
The -glycan Glycoprotein Deglycosylation Complex (Gpd) from Deglycosylates Human IgG
C.
canimorsus 5 has the capacity to grow at the expenses of glycan moieties from host cells N-glycoproteins. Here, we show that C. canimorsus 5 also has the capacity to deglycosylate human IgG and we analyze the deglycosylation mechanism. We show that deglycosylation is achieved by a large complex spanning the outer membrane and consisting of the Gpd proteins and sialidase SiaC. GpdD, -G, -E and -F are surface-exposed outer membrane lipoproteins. GpdDEF could contribute to the binding of glycoproteins at the bacterial surface while GpdG is a endo-β-N-acetylglucosaminidase cleaving the N-linked oligosaccharide after the first N-linked GlcNAc residue. GpdC, resembling a TonB-dependent OM transporter is presumed to import the oligosaccharide into the periplasm after its cleavage from the glycoprotein. The terminal sialic acid residue of the oligosaccharide is then removed by SiaC, a periplasm-exposed lipoprotein in direct contact with GpdC. Finally, most likely degradation of the oligosaccharide proceeds sequentially from the desialylated non reducing end by the action of periplasmic exoglycosidases, including β-galactosidases, β-N-Acetylhexosaminidases and α-mannosidases.
Published in the journal:
. PLoS Pathog 7(6): e32767. doi:10.1371/journal.ppat.1002118
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002118
Summary
C.
canimorsus 5 has the capacity to grow at the expenses of glycan moieties from host cells N-glycoproteins. Here, we show that C. canimorsus 5 also has the capacity to deglycosylate human IgG and we analyze the deglycosylation mechanism. We show that deglycosylation is achieved by a large complex spanning the outer membrane and consisting of the Gpd proteins and sialidase SiaC. GpdD, -G, -E and -F are surface-exposed outer membrane lipoproteins. GpdDEF could contribute to the binding of glycoproteins at the bacterial surface while GpdG is a endo-β-N-acetylglucosaminidase cleaving the N-linked oligosaccharide after the first N-linked GlcNAc residue. GpdC, resembling a TonB-dependent OM transporter is presumed to import the oligosaccharide into the periplasm after its cleavage from the glycoprotein. The terminal sialic acid residue of the oligosaccharide is then removed by SiaC, a periplasm-exposed lipoprotein in direct contact with GpdC. Finally, most likely degradation of the oligosaccharide proceeds sequentially from the desialylated non reducing end by the action of periplasmic exoglycosidases, including β-galactosidases, β-N-Acetylhexosaminidases and α-mannosidases.
Introduction
Capnocytophaga are capnophilic Gram negative bacteria that belong to the family of Flavobacteriaceae in the phylum Bacteroidetes and colonize the oral cavity of diverse mammals including humans [1], [2]. Capnocytophaga canimorsus, a usual member of dog's mouths flora [3], [4], was discovered in 1976 [5] in patients that underwent dramatic infections after having been bitten, scratched or simply licked by a dog. These infections occur, worldwide, with an approximate frequency of one per million inhabitants per year. They generally begin with flu symptoms and evolve in a few days into fulminant septicaemia and peripheral gangrene with a mortality as high as 40% [5], [6], [7], [8], [9]. A few recent observations help understanding the high aggressiveness of C. canimorsus for humans. First, C. canimorsus are able to escape complement killing and phagocytosis by human polymorphonuclear leukocytes (PMN's) [10], [11]. They also escape detection and phagocytosis by macrophages, which results in a lack of release of pro-inflammatory cytokines [12]. In addition to this passive evasion from innate immunity, 60% of the strains are able to block the killing of Escherichia coli phagocytosed by macrophages [3], [10] and some strains even block the onset of pro-inflammatory signalling induced by an E. coli lipopolysaccharide (LPS) stimulus [12]. The molecular bases of these immunosuppressive mechanisms are not understood yet. However, their study led to the serendipitous discovery that the fastidious C. canimorsus grow readily upon direct contact with mammalian cells including phagocytes. This property was found to be dependent on a sialidase (SiaC) allowing C. canimorsus to harvest amino sugars of glycan chains from host cell glycoproteins [13]. Recently, we reported the complete 2,571,405-bp genome sequence and the surface proteome of strain Cc5 (Manfredi et al., submitted). Among others, this study unravelled the existence of 13 complex feeding systems encoded by polysaccharide utilization loci (PULs), a hallmark of the Cytophaga-Flavobacteria-Bacteroides (CFB) group [14], [15]. The archetype of these systems is the Sus system, pioneered by the laboratory of A. Salyers and allowing Bacteroides thetaiotaomicron to forage starch. It is composed of the surface-exposed SusCDEF protein complex [15], [16] and the SusAB periplasmic proteins [17]. SusC resembles a TonB-dependent transporter essential for energy-dependent import of starch oligosaccharides into the periplasm [18] while SusD is a α-helical starch-binding lipoprotein [19], [20]. SusE and SusF are other surface-exposed lipoproteins that reinforce starch binding [17]. Finally, the outer membrane α-amylase SusG hydrolyses surface-bound starch [19]. B. thetaiotaomicron has 88 of these PULs, identified essentially by the presence of a pair of adjacent susC-like and susD-like alleles. Interestingly, expression of some PULs is upregulated in the presence of mucin O-glycans or glycosaminoglycans (GAGs), indicating that B. thetaiotaomicron also forages on host glycans, primarily the O-glycosylated mucin [14] but these glycoprotein foraging systems have not been characterized so far. Although Streptococcus oralis, a firmicute from the human oral flora and S. pneumoniae have been shown to remove and metabolize N-linked complex glycans of human glycoproteins [21], [22], [23], no Sus-like-N-linked glycan foraging system has been described in detail. One of the 13 Sus-like systems from C. canimorsus 5 is involved in the capacity to grow on mammalian cells and to deglycosylate glycoproteins. It is encoded by chromosome locus PUL5 and accounts for 12% of the Cc5 surface proteins. Since it contributes to survival in mice. it can be seen as a new type of bacterial virulence factor (Manfredi et al., submitted). Here, we present a detailed molecular characterization of this PUL5-encoded foraging complex and we show that it deglycosylates human immunoglobulins G (IgG). Among others, we show that it cleaves N-linked glycan moieties between two GlcNAc residues and we show its functional relation with sialidase.
Results
Genetic analysis of the PUL5 locus
PUL5 consists of the five genes Ccan_08700–Ccan_08740. Ccan_08700 encodes a SusC-like integral outer membrane (OM) protein presumably forming a pore in the OM while Ccan_08710 is a SusD-like protein presumably involved in substrate binding [20]. Since the locus was shown to confer the capacity to deglycosylate proteins (Manfredi et al., submitted), we named the five genes gpd (for glycoprotein deglycosylation) and we called gpdC and gpdD the genes encoding homologs to SusC and SusD, respectively. The five gpd genes seem to be organized as an operon in the order gpdC, gpdD, gpdG, gpdE and gpdF (Fig. 1A). GpdG is predicted to be an endo-β-N-acetylglucosaminidase and GpdE has similarities with the Concanavalin A-like lectins/glucanases superfamily on its 108 C-terminal amino acids and could have a substrate-binding role analogous to that of GpdD. Finally, GpdF shows homology to the galactose-binding domain-like superfamily on its 136 C-terminal amino acids suggesting again a role in glycan binding.

In order to investigate what is the function of the individual Gpd proteins we constructed single gpd genes knockout strains. None of the knockout mutants was significantly affected in its growth on blood agar plates. In contrast, deletion of any of the gpdC, -D, -G or -E genes led to a clear reduction of growth on HEK293 cells while deletion of gpdF had only a slight effect (Fig. 1B). Complementation of the deleted genes with plasmid-borne genes expressed from the natural gpdC promoter completely restored growth to the wildtype (wt) level indicating that none of the mutation was polar.
In order to determine whether the reduced growth of the mutants was due to a defect in protein deglycosylation, we incubated wt Cc5 bacteria and the gpd mutant bacteria with fetal calf serum protein fetuin, taken as a standard glycoprotein. Fetuin contains 3 O-linked glycans (accounting for 20% of fetuin-bond carbohydrates) and 3 N-linked glycans (80% of fetuin-bond carbohydrates) [24]. We monitored glycosylation by staining with Sambucus nigra agglutinin (SNA), a lectin that recognizes terminal sialic acids on glycans. As shown in Fig. 1C, fetuin that had been incubated with wt Cc5 reacted much less with SNA and appeared as two, still sialylated smaller degradation products. As shown by Manfredi et al. (submitted), this indicated that partial deglycosylation had occurred and progressed further than a simple desialylation. In contrast, fetuin that was incubated with the gpdC, -D, -G and -E mutant bacteria was unaffected, indicating that no desialylation occurred in the absence of these gpd genes, although sialidase SiaC [13] was not directly affected. Fetuin incubated with the gpdF mutant showed a slight desialylation indicating that fetuin deglycosylation was not completely abolished as with the other mutants. Fetuin glycosylation was also monitored by immuno-blotting with anti-fetuin antibodies. As shown in Fig. 1D, the size of fetuin was shifted down after incubation with wt Cc5 bacteria while the protein migration rate was unchanged after incubation with the gpdC, -D, -G and -E mutant bacteria. After incubation with gpdF mutant bacteria, fetuin did undergo a size shift but not as marked as when incubated with wt bacteria. Taken together these results indicate that partial fetuin deglycosylation was strictly dependent on the activity of proteins GpdC, -D, -G, -E and, to a lesser extend -F. Finally, our data strongly suggest that the defect in growth of the gpd mutants onto HEK293 cells was due to a defect in the ability to deglycosylate host glycoproteins.
GpdG is an endo-β-N-acetylglucosaminidase
GpdG is annotated as an endo-β-N-acetylglucosaminidase (Manfredi et al., submitted), i.e an endo-glycosidase that cleaves N-linked glycan structures at the base of the glycan in between two GlcNAc molecules. Hence, it should leave one GlcNac molecule attached to the protein. Fetuin is reported to be glycosylated on the three asparagine residues N99, N156 and N176 [24]. Analysis by liquid chromatography-mass spectrometry (LC-MS) of trypsin-digested fetuin showed that the main glycosylation site resides on N156. The m/z is in accordance with the peptide LCPDCPLLAPLNDSR carrying the GlcNAc5Man3Gal3Sial3 sugar (Fig. 2A). N176 was found to carry a sugar with a Hex6HexNAc5NeuAc4 composition, but its site occupancy was much lower than N156. Only trace amounts of glycans were found attached to N99. After incubation of fetuin with wt Cc5 bacteria, LC-MS analysis revealed the presence of a peptide whose mass indicated that only one GlcNAc moiety remained linked to N156 (Fig. 2B). The fragmentation spectrum of this peptide fully confirmed the presence of the GlcNAc moiety on N156 (Fig. 2C). Due to the low site occupancy of N176, deglycosylation of N176 to the GlcNAc moiety was too weak to be detected. The conversion of GlcNAc5Man3Gal3Sial3 to GlcNAc on N156 suggests an endo-β-N-acetylglucosaminidase dependent deglycosylation.

To confirm that fetuin deglycosylation was due to the Gpd complex activity and in particular to the GpdG glycosyl hydrolase activity, we then analysed fetuin after incubation with the gpdG knockout bacteria. Fetuin incubated in the presence of these mutant bacteria turned out to remain fully glycosylated (Fig. 2D) indicating that no cleavage occurred in the absence of the enzyme.
The sequence of GpdG was then compared to those of two endo-β-N-acetylglucosaminidases, namely EndoS from Streptococcus pyogenes capable of deglycosylating N-linked glycans from the γ chain of human immunoglobulins [25], and EndoF from Flavobacterium meningosepticum capable of cleaving off high-mannose and complex glycan N-linked from several glycoproteins including immunoglobulins [26]. It appeared that a chitinase motif present in these two enzymes was conserved in GpdG (FDGFDIDWE). In order to further confirm the endo-β-N-acetylglucosaminidase activity of GpdG we substituted the essential E205 residue [26] with a glycine and tested the growth on HEK293 cells of the gpdG mutant strain expressing in trans the GpdG catalytic mutant. As shown in Fig. 3A, the GpdG catalytic mutant was impaired in growth. We then tested the fetuin deglycosylation ability of the GpdG catalytic mutant. As shown by the lectin staining in Fig. 3B and by the immuno-blotting in Fig. 3C, bacteria endowed with the GpdG catalytic mutant were completely impaired in fetuin deglycosylation. We conclude from all these experiments that GpdG is an endo-β-N-acetylglucosaminidase.
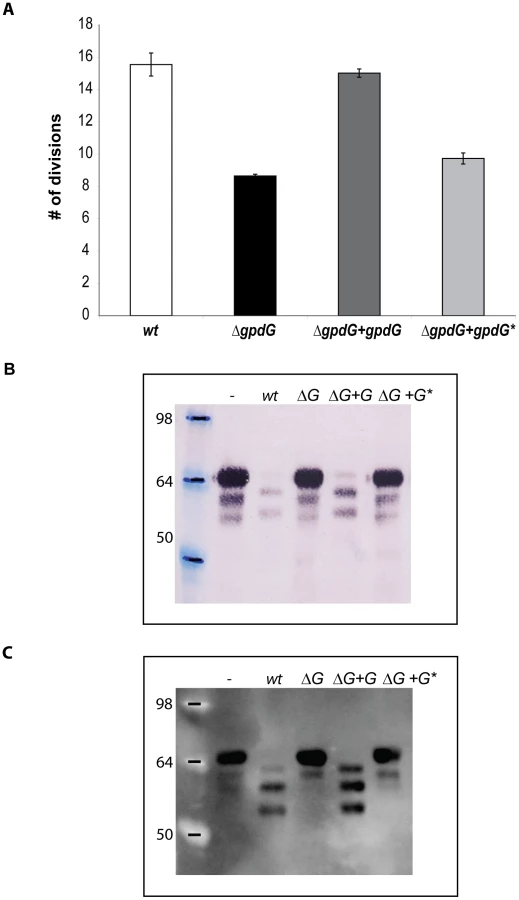
The Gpd complex deglycosylates human IgG
Since GpdG has the same chitinase motif as EndoF and EndoS, known to deglycosylate N-linked glycans from the γ chain of human IgGs [25], [26], we tested whether the Gpd complex would also be able to deglycosylate the heavy chain of IgGs. Cleavage of the N297-linked glycan moiety by EndoS was shown to determine a size shift of ∼3 KDa [25]. After incubation of purified human IgG with wt Cc5 bacteria, the molecular mass of the γ chain underwent a slight size shift (Fig. 4A and B) while the mass of the light chains was unchanged (Fig. 4A). In contrast incubation with ΔgpdG knockout bacteria did not alter the γ chain size indicating that the cleavage was GpdG dependent. To confirm that the size reduction of the γ chain was due to the removal of the glycan moiety, IgG was stained with SNA. As shown in Fig. 4C, the SNA signal of the γ chain was significantly reduced after incubation with wt Cc5. In contrast the γ chains remained fully glycosylated after incubation with ΔgpdG bacteria. Analysis by LC-MS of trypsin-digested IgG showed that N297 from this sample of IgG mainly bears a GlcNAc4Man3Gal2Sial1Fuc1 chain or a GlcNAc4Man3Gal2Fuc1 chain (Fig. 5A–B). After incubation of this sample of IgG with wt Cc5 bacteria, LC-MS analysis revealed the presence of a peptide whose mass indicated that only a GlcNAc1Fuc1 moiety remained linked to N297 (Fig. 5C). The fragmentation spectrum of this peptide fully confirmed the presence of the GlcNAc1Fuc1 moiety on N297 (Fig. 5D). LC-MS analysis of IgG after incubation with ΔgpdG bacteria revealed exactly the same profile as in the untreated sample indicating that no deglycosylation has occurred (Fig. 5E–F). The conversion of GlcNAc4Man3Gal2Sial1Fuc1 and GlcNAc4Man3Gal2Fuc1 to GlcNAc1Fuc1 on N297 indicates an endo-β-N-acetylglucosaminidase (GpdG) dependent deglycosylation. These data indicated that, like F. meningosepticum and S. pyogenes, C. canimorsus has the capacity to deglycosylate IgGs.
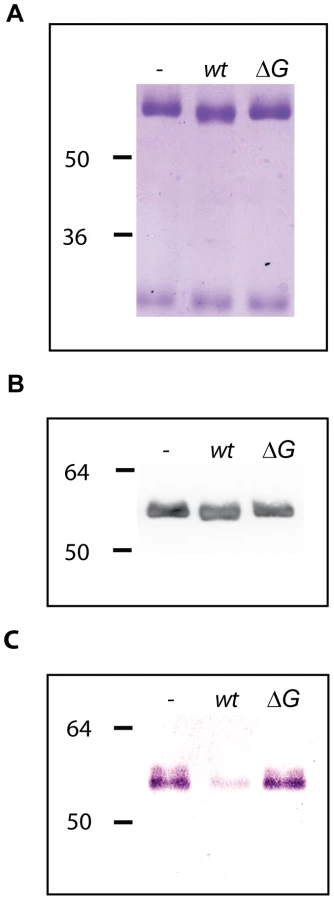

GpdD, -G, -E and -F are lipoproteins and lipid modification is fundamental for the complex activity
The GpdD, -G, -E and –F proteins belong to the OM and surface proteomes of Cc5 (Manfredi et al., submitted). In addition, these proteins are endowed with a signal peptidase II consensus signal peptide. Altogether, this suggests that they could be lipoproteins anchored to the outer leaflet of the outer membrane and exposed at the surface of the bacterium (Manfredi et al., submitted). In order to determine whether the lipidation of the Gpd proteins is required for their function, we generated soluble periplasmic versions of GpdD and GpdG by substituting the cystein residue of the lipobox with a glycine. We then tested the ability of the periplasmic variants of GpdD and GpdG to complement the growth deficiency of the gpdD and gpdG knockout strains on HEK293 cells. As shown in Fig. 6, both the GpdD and GpdG periplasmic variant were unable to complement the growth deficiency indicating that lipid modification is necessary for the proper localization and function of the proteins. This conclusion was reinforced by the fact that bacteria endowed with periplasmic GpdD or GpdG were unable to deglycosylate fetuin (Fig. 6). Hence, we infer that GpdD and GpdG are lipoproteins that are anchored in the outer leaflet of the outer membrane and exposed to the bacterial surface. The same presumably applies to GpdE and GpdF since they have also a lipobox and they are also part of the surface proteome (Manfredi et al., submitted).

The Gpd proteins form a deglycosylation complex associated with sialidase
In order to assay whether the five Gpd proteins interact with each other to form a complex at the bacterial surface, we performed a two-step affinity purification with a His-Strep tagged version of GpdC. Analysis by immuno-blot and mass spectrometry (Fig. 7) of the purified fraction revealed the presence, together with GpdC, of GpdD, -G, -E and –F, indicating a stable interaction between all these proteins. Furthermore, six other proteins, among which SiaC (Fig. 7), co-purified with the complex.

Sialidase is a periplasmic lipoprotein that interacts with GpdC
SiaC has been previously shown [13] to be essential to sustain growth of Cc5 in the presence of eukaryotic cells due to its role in the glycoprotein deglycosylation process. We thus focused our attention on the sialidase-Gpd complex interaction. The co-purification of SiaC with GpdC strongly suggested that SiaC is associated to the Gpd complex, although it is encoded far away from PUL5. However, unlike the Gpd proteins, sialidase was not identified in the surface proteome of Cc5 (Manfredi et al., submitted). On the other hand, earlier immunofluorescence assays suggested that sialidase is localized on the bacterial surface and removal of the signal sequence of sialidase prevented growth on cells [13]. In order to better understand the interplay between SiaC and Gpd proteins in the glycoprotein deglycosylation process, we decided to clarify its localization.
Since the sialidase sequence analysis revealed the presence of a signal peptide with a lipobox in the N-terminal sequence, we first sought to determine whether SiaC is a lipoprotein. We incubated Cc5 and sialidase mutant (siaC) bacteria encoding SiaCC17Y in the presence of tritiated palmitate and analyzed the total proteins by SDS-PAGE and fluorography (Fig. 8A). Sialidase appeared indeed to be lipidated and the C17Y mutation completely prevented this lipid modification. The analysis of outer membrane proteins isolated by sarcosyl extraction confirmed that sialidase but not its C17Y variant was associated with the OM (Fig. 8B). We conclude from these experiments that SiaC is a lipoprotein anchored into the outer membrane.

In order to define whether it is exposed towards the outside like GpdDGEF or towards the periplasm, we tested whether the periplasmic SiaC17Y could restore the growth deficiency of the siaC mutant strain. In contrast to what was observed for GpdD and GpdG, expression of SiaC17Y in trans did fully restore the growth defect (Fig. 8C) indicating that the localization of sialidase in the periplasm and the absence of association with the outer membrane did not prevent its function. This data pointed to the direction of a periplasmic localization of SiaC rather than a surface-exposed localization as was previously suggested [13].
The association between sialidase and the Gpd complex obviously suggests that the two work cooperatively. This was already suggested by the fact that the gpd mutant bacteria did not remove the terminal sialic acid residues from fetuin, although SiaC was functional in these mutants (Fig. 1C). We then tested the ability of the siaC knockout bacteria to deglycosylate fetuin. SNA lectin staining (Fig. 8D) and immuno-blotting (Fig. 8E) clearly showed the same fetuin deglycosylation pattern for the wt and siaC mutant bacteria. These results indicate that the endo-cleavage of fetuin N-glycans, operated by the Gpd complex is completely independent from the activity of SiaC. However, the evidence that SiaC activity is essential for growth on Hek293 cells (Fig. 8C), suggests that removal of the glycan terminal sialic acid is nevertheless a crucial step for the subsequent glycan catabolism process. This indicates that the Gpd complex acts upstream of SiaC. Since the Gpd complex includes the GpdC porin-like protein, this sequential order is perfectly compatible with a periplasmic localization of sialidase. Sialic acid removal would thus occur in the periplasm after the glycan has been cleaved off and transported through the GpdC OM channel.
If this model was correct, the interaction between the periplasmic SiaC and the GpdC complex could only occur through a direct interaction with GpdC, since the other Gpd proteins are surface exposed. To test this prediction, we expressed a C-terminally Strep-His double tagged GpdC in a gpdCDGE multi knockout strain and we performed a two-step affinity purification of GpdC. The analysis by immuno-blotting (Fig. 8F) of the fractions eluted after the second purification step showed that SiaC did indeed co-purify with GpdC indicating that SiaC and GpdC do indeed interact directly with each other. The complete deglycosylation complex would thus consist of the surface-exposed lipoproteins GpdDGEF and the periplasm-exposed lipoprotein SiaC, all of them associated to the porin-like GpdC (Fig. 8).
Discussion
Our previous work has shown that C. canimorsus deglycosylates surface glycoproteins from the host and sustains its growth on the glycan moieties [13]. Here, we showed that this deglycosylating activity is achieved by the joined action of the PUL5-encoded Gpd complex and sialidase [13]. PUL5 consists of the five gpdCDGEF genes. GpdC, an homolog of the archetypal SusC [17], likely represents the specific OM porin of the system. GpdD is an homolog of SusD, a starch-binding protein [16], [20] and hence most likely a glycoprotein-binding protein. On the basis of their annotation, we propose that GpdE and GpdF are also glycan-binding proteins. GpdG was annotated as an endo-β-N-acetylglucosaminidase (Manfredi et al., submitted) and this annotation was shown to be correct. Indeed mass spectrometry analyses demonstrated that GpdG deglycosylates the tribranched GlcNAc5Man3Gal3Sial3 glycan structure linked to N156 from the model glycoprotein fetuin, leaving one GlcNac residue on the protein. GpdDGEF were predicted to be lipoproteins (Manfredi et al., submitted). Replacement of the critical cysteine of the lipoprotein signal peptide from GpdD and GpdG completely abolished the deglycosylating activity, indicating that a periplasmic location did not sustain the activity. These data, together with the fact that the two proteins belong to the surface proteome indicate that these two lipoproteins are exposed to the surface and not to the periplasm. We assume the same is true for GpdE and F since, like GpdD, they are thought to bind glycans, they contain a lipobox and they belong to the surface proteome. Interestingly, all the five Gpd proteins could be co-purified with the porin-like GpdC, indicating that they all form one single complex at the bacterial surface. Unexpectedly, not only GpdD, -G, -E and -F co-purified with GpdC but also SiaC. Although SiaC was known to be part of the catabolic process, SiaC is not encoded together with GpdCDGEF (Manfredi et al., submitted) and it was not anticipated that the interaction would be so close. SiaC turned out to be also a lipoprotein but, unlike GpdD and GpdG, it was still functional when it was directed to the periplasm, unlipidated. We inferred from this observation that, contrary to our initial report, SiaC is a periplasm-oriented lipoprotein. Thus, the observations presented here suggest the model illustrated in Fig. 9: The surface-exposed GpdCDEF complex captures the N-linked complex glycan moieties of glycoproteins, which are then detached from the protein by GpdG and internalized by GpdC. As soon as they reach the periplasm, SiaC removes the terminal sialic acid. This sequence of events is strongly supported by the observation that gpd mutant bacteria do not desialylate fetuin, although SiaC is functional in these mutants (Fig. 1). After desialylation, the oligosaccharide would be sequentially degraded by periplasmic exoglycosidases and the monosaccharides would be transferred to the cytosol. This last step of the model is supported by the fact that the genome encodes three putative β-galactosidases (Ccan_01530, Ccan_15520, Ccan_17480), five putative β-N-acetylhexosaminidases (Ccan_03860, Ccan_04040, Ccan_16820, Ccan_17870, Ccan_20090) and four putative α-mannosidases (Ccan_00510, Ccan_01900, Ccan_04050 and Ccan_16220), all of them endowed with a signal peptide I or II, and none of them surface exposed (Manfredi et al., submitted). The β-galactosidase and α-mannosidase activities were confirmed in the crude extract (data not shown). The three β-galactosidases seemed actually redundant since they could all be individually knocked out without affecting the growth on cells (data not shown).

This global model strikingly reminds the archetypal Sus system shown to consist of one single complex made of SusCDEF [16]. It is thought that SusG, an endo-acting enzyme, generates internal cuts in a bound starch molecule and releases oligosaccharides larger than maltotriose, which are then transported by SusC into the periplasmic compartment. In the periplasm, glycoside hydrolases SusA and SusB then degrade the oligosaccharides into their component sugars prior to final transport to the cytosol [27], [28]. The two systems are thus remarkably conserved, although they adapted to different complex saccharides.
To our knowledge, the Gpd system is the first Sus-like system devoted to foraging N-linked glycoproteins. It contributes to sustain growth of C. canimorsus at the expenses of cultured cells (Manfredi et al., submitted). Since C. canimorsus has 13 PULs (Manfredi et al., submitted), it is very likely that some of them could be devoted to the harvest of O-linked glycans, but this activity has not been identified thus far. The best approach would probably be to look for upregulation in the presence of O-linked glycoproteins, as was done in B. thetaiotaomicron [29]. Deglycosylation of N-linked glycans is not unprecedented among pathogens and commensals. As mentioned earlier, two streptococci, S. pyogenes and S. oralis have this remarkable property. In the case of S. pyogenes, this activity is exerted towards IgGs by secreted endoglycosidase EndoS and it does not seem to play a major role in nutrient acquisition [25]. In contrast, in S. oralis, the activity was shown to sustain growth [30]. It is interesting to notice that S. oralis, like C. canimorsus, is emerging as an important opportunistic pathogen originating from the oral flora. This commonality between two very different bacteria from the same ecosystem suggests first that the capacity to deglycosylate host proteins is a favourable trait in the mouth ecosystem and, second, could favour opportunistic infections. Deglycosylation of IgGs is very likely to contribute to a generalized infection as discussed by Collin and Olsen [25] but, for C. canimorsus, one cannot exclude that deglycosylation of other host proteins would also significantly contribute to pathogenesis.
Our data demonstrate that PUL-encoded lipoproteins are surface-exposed. Prolipoproteins are exported through the Sec pathway and then acylated at the periplasmic leaflet of the inner membrane (IM) by the sequential action of glyceryl transferase, O-acyl transferase(s) and prolipoprotein signal peptidase (signal peptidase II). A mature lipoprotein harbours as a first aminoacid a cysteine residue that is lipid modified with a N-Acyl diacyl Glyceryl group which serves to anchor the protein to the IM. In Gram-negative bacteria some lipoproteins are destined for the OM. These proteins are extracted from the IM, transported across the periplasm and inserted in the inner leaflet of the OM by the Lol pathway (for review see refs [31], [32]. Insertion of lipoproteins into the outer leaflet of the OM is however established in some pathogens like Borrelia but the pathway is neither well documented nor well understood [32]. Since bacteria from the Cytophaga-Flavobacteria-Bacteroides group massively insert lipoproteins in the outer leaflet of the OM, we postulate that they have an original system dedicated to the transport of lipoproteins across the OM but this system still needs to be identified and investigated.
Materials and Methods
Bacterial strains and growth conditions
Conventional bacterial growth conditions and selective agents
The strains used in this study are listed in Table 1. Escherichia coli strains were routinely grown in LB broth at 37°C. C. canimorsus bacteria were routinely grown on heart infusion agar (Difco) supplemented with 5% sheep blood (Oxoid) for 2 days at 37°C in the presence of 5% CO2. To select for plasmids, antibiotics were added at the following concentrations: 10 µg/ml erythromycin (Em), 10 µg/ml cefoxitin (Cf), 20 µg/ml gentamicin (Gm), 100 µg/ml ampicillin (Ap) and 50 µg/ml kanamycin (Km).

Growth of Cc5 bacteria on HEK293 cultured cells
Human Embryonic Kidney 293 cells (HEK293) were cultured in DMEM (Invitrogen) with 10% (v/v) fetal calf serum (Invitrogen) and 1 mM sodium pyruvate. Cells were grown in medium without antibiotics in a humidified atmosphere enriched with 5% CO2 at 37°C. Bacteria were harvested by gently scraping colonies off the agar surface and resuspended in PBS. A total of 4×104 bacteria were incubated with 2×105 HEK293 cells (MOI = 0.2) in a final volume of 1 ml medium devoid of antibiotics for 23 h.
Mutagenesis and allelic exchange
Mutagenesis of Cc5 wt has been performed has described in ref [33] with slight modifications. Briefly, replacement cassettes with flanking regions spanning approximately 500 bp homologous to direct gpd framing regions were constructed with a three-fragment overlapping-PCR strategy. First, two PCRs were performed on 100 ng of of Cc5 genomic DNA with primers A and B (Table 2) for the upstream flanking regions and with primers C and D for the downstream regions. Primers B and C contained an additional 5′ 20-nucleotide extension homologous to the resistance ermF insertion cassette. The ermF resistance cassette was amplified from plasmid pMM106 DNA with primers 5502 and 5503. All three PCR products were cleaned and then mixed in equal amounts for PCR using Phusion polymerase (Finnzymes). The initial denaturation was at 98°C for 2 min, followed by 12 cycles without primers to allow annealing and elongation of the overlapping fragments (98°C for 30 s, 50°C for 40 s, and 72°C for 2 min). After the addition of external primers (A and D), the program was continued with 20 cycles (98°C for 30 s, 50°C for 40 s, and 72°C for 2 min 30 s) and finally 10 min at 72°C. Final PCR products consisted in gpd::ermF insertion cassettes and were then digested with PstI and SpeI for cloning into the appropriate sites of the C. canimorsus suicide vector pMM25. Resulting plasmids were transferred by RP4-mediated conjugative DNA transfer from E. coli S17-1 to C. canimorsus 5 to allow integration of the insertion cassette. Transconjugants were then selected for presence of the ermF cassette, checked for sensitivity to cefoxitin and the deleted regions were sequenced.

Construction of complementation and expression plasmids
Plasmid pPM1, used for complementation and expression of the Gpd proteins, is a derivative of the E. coli - C. canimorsus shuttle vector pMM47A.1 [33]. pMM47A.1 ermF promoter region was cleaved with SalI and NcoI and the 117 nucteotides upstream the gpdC starting codon sequence, containing the putative gpdC promoter, was cloned using the same restriction sites. Full length gpdC, -D, -G, -E and -F were amplified with the specific primers listed in Table 3 and cloned into plasmid pPM1 into NcoI and XbaI restriction sites leading to the insertion of a glycine at position 2.

The E205G substitution inactivating the catalytic site of GpdG was introduced by site directed mutagenesis by overlapping PCR using primers 5008/6061 and 6060/6055 and cloned in pPM1 using NcoI and XbaI restriction sites leading to plasmid pFR10 (gpdG*). The C17G substitution of GpdD was introduced by site directed mutagenesis amplifying by PCR using primers 6056 and 6057 and cloning NcoI/XbaI in pPM1 leading to plasmid pFR8.
The C21G substitution of GpdG was introduced by site directed mutagenesis amplifying by PCR using primers 6054 and 6055 and cloning NcoI/XbaI in pPM1 leading to plasmid pFR9.
The C17Y substitution of SiaC was introduced by site directed mutagenesis amplifying by inverse PCR using primers 5045 and 5046 using as pMM52 as template leading to plasmid pMM121.1.
C-terminal His-Strep double tagged gpdC was amplified by two-step overlapping PCR using primers 5081, 5467 and 5530 and cloned in pMM47.A using SalI and SpeI restriction sites leading to plasmid pPM3.
Fetuin deglycosylation analyses and lectin stainings
Bacteria were collected from blood agar plates and resuspended in PBS at OD600 = 1. 100 µl of bacterial suspensions were then incubated with 100 µl of a fetal calf serum fetuin (Sigma F2379) solution (0.1 g.l−1) for 120 minutes at 37°C. As negative control, 200 µl of 1∶2 diluted fetuin solution alone was incubated for 120 minutes at 37°C. Samples were then centrifuged for 5 min at 13000× g, supernatant collected and loaded in a 12% SDS gel. Samples were analyzed by immunoblotting (Fetuin, Rabbit anti-Bovine RIA, UCBA699/R1H, ACCURATE CHEMICAL & SCIENTIFIC CORPORATION) and lectin stainings were performed with Sambucus nigra lectin (SNA) according to manufacturer recommendations (DIG Glycan Differentiation Kit, 11210238001, Roche).
Human IgG deglycosylation analyses and lectin stainings
Bacteria were collected from blood agar plates and resuspended in PBS at OD600 = 1. 100 µl of bacterial suspensions were then incubated with 100 µl of a purified human IgG (Invitrogen, 02-7102) solution (0.5 g.l−1) for 180 minutes at 37°C. As negative control, 200 µl of 1∶2 diluted IgG solution alone was incubated for 180 minutes at 37°C. Samples were then centrufiged for 5 min at 13000× g, supernatant collected and and loaded in a 12% SDS gel. Samples were analyzed by Coomassie blue staining, immunoblotting [Goat Anti-Human IgG (Fc specific)-FITC antibody, F9512 Sigma] and lectin stainings were performed with SNA according to manufacturer recommendations (DIG Glycan Differentiation Kit, 11210238001, Roche).
Fetuin and IgG mass spectrometric analyses
Fetuin (Sigma F2379) and human IgG (Invitrogen, 02-7102) were reduced with 10 mM TCEP at 37°C for 1 hour and alkylated with 50 mM iodoacetamide for 15 min at room temperature. Fetuin and IgG were digested with trypsin at an enzyme to protein ratio of 1∶50 (w/w) at 37°C overnight. The peptides were desalted on C18 StageTips (Thermo Fisher Scientific, Reinach, Switzerland) according to the manufacurer's recommendations. The fetuin and IgG peptides were analysed on an LTQ Orbitrap instrument (Thermo Fisher, San José, CA, USA) coupled to an Agilent 1200 nano pump according to (Manfredi et al. submitted).
Outer membrane protein purification
Bacteria were collected from blood agar plates and resuspended in 3 ml ice cold HEPES 10 mM (pH 7.4) at OD600 = 1. Bacterial suspensions were then sonicated on ice until they turned clear and spun at 15600× g for 2 minutes at 4°C. Supernatants were transferred and centrifuged again for 30 minutes at 15600× g at 4°C. Pellets were resuspended in 2 ml HEPES 10 mM with 1% sarcosyl (N-Lauroylsarcosine sodium salt, Sigma) and incubated at room temperature for 30 minutes. Finally, samples were centrifuged at 15600 g for 30 min at 4°C and pellet resuspended in 0.1 ml HEPES. Samples were checked for quality and quantity on silver stained SDS-PAGE and analysed by MS/MS.
Gpd proteins and sialidase co-purification
Cc5 ΔgpdC bacteria harbouring plasmid pPM3, expressing a C-terminal His-Strep double tagged GpdC, or harbouring plasmid pPM2, expressing GpdC without any tag (Mock), were grown for 2 days at 37°C in the presence of 5% CO2 on sheep blood agar plates. Bacteria from 6 plates were scraped and lysed in 35 ml of 25 mM Tris-HCl, 150 mM NaCl, 0.2% triton, 1% NP-40%, 1% sodium deoxycholate, pH 7.6.
For His affinity purification, the lysates were clarified by centrifugation (10 min at 18500 g at RT) and the supernatant was diluted 1∶2 in PBS, 10 mM Imidazole, in the presence of proteinase inhibitor (cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail Tablets, Roche). 3.5 ml of 50% slurry Chelating sepharose Fast Flow beads (GE Healthcare) was first coupled to Ni2+ according to the manufacturer instructions and then 1.75 ml of resin was added to the solution and incubated overnight at 4°C on a rotating wheel. The solution was then loaded into a column and the resin washed first with 25 column volumes (CV) of high salt buffer (50 mM Tris, 500 mM NaCl, pH 8) and then with 5 CV of low salt buffer (50 mM Tris, 100 mM NaCl, pH 8). Proteins were then eluted from the resin with 2 CV of elution buffer (50 mM Tris, 100 mM NaCl, 350 mM Imidazole, pH 8). The material eluted from the Ni2+ column was then diluted 1∶2 in PBS and 1 ml of 50% slurry (0.5 ml CV) Strep-Tactin Superflow resin (IBA, cat No: 2-1206-002) was added. The solution was then incubated overnight at 4°C on a rotating wheel. The solution was then loaded into a column and the flow through reloaded into the resin 2 more times. The resin was then washed 4 times with 10 CV of Buffer W (100 mM Tris, 150 mM NaCl, 1 mM EDTA, pH 8) and proteins eluted in 3 steps with 0.5 ml elution buffer (100 mM Tris, 150 mM NaCl, 1 mM EDTA, 2.5 mM desthiobiotin, pH 8). The proteins present in the elution fractions were identified by MS and immunoblotting, using anti-His for GpdC detection, anti-GpdG and anti-SiaC .
GpdC-sialidase co-purification was performed exactly as described above using Cc5 ΔPUL5 bacteria harbouring pPM3 plasmid or harbouring plasmid pPM2 (Mock). Proteins present in the elution fractions were identified by immunoblotting with anti-Strep antibodies to detect GpdC and anti-SiaC.
In vivo radiolabeling with [3H] palmitate, immuno-precipitation and fluorography
Bacteria were inoculated to HeLa epithelial cells (ATCC CCL-2) in complete DMEM at 37°C with 5% CO2 at a moi of 20. 15–16 h post infection, [9,10-3H] palmitic acid (48 Ci/mmol; Perkin-Elmer Life Sciences) was added to a final concentration of 50 µCi/ml and incubation was continued for 8–9 h, by which time the bacterial culture had reached approximately 108 bacteria/ml as described elsewhere [13]. Supernatants of 2×1 ml were collected without detaching epithelial cells from the wells. Bacteria corresponding to approximately 2×108 cfu were then collected by centrifugation and pellets were combined from 2 ml and stored at −20°C until they were processed. Pellets were resuspended in 0.1 ml PBS TritonX 1% to lyze bacteria and sialidase was immuno-precipitated by addition of 10 µl rabbit polyclonal anti-SiaC for 1 h at RT on a rotating wheel. Protein A agarose slurry (Sigma) was then added in equal amounts for 30 min under constant rotation at RT. Samples were then centrifuged at 14000× g for 2 min at RT, supernatant was discarded and pellets were washed with 0.5 ml PBS 0.1% Triton which was repeated 4 times. Captured proteins were eluted by addition of 50 µl Lämmli buffer (1% SDS, 10% glycerol, 50 mM dithiothreitol, 0.02% bromophenol blue, 45 mM Tris, pH 6.8) for 5 min at 85°C. Samples were centrifuged again and supernatant was carefully separated from the agarose beads and loaded on SDS PAGE gels using 10% polyacrylamide. After gel electrophoresis, gels were fixed in 25∶65∶10 isopropanol∶water∶acetic acid overnight and subsequently soaked for 30 min in Amplify (Amersham). Gels were vacuum dried and exposed to SuperRX autoradiography film (Fuji) for 13days until desired signal strength was reached.
Zdroje
1. KolenbranderPEPalmerRJJrPeriasamySJakubovicsNS 2010 Oral multispecies biofilm development and the key role of cell-cell distance. Nat Rev Microbiol 8 471 480
2. FrandsenEVPoulsenKKononenEKilianM 2008 Diversity of Capnocytophaga species in children and description of Capnocytophaga leadbetteri sp. nov. and Capnocytophaga genospecies AHN8471. Int J Syst Evol Microbiol 58 324 336
3. MallyMParozCShinHMeyerSSoussoulaLV 2009 Prevalence of Capnocytophaga canimorsus in dogs and occurrence of potential virulence factors. Microbes Infect 11 509 514
4. BlanchePBlochESicardD 1998 Capnocytophaga canimorsus in the oral flora of dogs and cats. J Infect 36 134
5. BoboRANewtonEJ 1976 A previously undescribed gram-negative bacillus causing septicemia and meningitis. Am J Clin Pathol 65 564 569
6. PersCGahrn-HansenBFrederiksenW 1996 Capnocytophaga canimorsus septicemia in Denmark, 1982–1995: review of 39 cases. Clin Infect Dis 23 71 75
7. Le MoalGLandronCGrollierGRobertRBurucoaC 2003 Meningitis due to Capnocytophaga canimorsus after receipt of a dog bite: case report and review of the literature. Clin Infect Dis 36 e42 46
8. WestwellAJKerrKSpencerMBHutchinsonDN 1989 DF-2 infection. BMJ 298 116 117
9. BailieWEStoweECSchmittAM 1978 Aerobic bacterial flora of oral and nasal fluids of canines with reference to bacteria associated with bites. J Clin Microbiol 7 223 231
10. MeyerSShinHCornelisGR 2008 Capnocytophaga canimorsus resists phagocytosis by macrophages and blocks the ability of macrophages to kill other bacteria. Immunobiology 213 805 814
11. ShinHMallyMMeyerSFiechterCParozC 2009 Resistance of Capnocytophaga canimorsus to killing by human complement and polymorphonuclear leukocytes. Infect Immun 77 2262 2271
12. ShinHMallyMKuhnMParozCCornelisGR 2007 Escape from immune surveillance by Capnocytophaga canimorsus. J Infect Dis 195 375 386
13. MallyMShinHParozCLandmannRCornelisGR 2008 Capnocytophaga canimorsus: a human pathogen feeding at the surface of epithelial cells and phagocytes. PLoS Pathog 4 e1000164
14. MartensECChiangHCGordonJI 2008 Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 4 447 457
15. MartensECKoropatkinNMSmithTJGordonJI 2009 Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J Biol Chem 284 24673 24677
16. ChoKHSalyersAA 2001 Biochemical analysis of interactions between outer membrane proteins that contribute to starch utilization by Bacteroides thetaiotaomicron. J Bacteriol 183 7224 7230
17. ShipmanJABerlemanJESalyersAA 2000 Characterization of four outer membrane proteins involved in binding starch to the cell surface of Bacteroides thetaiotaomicron. J Bacteriol 182 5365 5372
18. ReevesARD'EliaJNFriasJSalyersAA 1996 A Bacteroides thetaiotaomicron outer membrane protein that is essential for utilization of maltooligosaccharides and starch. J Bacteriol 178 823 830
19. ReevesARWangGRSalyersAA 1997 Characterization of four outer membrane proteins that play a role in utilization of starch by Bacteroides thetaiotaomicron. J Bacteriol 179 643 649
20. KoropatkinNMMartensECGordonJISmithTJ 2008 Starch catabolism by a prominent human gut symbiont is directed by the recognition of amylose helices. Structure 16 1105 1115
21. ByersHLTarelliEHomerKABeightonD 1999 Sequential deglycosylation and utilization of the N-linked, complex-type glycans of human alpha1-acid glycoprotein mediates growth of Streptococcus oralis. Glycobiology 9 469 479
22. BurnaughAMFrantzLJKingSJ 2008 Growth of Streptococcus pneumoniae on human glycoconjugates is dependent upon the sequential activity of bacterial exoglycosidases. J Bacteriol 190 221 230
23. KingSJHippeKRWeiserJN 2006 Deglycosylation of human glycoconjugates by the sequential activities of exoglycosidases expressed by Streptococcus pneumoniae. Mol Microbiol 59 961 974
24. GreenEDAdeltGBaenzigerJUWilsonSVan HalbeekH 1988 The asparagine-linked oligosaccharides on bovine fetuin. Structural analysis of N-glycanase-released oligosaccharides by 500-megahertz 1H NMR spectroscopy. J Biol Chem 263 18253 18268
25. CollinMOlsenA 2001 EndoS, a novel secreted protein from Streptococcus pyogenes with endoglycosidase activity on human IgG. EMBO J 20 3046 3055
26. ElderJHAlexanderS 1982 endo-beta-N-acetylglucosaminidase F: endoglycosidase from Flavobacterium meningosepticum that cleaves both high-mannose and complex glycoproteins. Proc Natl Acad Sci U S A 79 4540 4544
27. D'EliaJNSalyersAA 1996 Contribution of a neopullulanase, a pullulanase, and an alpha-glucosidase to growth of Bacteroides thetaiotaomicron on starch. J Bacteriol 178 7173 7179
28. KitamuraMOkuyamaMTanzawaFMoriHKitagoY 2008 Structural and functional analysis of a glycoside hydrolase family 97 enzyme from Bacteroides thetaiotaomicron. J Biol Chem 283 36328 36337
29. KoropatkinNMartensECGordonJISmithTJ 2009 Structure of a SusD homologue, BT1043, involved in mucin O-glycan utilization in a prominent human gut symbiont. Biochemistry 48 1532 1542
30. ByersHLTarelliEHomerKAHambleyHBeightonD 1999 Growth of Viridans streptococci on human serum alpha1-acid glycoprotein. J Dent Res 78 1370 1380
31. BosMPRobertVTommassenJ 2007 Biogenesis of the gram-negative bacterial outer membrane. Annu Rev Microbiol 61 191 214
32. Kovacs-SimonATitballRWMichellSL 2010 Lipoproteins of bacterial pathogens. Infect Immun 79 548 561
33. MallyMCornelisGR 2008 Genetic tools for studying Capnocytophaga canimorsus. Appl Environ Microbiol 74 6369 6377
34. SimonRPrieferUPuhlerA 1983 A Broad Host Range Mobilization System for In Vivo Genetic Engineering: Transposon Mutagenesis in Gram Negative Bacteria. Nat Biotech 1 784 791
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- High Affinity Nanobodies against the VSG Are Potent Trypanolytic Agents that Block Endocytosis
- Structural and Mechanistic Studies of Measles Virus Illuminate Paramyxovirus Entry
- Sporangiospore Size Dimorphism Is Linked to Virulence of
- The Binding of Triclosan to SmeT, the Repressor of the Multidrug Efflux Pump SmeDEF, Induces Antibiotic Resistance in
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy