) (Figure 1b). A similar 5.6-fold change in the rate with which virions lost infectivity in the presence of E16 was observed using fully-infectious WNV (Figure 1c). Incubation of RVPs or virus with high concentrations of the DENV-specific MAb 3H5 did not impact WNV infectivity, indicating a requirement for virus-reactive antibody (Figure 1b and c). These results were not explained by increased binding of virions to tissue-culture plastic during prolonged incubation with antibody (p = .32, n = 5; Figure S2a), the action of complement (p = .17, n = 5; Figure S2b), or antibody-mediated aggregation (E16 Fab experiments discussed below; Figure 2b). Additionally, the rapid reduction in RVP infectivity in the presence of virus-specific antibody was still observed in the presence of a broad spectrum of protease inhibitors (p = .74, n = 2; Figure S2c), suggesting that the loss of infectivity cannot be explained by physical destruction of RVPs due to contaminating proteases.


A kinetic aspect of neutralization by the WNV-specific MAb E16
We next evaluated the impact of kinetics on neutralization by E16 in a series of dose-response studies. WNV RVPs were incubated with serial four-fold dilutions of E16 at room temperature for one hour, followed by a shift to 37°C for the indicated times prior to the addition of target cells (Figure 2a). The dose-response profile of E16 obtained when cells were added immediately after the room temperature incubation required to achieve steady-state binding revealed the expected relatively steep sigmoidal curve (Figure 2a; reference curve) [38]. Dose-response curves obtained from samples incubated for varying lengths of time at 37°C prior to the addition of cells differed from the reference curve in several respects. First, the percentage of cells infected in the presence of low concentrations of antibody (the top of the sigmoidal dose-response curve) decreased as a function of increased incubation at 37°C, corresponding to the intrinsic decay of virus particles observed in the kinetic studies described above (Figure 1, Figure 2a; left panel). Second, increasing the length of incubation at 37°C shifted the EC50 to lower concentrations of antibody (Figure 2a; right panel (n = 19, p<0.0001)). Analysis of changes in infectivity over time revealed a 5.8-fold decrease in the apparent half-life at ∼10 ng/ml E16 relative to the antibody-independent intrinsic decay for RVPs alone (n = 18 independent dose-response experiments, p<0.0001), similar to the results obtained using the single antibody-concentration studies described in Figure 1. Time-dependent changes in dose-response curves were also observed with Fab fragments of E16 (∼3.8-fold decrease in EC50 after 24 hr incubation, n = 2) (Figure 2b), indicating the phenomenon does not reflect antibody-mediated virus aggregation.
Kinetic aspects of the antibody dependent enhancement (ADE) profile of MAb E16
ADE describes the dramatic increase in infection of Fcγ-receptor-expressing cells in the presence of non - or weakly-neutralizing quantities of antibody and has been linked to severe clinical outcomes following secondary DENV infections of humans (reviewed in [50]). Antibody-mediated neutralization and ADE are two phenomena related by the number of antibodies bound to the virion [38], [51], therefore kinetic changes in the neutralization activity of an antibody should have a corresponding impact on the ADE profile as well. To explore this, kinetic dose-response curves were set up as described above, except using FcγIIb-expressing K652 target cells. Infection of K562 cells occurs through Fcγ-receptor-mediated uptake of antibody-virus immune complexes; in the absence of antibody, these cells are refractory to infection due to an inability to efficiently bind WNV. When immune complexes were added to cells immediately following the room temperature incubation required to achieve steady-state binding (Figure 3; reference curve), the expected bell shaped dose-response profile was observed. Incubation of antibody-virion complexes at 37°C prior to the addition of cells resulted in a marked change in the shape of the ADE profile. A reduction in the magnitude of ADE was observed over time, consistent with a decrease in the number of infectious virions in solution due to intrinsic decay (Figure 3; left panel). Notably, prolonged incubation resulted in a reduction in the concentration of E16 at which maximal ADE was observed (Figure 3; right panel), corresponding to the reduction in the EC50 observed above (Figure 2a; right panel).

Because the kinetic neutralization and enhancement experiments presented above were designed to allow for steady-state binding of the virion by antibody prior to incubation at 37°C (validated in Figure S1), changes in virus infectivity over time should not reflect continued engagement of individual virions by increasing numbers of antibody molecules. However, an important assumption of this model is that the number of epitopes on the virion does not change.
Dynamic motion changes epitope accessibility in a time - and temperature-dependent manner
To explore the role of dynamic motion of the virion in WNV neutralization, we took advantage of the fact that the neutralizing activity of several classes of antibodies is significantly limited by epitope accessibility [21], [39], [40]. MAb E53 is a high affinity DII-fusion loop-reactive antibody that has a limited capacity to neutralize mature virions because its epitope is buried on the surface of the virus particle. Even in the presence of saturating concentrations of antibody, E53 does not bind mature virions enough times to exceed the stoichiometric requirements for neutralization [21]. In support, structural studies revealed that E53 Fab fragments were only capable of binding E proteins of the heterotrimeric spikes present on immature virions [52]. For E53 to engage a mature virion with a stoichiometry that exceeds the neutralization threshold, changes in the accessibility of its otherwise cryptic epitopes would be required.
In agreement with our previous findings [21], MAb E53 failed to significantly neutralize a homogeneous population of mature WNV RVPs when antibody-virion complexes were added to cells following a one hour incubation at room temperature (Figure 4a and b, reference curves). Incubation at 37°C for increasing time intervals prior to infection revealed a gradual increase in antibody potency, with more than half the virions incubated at 37°C for 26 hours becoming susceptible to neutralization (Figure 4a). Because the E53 epitope is not accessible on the highly ordered surface of the mature virion [52], the increase in potency requires changes in exposure of the fusion loop epitope over time as the E proteins on the surface of the virion sample different conformations at equilibrium. Binding of E53 has the potential to stabilize the epitope in an exposed conformation that occurs only transiently in the absence of antibody. That significant neutralization requires considerable time at 37°C likely reflects the relatively large number of epitopes that must become accessible on the average virion in order to exceed the neutralization threshold (Figure S3). If increased neutralization by E53 reflects dynamic movement of the virion, one would predict it would be both time - and temperature-dependent. To test this, we performed additional experiments with longer incubation times and a range of temperatures. Increasing the temperature to 40°C enhanced neutralization as compared to 37°C (Figure 4b, compare green to orange curves), whereas decreasing the temperature to 33°C reduced neutralization capacity (Figure 4b, compare green to blue curves). Similar results were observed for the E-DI specific MAb E121 shown previously to bind an epitope that is poorly accessible on the mature virion (Figure 4c) [21], [39].

Viral dynamics broadly impact antibody-mediated neutralization of flaviviruses
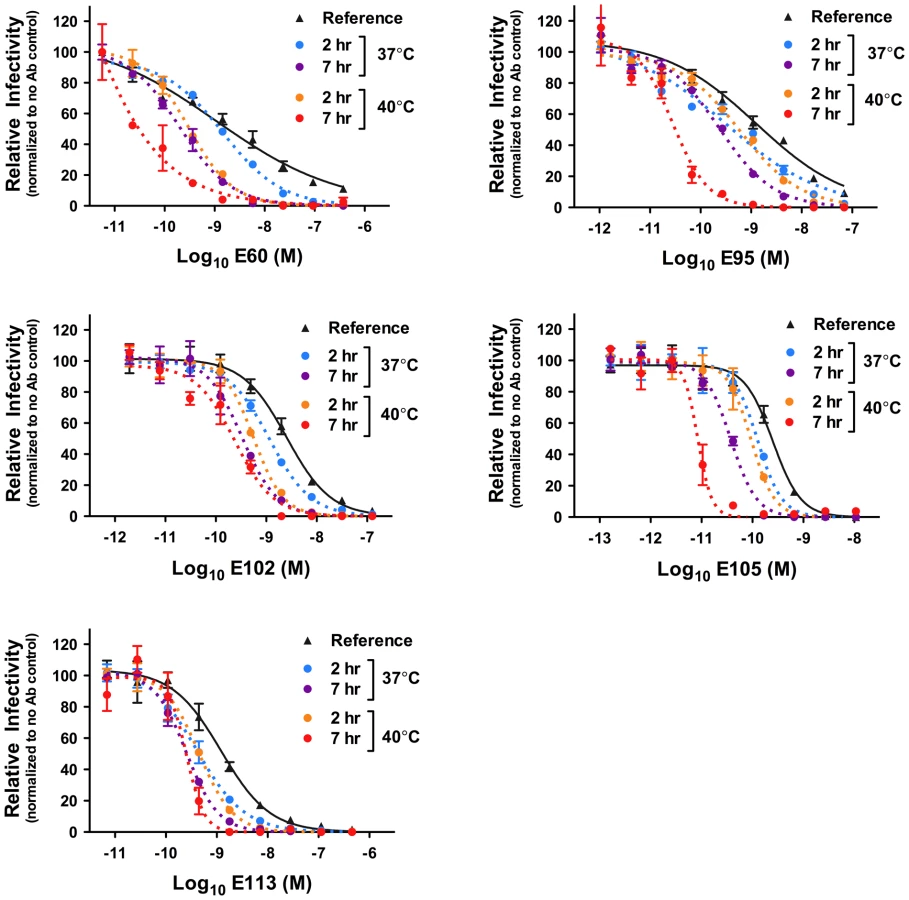
To investigate whether kinetic effects on antibody-mediated neutralization are a common phenomenon, we performed additional studies using a panel of 12 WNV MAbs. In each case, we observed time - and temperature-dependent increases in neutralization, similar to our results with E16, E53, and E121 (Figure 5). Collectively, the 15 WNV MAbs used in the current study bind epitopes distributed across all three domains of the E protein and are characterized by varying degrees of functional potency [39], [53]. We next performed experiments using previously characterized immune sera from five recipients of a phase I clinical trial of a live attenuated WNV vaccine [21] (A. Durbin, S. Whitehead, and colleagues, unpublished data). After a one hour room temperature incubation to allow for steady-state binding, virus-sera mixtures either were used to immediately infect cells (Figure 6, reference curves) or incubated an additional 23 hours at 37°C or 40°C before infection (Figure 6, blue and orange curves, respectively). We found that the polyclonal mixtures of antibody present in immune sera behaved in a manner similar to MAb of defined specificity.


To extend our observations to another flavivirus, we performed analogous studies with DENV serotype 1 (DENV-1) RVPs and a panel of previously characterized DENV-1 E protein-reactive MAbs [53], [54]. After incubating RVPs and antibody at 37°C for one hour to allow steady-state binding to occur, RVP-antibody complexes were either immediately used to infect cells (Figure 7, reference curves) or further incubated for two or seven additional hours at 37°C and 40°C before infection (Figure 7, dotted curves). Because it was established previously that some antibodies do not bind DENV efficiently at low temperatures [46], incubation at 37°C was used to establish steady-state binding (Figure S1). As observed in our studies with WNV, time - and temperature-dependent increases in neutralization potency were observed (Figure 7). Interestingly, shorter incubation times were necessary as compared to experiments with WNV, due to a more rapid intrinsic decay rate of DENV as compared to WNV, which has previously been reported [49]. This was also demonstrated in parallel studies of WNV and DENV-1 RVPs with the cross-reactive MAb E60; significant increases in neutralization were observed more rapidly with DENV than WNV (Figure S4). Altogether, these results suggest that DENV exists in a more dynamic state than WNV.
Discussion
The existence of high-resolution structures of the E proteins of flaviviruses, and their arrangement on the surface of the virion at different stages of the viral lifecycle, have been a powerful tool for studying how these viruses interact with the host (reviewed in [6], [26]). Interpretation of these structures in the context of biological systems is complicated by the fact that they represent the average state of what is likely a very dynamic ensemble of conformations sampled by virions at equilibrium. The structural dynamics of non-enveloped viruses have been studied extensively (reviewed in [45], [55]). Limited proteolysis studies of the Flock house virus strongly suggest the capsid proteins “breathe”, allowing proteases access to internal structures predicted to be inaccessible on an intact and non-dynamic virion [47]. Furthermore, drugs that prevent the dynamic movement of the picornavirus human rhinovirus 14 have been shown to effectively inhibit infectivity [48]. While the dynamics of enveloped viruses have been studied less extensively, examples of temperature - and time-dependent antibody reactivity have been described for several viruses [46], [56], [57]. The DENV group-reactive neutralizing MAb 1A1D-2 binds an epitope on the A-strand of E-DIII that is poorly accessible on proteins proximal to each of the three symmetry axes of the mature virion. Binding of this MAb is temperature-dependent (does not occur at 4°C) and appears to trap E proteins on the virion surface in a conformation distinct from the herringbone icosahedral arrangement of the mature virion [46].
Overall, prior studies implicating a kinetic component of antibody-mediated neutralization have focused on either a single MAb (DENV MAb 1A1D-2, influenza MAb Y8-10C2) or a panel of MAbs that recognizes a similar epitope (MAbs specific for the membrane-proximal external region (MPER) of HIV gp41), with the implication that such antibodies were atypical in their ability to bind buried or inaccessible epitopes [57], [58], [59]. In this study we provide functional evidence identifying the widespread impact of the dynamic movement of flaviviruses on neutralization by all antibodies that bind the E protein; a kinetic aspect of neutralization appears to be the rule rather than the exception.
Our data suggest that the structural dynamics of virions has the potential to modulate the potency of all antibodies that bind E proteins arrayed on the surface of the virion through changes in epitope accessibility. This is illustrated most dramatically by antibodies that bind poorly accessible determinants on the mature virion, such as the WNV DII-fusion loop-reactive MAb E53. Neutralization of mature WNV by E53 is restricted by the number of times the antibody can bind the virion. E53 binds the E protein only when associated with prM as heterotrimeric spikes that project off the surface of the immature virus particle [21], [52]. While homogeneous populations of mature WNV are not efficiently neutralized by E53 when assayed using conventional approaches, increasing the time the virus is incubated with antibody resulted in a marked increase in neutralization activity (>100-fold) (Figure 4a and b). Because the virus cannot revert to an immature configuration once prM cleavage occurs and the virion is released from the cell [15], this dramatic increase in neutralization can only be explained by exposure of the DII-fusion loop epitope through dynamic motion of viral E proteins.
The neutralization activity of every monoclonal (n = 20) and polyclonal (n = 5) antibody assayed in this study was enhanced by increasing the time the virion was exposed to antibody prior to the addition of target cells, including antibodies previously demonstrated to be non - or weakly-neutralizing using standard assays. This reflects the fact that as the number of accessible epitopes on the individual virion increases as a consequence of dynamic motion, the fraction of them that must be bound in order to exceed the stoichiometric threshold (percent occupancy) is reduced; neutralization can then occur at lower concentrations of antibody. However, the magnitude of this kinetic effect was not uniform among antibodies localizing to different epitopes. This may reflect differences in the number of times an antibody binds the average state of the virion relative to the threshold number of antibodies required for neutralization, as well as the rate at which additional epitopes are made available for binding (Figure S3). In contrast to the cryptic nature of the DII-fusion loop epitope recognized by E53, the potently neutralizing MAb E16 is specific for a relatively accessible determinant displayed on the lateral ridge of DIII [53]. Cryo-electron microscopy studies indicate this antibody binds 120 out of 180 E proteins incorporated into mature virions; the remaining 60 E proteins proximal to the five-fold symmetry axis of the virion cannot be bound due to steric conflicts among the tightly clustered DIII epitopes [27], [28]. An increase in the accessibility of these additional epitopes on the virion through dynamic motion would translate into a modest reduction in the occupancy requirements for neutralization by E16, in agreement with the 4.0-fold increase in antibody potency (n = 11, range 2.3–6.8) observed in our studies after ∼24 hours incubation. A similar 3.8-fold increase after ∼24 hours was observed when E16 Fab fragments were used (n = 2, range 3.2–4.5).
The dynamic motion of virions has the potential to increase antibody potency by providing access to otherwise cryptic antibody-binding determinants. Of note, mapping studies suggest that many epitopes on the mature virion are poorly accessible for antibody binding [38], [39], [40], [46]. Antibodies do not induce viral breathing, but rather stabilize conformations of the E protein that exist as part of the ensemble of conformations sampled by the virion at equilibrium. The longer the virion remains exposed to antibody, the more opportunities exist for engagement of an otherwise inaccessible epitope, allowing for time-dependent increases in the stoichiometry with which antibodies decorate virions. If changes in epitope accessibility are the underlying mechanism of the kinetic aspects of neutralization, there should be a limit to the increase in potency observed over time. Eventually, dynamic virion structures should expose all potential epitopes, and these will become fixed in place by antibody binding, yielding a neutralization profile determined by the relationship between antibody occupancy and the stoichiometric threshold. In support of this, increases in neutralization and changes in the ADE curves for E16 no longer occurred when incubations longer than 24 hours were performed (Figure S5).
In addition to exposing more epitopes for antibody binding, time-dependent changes in the antigenic surface of the virus particle may also allow engagement of the virion with increased affinity, via bivalent interactions among E proteins in conformations not present in the average state, as well as cooperative effects during antibody binding. That the kinetic impact on neutralization by E16 was observed using both intact antibodies and Fab fragments incapable of cross-linking virions indicates that increases in antibody potency do not reflect antibody-mediated aggregation among virions. Importantly, all of the experiments included in our study were performed using conditions of antibody excess, and yielded results that were independent of the concentration of virus in the assay. In contrast, the aggregation of antigens by antibodies is dependent on the antibody-antigen ratio.
Our results suggest that changes in the antibody-mediated neutralization of DENV occur more rapidly than with WNV. One interpretation of this result is that DENV virions are more dynamic than those of WNV, allowing more rapid access to otherwise inaccessible determinants. In the absence of antibody, preparations of DENV become less infectious at a faster rate than observed with WNV, consistent with prior studies [49]. Additionally, kinetic changes in neutralization with the cross-reactive MAb E60 occur at a faster rate with DENV-1 than WNV RVPs when compared in parallel studies (Figure S4). While we do not yet understand, in molecular terms, why viruses lose infectivity over time, one possibility is that the intrinsic decay of flaviviruses is a consequence of structural dynamics. Viruses sampling multiple conformations in dynamic equilibrium may not always return to the average state because moving backwards may no longer be the most energetically favorable path. Additional evidence of time-dependent structural changes to the virus population is demonstrated by differences in the intrinsic decay rate of WNV observed when different cell types are used to measure infectivity. The rate of decay of viruses was ∼2.7-fold more rapid when assayed on Raji-DC-SIGNR cells as compared to a K562 cell line expressing the same attachment protein (n = 6, p<0.0001). Thus, the observed intrinsic decay cannot be attributed solely to the physical destruction of the virus, and suggests the additional possibility that not all conformations in a heterogeneous ensemble of virions are equally infectious on different cell types. Of interest, E proteins on individual virions in conformations that may no longer contribute functionally to fusion may also stably expose a different array of epitopes.
Our data suggest that the circumstances of antibody-virion interactions may significantly impact the fate of the virion immune complex. Standard in vitro neutralization assays for flaviviruses generally include a short pre-incubation (∼1 hr) of antibody and virus prior to infection of target cells; this incubation presumably allows the binding reaction between antibody and cognate epitope to reach steady-state. However, depending on the extent to which a virion is structurally dynamic (which controls the rate at which epitopes may become transiently accessible), the target cell type, and the volume of infection in vitro, this presumption may be inaccurate. Because increases in the neutralization activity of DENV-reactive antibodies that bind dynamically exposed epitopes occur rapidly (within two hours) (Figure 7), the interaction of DENV with antibodies may never truly reach steady-state. From this perspective, the length of time antibody is incubated with DENV is a variable that cannot be ignored. While antibody-mediated neutralization activity measured in vitro using standard plaque reduction neutralization tests (PRNT) generally correlates with protection in vivo [39], [53], [54], [60], [61], [62], this is an imperfect relationship. Antibodies with limited neutralization activity have been shown to protect in animal models of flavivirus infection [39], [54], [62]. While this may reflect the direct contributions of effector functions of antibodies in vivo, it is also possible that existing assays of the functional properties of antibodies have limitations. Considering the contribution of the structural dynamics of the virion when designing neutralization studies merits a systematic evaluation.
The impact of the dynamic exposure of viral epitopes in vivo remains uncertain. Virtually nothing is known about the relevant concentrations and volumes that govern antibody-virion interactions in the tissues where many of the key events in the pathogenesis of these viruses occur. Kinetic changes in neutralization occur gradually over time as dynamic motion provides new opportunities for engagement of virions with a stoichiometry sufficient for neutralization (Figure S3). Thus the rate of virus entry in vivo is also an important, yet unknown, parameter that defines the extent to which this phenomenon will contribute to protection of the host. Of interest, the kinetics of WNV binding to target cells in vitro occurred rather slowly (with maximal binding requiring ∼3 hours) even in the presence of the high affinity attachment factor DC-SIGNR (Figure S6). As DENV appears to be extremely dynamic, with kinetic effects on neutralization observed almost immediately, the impact of viral breathing on neutralization in vivo cannot be discounted. Because the kinetics of neutralization are increased by an elevated temperature, it is interesting to speculate that certain classes of antibodies, such as those recognizing the fusion loop epitope commonly observed in infected individuals, may function better than previously anticipated in the context of the febrile response. Resolving this question awaits the development of approaches to quantitatively and directly measure antibody-mediated neutralization in vivo.
Neither proteins, nor intact virions, are static structures. Our findings are consistent with a model in which the dynamic motion of flaviviruses provides an opportunity for antibodies to engage virions at otherwise inaccessible epitopes to reach a stoichiometry sufficient for neutralization. Given time, all of the E protein-reactive antibodies investigated were able to block virus infection, even those described originally as non-neutralizing using conventional assays [39]. These results add to the complexity of our understanding of the functional properties of antibodies and suggest new avenues of investigation and analysis into the widespread and unappreciated impact of the dynamic motion of virions as moving targets for antibody recognition.
Materials and Methods
Cell lines
Cell lines were maintained at 37°C in the presence of 7% CO2. HEK-293T cells were grown in complete Dulbecco's modified Eagle medium (DMEM) containing Glutamax (Invitrogen, Carlsbad, CA) and supplemented with 10% fetal bovine serum (FBS; (HyClone, Logan, UT)) and 100 U/ml penicillin-streptomycin (PS; (Invitrogen, Carlsbad, CA)). K562 and Raji cell lines were maintained in RPMI-1640 medium containing Glutamax (Invitrogen, Carlsbad, CA) and supplemented with 10% FBS and 100 U/ml PS.
WNV and DENV immune sera
Neutralization studies were performed using sera obtained from recipients of a candidate WNV vaccine. Sera from five participants of a Phase I double-blinded, placebo-controlled study designed to evaluate the safety and immunogenicity of a single dose of live attenuated WNV/DENV-4 vaccine were obtained and characterized previously [21]. The clinical study was conducted at the Center for Immunization at the Johns Hopkins Bloomberg School of Public Health under an investigational new drug application reviewed by the United States Food and Drug Administration.
Production of WNV and DENV RVPs
Reporter virus particles (RVPs) are pseudo-infectious virions produced by genetic complementation of a WNV sub-genomic replicon with the structural genes in trans. Flavivirus replicons do not encode intact genes for the three structural proteins of the virus, thus RVPs that encapsidate these sub-genomic RNAs are capable of only a single round of infection. WNV and DENV RVPs have been used extensively to characterize the functional properties of anti-flavivirus antibodies [21], [38], [39], [42], [63], [64].
RVPs were produced by transfection of HEK-293T cells with DNA plasmids encoding the structural genes and a sub-genomic WNV replicon as described previously [38], [65]. Standard preparations of WNV RVPs were produced using plasmids encoding a GFP-expressing replicon, WNV C-prM-E, and pcDNA3.1 with a 1∶3∶0.5 ratio by mass. Because standard preparations of WNV RVPs retain detectable amounts of uncleaved prM, homogeneous populations of mature WNV RVPs were produced using the plasmids above except that a plasmid encoding human furin was used in place of pcDNA3.1 to promote efficient cleavage of prM [21]. Over-expression of furin in this context has been shown to reduce the amount of uncleaved prM protein in populations of RVPs to levels that are no longer detectable by Western blot [21], [66]. Standard RVPs composed of the structural genes of the DENV Western Pacific strain (serotype I: genotype IV) were produced as described above by substituting a plasmid encoding DENV structural genes in place of the WNV construct [49]. All transfections were performed using Lipofectamine LTX (Invitrogen, Carlsbad, CA) in accordance with the manufacturer's instructions. RVPs were harvested at 48 h post-transfection, filtered through a 0.22 µM filter, and frozen in aliquots at −80°C.
Determining the infectious titer of RVPs
Because the relationship between infected cells and virus dose is typically not linear for flaviviruses, the titers of all RVP stocks were measured by serial dilution. Serial two-fold dilutions of RVP-containing supernatants were used to infect Raji cells that express the attachment factor DC-SIGNR. Infection was measured 48 h post-infection by flow cytometry. Only data obtained from the linear portion of the resulting virus dose-response curves was used for analysis.
Measurements of kinetic aspects of antibody-mediated neutralization and ADE
Single concentration studies
WNV RVP stocks were diluted in the presence or absence of 10 ng/ml MAb E16 or 200 ng/ml MAb 3H5 in a total volume of ∼7 ml and pre-incubated for one hour at room temperature to allow the binding reaction to reach equilibrium. The virus mixtures were further incubated at 37°C for up to four days, during which 450 µl samples were removed at indicated times and frozen for analysis. Two-fold dilutions of RVPs from each sample were used to infect Raji-DC-SIGNR cells at 37°C (200 µl total volume). Infectivity was determined 48 h post-infection by flow cytometry. Using values corresponding to the linear portion of the curve, infectivity was normalized to levels obtained prior to incubation at 37°C (but after the initial room temperature incubation) and fitted to a single-phase exponential decay to obtain the half-life (GraphPad Prism; GraphPad Software, La Jolla, CA).
Dose-response studies
To assess the kinetic aspects of neutralization over a range of antibody concentrations, WNV RVP stocks were diluted into the linear range of infectivity and incubated with serial dilutions of the indicated MAbs, Fab fragments, or polyclonal sera. To ensure our experiments were performed under conditions of antibody excess, antibody dose-response studies were performed using multiple dilutions of RVPs. These studies confirmed that the concentration of RVPs does not affect neutralization potency (EC50), as predicted by the assumptions of the law of mass action and the percentage law [65], [67]. Antibody-virus complexes were initially incubated for one hour at room temperature to allow the binding reaction to reach equilibrium, and then further incubated at 33°C, 37°C, or 40°C for incremental lengths of time, at which point the infectivity of virus-antibody complexes was determined by infecting Raji-DC-SIGNR target cells. Infections were carried out at 37°C (300 µl total volume) and infectivity was monitored by flow cytometry 48 h post-infection. In addition to the half-life data at each concentration of MAb or dilution of sera (calculated as described above), dose-response curves at each time point were analyzed by non-linear regression (variable slope) to predict the EC50 value using GraphPad Prism (GraphPad Software, La Jolla, CA). In a subset of experiments, K562 cells that express the activating human FcγIIb-receptor CD32a were used instead of Raji-DC-SIGNR cells in order to assess kinetic changes in ADE profiles. Neutralization studies with DENV-1 RVPs were performed analogous to WNV-Raji-DC-SIGNR infections, except the initial one hour incubation allowing for equilibrium was performed at 37°C instead of at room temperature.
Quantitative real-time PCR
The genomic RNA content of RVP populations was measured using a modification of a previously described protocol [68]. Briefly, RVP containing supernatants were treated with 100 U recombinant DNase I, followed by RNA isolation using the QiaAmp Viral RNA Mini kit per the manufacturer's instructions (Qiagen, Valencia, CA). Amplification of viral genomic RNA was accomplished using the Superscript III One-Step RT-PCR system (Invitrogen, Carlsbad, CA) and primers specific for the 3′ untranslated region of the WNV lineage II replicon [69].
Virus binding studies
Standard preparations of WNV RVPs were incubated with Raji-DC-SIGNR cells at 37°C in the absence or presence of the MAb E16 for incremental lengths of time to allow virus-cell binding to occur. Target cells were incubated in the presence of 20 mM NH4Cl to prevent virus fusion and genomic RNA replication. At the indicated times, cells were washed extensively with media and lysed. Total RNA was isolated using the QIAshredder and RNeasy Mini kits in accordance with the manufacturer's instructions (Qiagen, Valencia, CA). The relative amount of bound WNV RNA was enumerated by quantitative real-time PCR using a DNA molecular clone plasmid as a standard [49], [68]. The resulting kinetic data was fit to a one-phase association model using GraphPad Prism (GraphPad Software, La Jolla, CA).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. LindenbachBDThielHJRiceCM 2007 Flaviviridae: The Viruses and Their Replication. KnipeDMHowleyPM Fields Virology. 5th ed Philadelphia Lippincott-Williams & Wilkins 1101 1152
2. SejvarJJ 2007 The long-term outcomes of human West Nile virus infection. Clin Infect Dis 44 1617 1624
3. PlanitzerCBModrofJYuMYKreilTR 2009 West Nile virus infection in plasma of blood and plasma donors, United States. Emerg Infect Dis 15 1668 1670
4. Fernandez-GarciaMDMazzonMJacobsMAmaraA 2009 Pathogenesis of flavivirus infections: using and abusing the host cell. Cell Host Microbe 5 318 328
5. BrintonMA 2002 The molecular biology of West Nile Virus: a new invader of the western hemisphere. Annu Rev Microbiol 56 371 402
6. PereraRKuhnRJ 2008 Structural proteomics of dengue virus. Curr Opin Microbiol 11 369 377
7. MackenzieJMKhromykhAAPartonRG 2007 Cholesterol manipulation by West Nile virus perturbs the cellular immune response. Cell Host Microbe 2 229 239
8. RoosendaalJWestawayEGKhromykhAMackenzieJM 2006 Regulated cleavages at the West Nile virus NS4A-2K-NS4B junctions play a major role in rearranging cytoplasmic membranes and Golgi trafficking of the NS4A protein. J Virol 80 4623 4632
9. Laurent-RolleMBoerEFLubickKJWolfinbargerJBCarmodyAB 2010 The NS5 protein of the virulent West Nile virus NY99 strain is a potent antagonist of type I interferon-mediated JAK-STAT signaling. J Virol 84 3503 3515
10. Munoz-JordanJLSanchez-BurgosGGLaurent-RolleMGarcia-SastreA 2003 Inhibition of interferon signaling by dengue virus. Proc Natl Acad Sci U S A 100 14333 14338
11. AvirutnanPFuchsAHauhartRESomnukePYounS 2010 Antagonism of the complement component C4 by flavivirus nonstructural protein NS1. J Exp Med 207 793 806
12. ZhangYKaufmannBChipmanPRKuhnRJRossmannMG 2007 Structure of immature West Nile virus. J Virol 81 6141 6145
13. ZhangYCorverJChipmanPRZhangWPletnevSV 2003 Structures of immature flavivirus particles. EMBO J 22 2604 2613
14. YuIMZhangWHoldawayHALiLKostyuchenkoVA 2008 Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 319 1834 1837
15. LiLLokSMYuIMZhangYKuhnRJ 2008 The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science 319 1830 1834
16. AllisonSLSchalichJStiasnyKMandlCWKunzC 1995 Oligomeric rearrangement of tick-borne encephalitis virus envelope proteins induced by an acidic pH. J Virol 69 695 700
17. ElshuberSAllisonSLHeinzFXMandlCW 2003 Cleavage of protein prM is necessary for infection of BHK-21 cells by tick-borne encephalitis virus. J Gen Virol 84 183 191
18. GuirakhooFBolinRARoehrigJT 1992 The Murray Valley encephalitis virus prM protein confers acid resistance to virus particles and alters the expression of epitopes within the R2 domain of E glycoprotein. Virology 191 921 931
19. DavisCWNguyenHYHannaSLSanchezMDDomsRW 2006 West Nile virus discriminates between DC-SIGN and DC-SIGNR for cellular attachment and infection. J Virol 80 1290 1301
20. Rodenhuis-ZybertIAvan der SchaarHMda Silva VoorhamJMvan der Ende-MetselaarHLeiHY 2010 Immature dengue virus: a veiled pathogen? PLoS Pathog 6 e1000718
21. NelsonSJostCAXuQEssJMartinJE 2008 Maturation of West Nile virus modulates sensitivity to antibody-mediated neutralization. PLoS Pathog 4 e1000060
22. JunjhonJEdwardsTJUtaipatUBowmanVDHoldawayHA 2010 Influence of pr-M cleavage on the heterogeneity of extracellular dengue virus particles. J Virol 84 8353 8358
23. AllisonSLStiasnyKStadlerKMandlCWHeinzFX 1999 Mapping of functional elements in the stem-anchor region of tick-borne encephalitis virus envelope protein E. J Virol 73 5605 5612
24. MukhopadhyaySKimBSChipmanPRRossmannMGKuhnRJ 2003 Structure of West Nile virus. Science 302 248
25. KuhnRJZhangWRossmannMGPletnevSVCorverJ 2002 Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell 108 717 725
26. MukhopadhyaySKuhnRJRossmannMG 2005 A structural perspective of the flavivirus life cycle. Nat Rev Microbiol 3 13 22
27. KaufmannBNybakkenGEChipmanPRZhangWDiamondMS 2006 West Nile virus in complex with the Fab fragment of a neutralizing monoclonal antibody. Proc Natl Acad Sci U S A 103 12400 12404
28. NybakkenGEOliphantTJohnsonSBurkeSDiamondMS 2005 Structural basis of West Nile virus neutralization by a therapeutic antibody. Nature 437 764 769
29. PiersonTCFremontDHKuhnRJDiamondMS 2008 Structural insights into the mechanisms of antibody-mediated neutralization of flavivirus infection: implications for vaccine development. Cell Host Microbe 4 229 238
30. WhiteheadSSBlaneyJEDurbinAPMurphyBR 2007 Prospects for a dengue virus vaccine. Nat Rev Microbiol 5 518 528
31. RoehrigJT 2003 Antigenic structure of flavivirus proteins. Adv Virus Res 59 141 175
32. ColombageGHallRPavyMLobigsM 1998 DNA-based and alphavirus-vectored immunisation with prM and E proteins elicits long-lived and protective immunity against the flavivirus, Murray Valley encephalitis virus. Virology 250 151 163
33. PincusSMasonPWKonishiEFonsecaBAShopeRE 1992 Recombinant vaccinia virus producing the prM and E proteins of yellow fever virus protects mice from lethal yellow fever encephalitis. Virology 187 290 297
34. FalconarAK 1999 Identification of an epitope on the dengue virus membrane (M) protein defined by cross-protective monoclonal antibodies: design of an improved epitope sequence based on common determinants present in both envelope (E and M) proteins. Arch Virol 144 2313 2330
35. VazquezSGuzmanMGGuillenGChineaGPerezAB 2002 Immune response to synthetic peptides of dengue prM protein. Vaccine 20 1823 1830
36. DejnirattisaiWJumnainsongAOnsirisakulNFittonPVasanawathanaS 2010 Cross-reacting antibodies enhance dengue virus infection in humans. Science 328 745 748
37. Della-PortaAJWestawayEG 1978 A multi-hit model for the neutralization of animal viruses. J Gen Virol 38 1 19
38. PiersonTCXuQNelsonSOliphantTNybakkenGE 2007 The stoichiometry of antibody-mediated neutralization and enhancement of West Nile virus infection. Cell Host Microbe 1 135 145
39. OliphantTNybakkenGEEngleMXuQNelsonCA 2006 Antibody recognition and neutralization determinants on domains I and II of West Nile Virus envelope protein. J Virol 80 12149 12159
40. StiasnyKKiermayrSHolzmannHHeinzFX 2006 Cryptic properties of a cluster of dominant flavivirus cross-reactive antigenic sites. J Virol 80 9557 9568
41. GromowskiGDBarrettNDBarrettAD 2008 Characterization of Dengue Complex-specific Neutralizing Epitopes on the Envelope Protein Domain III of Dengue 2 Virus. J Virol 82 8828 8837
42. MehlhopENelsonSJostCAGorlatovSJohnsonS 2009 Complement protein C1q reduces the stoichiometric threshold for antibody-mediated neutralization of West Nile virus. Cell Host Microbe 6 381 391
43. WestawayEG 1965 The neutralization of arboviruses. II. Neutralization in heterologous virus-serum mixtures with four group B arboviruses. Virology 26 528 537
44. PeirisJSPorterfieldJS 1979 Antibody-mediated enhancement of Flavivirus replication in macrophage-like cell lines. Nature 282 509 511
45. WitzJBrownF 2001 Structural dynamics, an intrinsic property of viral capsids. Arch Virol 146 2263 2274
46. LokSMKostyuchenkoVNybakkenGEHoldawayHABattistiAJ 2008 Binding of a neutralizing antibody to dengue virus alters the arrangement of surface glycoproteins. Nat Struct Mol Biol 15 312 317
47. BothnerBDongXFBibbsLJohnsonJESiuzdakG 1998 Evidence of viral capsid dynamics using limited proteolysis and mass spectrometry. J Biol Chem 273 673 676
48. LewisJKBothnerBSmithTJSiuzdakG 1998 Antiviral agent blocks breathing of the common cold virus. Proc Natl Acad Sci U S A 95 6774 6778
49. Ansarah-SobrinhoCNelsonSJostCAWhiteheadSSPiersonTC 2008 Temperature-dependent production of pseudoinfectious dengue reporter virus particles by complementation. Virology 381 67 74
50. HalsteadSB 2003 Neutralization and antibody-dependent enhancement of dengue viruses. Adv Virus Res 60 421 467
51. MorensDMHalsteadSBLarsenLK 1985 Comparison of dengue virus plaque reduction neutralization by macro and “semi-micro’ methods in LLC-MK2 cells. Microbiol Immunol 29 1197 1205
52. CherrierMVKaufmannBNybakkenGELokSMWarrenJT 2009 Structural basis for the preferential recognition of immature flaviviruses by a fusion-loop antibody. EMBO J 28 3269 3276
53. OliphantTEngleMNybakkenGEDoaneCJohnsonS 2005 Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat Med 11 522 530
54. ShresthaBBrienJDSukupolvi-PettySAustinSKEdelingMA 2010 The development of therapeutic antibodies that neutralize homologous and heterologous genotypes of dengue virus type 1. PLoS Pathog 6 e1000823
55. JohnsonJE 2003 Virus particle dynamics. Adv Protein Chem 64 197 218
56. YewdellJWTaylorAYellenACatonAGerhardW 1993 Mutations in or near the fusion peptide of the influenza virus hemagglutinin affect an antigenic site in the globular region. J Virol 67 933 942
57. RuprechtCRKrarupAReynellLMannAMBrandenbergOF 2011 MPER-specific antibodies induce gp120 shedding and irreversibly neutralize HIV-1. J Exp Med 208 439 454
58. FinneganCMBergWLewisGKDeVicoAL 2002 Antigenic properties of the human immunodeficiency virus transmembrane glycoprotein during cell-cell fusion. J Virol 76 12123 12134
59. BinleyJMWrinTKorberBZwickMBWangM 2004 Comprehensive cross-clade neutralization analysis of a panel of anti-human immunodeficiency virus type 1 monoclonal antibodies. J Virol 78 13232 13252
60. RoehrigJTStaudingerLAHuntARMathewsJHBlairCD 2001 Antibody prophylaxis and therapy for flavivirus encephalitis infections. Ann N Y Acad Sci 951 286 297
61. MathewsJHRoehrigJT 1984 Elucidation of the topography and determination of the protective epitopes on the E glycoprotein of Saint Louis encephalitis virus by passive transfer with monoclonal antibodies. J Immunol 132 1533 1537
62. Sukupolvi-PettySAustinSKEngleMBrienJDDowdKA 2010 Structure and function analysis of therapeutic monoclonal antibodies against dengue virus type 2. J Virol 84 9227 9239
63. MehlhopEAnsarah-SobrinhoCJohnsonSEngleMFremontDH 2007 Complement protein C1q inhibits antibody-dependent enhancement of flavivirus infection in an IgG subclass-specific manner. Cell Host Microbe 2 417 426
64. OliphantTNybakkenGEAustinSKXuQBramsonJ 2007 Induction of epitope-specific neutralizing antibodies against West Nile virus. J Virol 81 11828 11839
65. PiersonTCSanchezMDPufferBAAhmedAAGeissBJ 2006 A rapid and quantitative assay for measuring antibody-mediated neutralization of West Nile virus infection. Virology 346 53 65
66. DavisCWMatteiLMNguyenHYAnsarah-SobrinhoCDomsRW 2006 The location of asparagine-linked glycans on West Nile virions controls their interactions with CD209 (dendritic cell-specific ICAM-3 grabbing nonintegrin). J Biol Chem 281 37183 37194
67. AndrewesCHElfordWJ 1933 Observations on anti-phage sera. I. ‘he percentage law’. Br J Exp Pathol 14 367 376
68. HannaSLPiersonTCSanchezMDAhmedAAMurtadhaMM 2005 N-linked glycosylation of west nile virus envelope proteins influences particle assembly and infectivity. J Virol 79 13262 13274
69. GeissBJPiersonTCDiamondMS 2005 Actively replicating West Nile virus is resistant to cytoplasmic delivery of siRNA. Virol J 2 53
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- High Affinity Nanobodies against the VSG Are Potent Trypanolytic Agents that Block Endocytosis
- Structural and Mechanistic Studies of Measles Virus Illuminate Paramyxovirus Entry
- Sporangiospore Size Dimorphism Is Linked to Virulence of
- The Binding of Triclosan to SmeT, the Repressor of the Multidrug Efflux Pump SmeDEF, Induces Antibiotic Resistance in
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy