
Pathogen Recognition Receptor Signaling Accelerates Phosphorylation-Dependent Degradation of IFNAR1
An ability to sense pathogens by a number of specialized cell types including the dendritic cells plays a central role in host's defenses. Activation of these cells through the stimulation of the pathogen-recognition receptors induces the production of a number of cytokines including Type I interferons (IFNs) that mediate the diverse mechanisms of innate immunity. Type I IFNs interact with the Type I IFN receptor, composed of IFNAR1 and IFNAR2 chains, to mount the host defense responses. However, at the same time, Type I IFNs elicit potent anti-proliferative and pro-apoptotic effects that could be detrimental for IFN-producing cells. Here, we report that the activation of p38 kinase in response to pathogen-recognition receptors stimulation results in a series of phosphorylation events within the IFNAR1 chain of the Type I IFN receptor. This phosphorylation promotes IFNAR1 ubiquitination and accelerates the proteolytic turnover of this receptor leading to an attenuation of Type I IFN signaling and the protection of activated dendritic cells from the cytotoxic effects of autocrine or paracrine Type I IFN. In this paper we discuss a potential role of this mechanism in regulating the processes of innate immunity.
Published in the journal:
. PLoS Pathog 7(6): e32767. doi:10.1371/journal.ppat.1002065
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002065
Summary
An ability to sense pathogens by a number of specialized cell types including the dendritic cells plays a central role in host's defenses. Activation of these cells through the stimulation of the pathogen-recognition receptors induces the production of a number of cytokines including Type I interferons (IFNs) that mediate the diverse mechanisms of innate immunity. Type I IFNs interact with the Type I IFN receptor, composed of IFNAR1 and IFNAR2 chains, to mount the host defense responses. However, at the same time, Type I IFNs elicit potent anti-proliferative and pro-apoptotic effects that could be detrimental for IFN-producing cells. Here, we report that the activation of p38 kinase in response to pathogen-recognition receptors stimulation results in a series of phosphorylation events within the IFNAR1 chain of the Type I IFN receptor. This phosphorylation promotes IFNAR1 ubiquitination and accelerates the proteolytic turnover of this receptor leading to an attenuation of Type I IFN signaling and the protection of activated dendritic cells from the cytotoxic effects of autocrine or paracrine Type I IFN. In this paper we discuss a potential role of this mechanism in regulating the processes of innate immunity.
Introduction
Cytokines that belong to the family of Type I interferons (IFNs, including IFNα/β), play an important role in innate immunity [1], [2], [3], [4]. These cytokines are produced by various cell types including dendritic cells (DCs), which are equipped with a diverse set of specialized pathogen recognition receptors (PRR) that recognize a variety of pathogens. Activation of PRR in DCs is known to induce the production of numerous cytokines including inflammatory cytokines and IFNα/β. The latter cytokines modulate the subsequent antigen-specific adaptive immune responses and mount the overall host defenses against infection and injury (reviewed in [5], [6], [7]).
Type I IFN mediates its effects through the stimulation of the Type I IFN receptor (which consists of IFNAR1 and IFNAR2 chains) and the subsequent activation of the Janus kinases TYK2 and JAK1, phosphorylation of STAT1/2 proteins, and ensuing trans-activation of a plethora of interferon-stimulated genes. The products of these genes directly suppress the spread of some pathogens (e.g., viruses) and cooperate with inflammatory cytokines in DCs to promote antigen presentation linking innate and adaptive immunity (reviewed in [8], [9], [10]). Some of these genes also elicit pronounced anti-proliferative and pro-apoptotic effects [10]. Conversely, cells that produce IFNα/β have to survive in this environment and have to be protected from autocrine/paracrine IFNα/β [11].
Accordingly, Type I IFNs play a dynamic role in homeostasis and in the function of DCs [12]. Whereas IFNα/β contribute to the maturation and activation of DCs [13], [14], [15], these cytokines are also known to decrease the viability of IFNα/β-producing DCs [4], [12], [16]. Additional mechanisms of negative regulation are expected to prevent a hypothetical scenario where IFNα/β-stimulated maturation of DCs and subsequent production of more of IFNα/β by these DCs spirals out of control and leads to a hyperactivation of pathways induced by Type I IFN. Such hyperactivation of the IFNα/β pathways might be detrimental, not only to a population of specific IFN-producing cells, but also to the entire host because it leads to autoimmune disorders. A key role of IFNα/β in pathogenesis of such disorders, including psoriases, systemic lupus erythematosus and Type 1 diabetes mellitus, has been thoroughly documented [17], [18], [19], [20]. Therefore, developing an understanding of the mechanisms, by which IFNα/β responses are kept in check, is of medical importance.
All responses to IFNα/β require the presence of the Type I IFN receptor at the cell surface [21]. Upon interacting with the ligands, this receptor not only conducts the forward signaling through the JAK-STAT pathway but also undergoes rapid downregulation [22], [23]. This downregulation is driven by the phosphorylation-dependent ubiquitination, endocytosis, and degradation of the IFNAR1 chain [24], [25]. Ubiquitination of IFNAR1 is facilitated by the SCFβTrcp E3 ubiquitin ligase, which is recruited to the receptor upon phosphorylation on serine residues (S535 in humans, S526 in mice) within IFNAR1's degron [25], [26]. Such phosphorylation can be induced by a ligand in a TYK2 activity-dependent manner [27] through the activation of protein kinase D2 [28]. This ligand-inducible pathway limits the extent of IFNα/β signaling in the cells that have already been exposed to these cytokines.
We have recently discovered an alternative (TYK2 activity-independent) signaling pathway that leads to S535 phosphorylation and to the subsequent ubiquitination and downregulation of IFNAR1 in the absence of a ligand. As a result, IFNAR1 is being removed from the surface of those cells, which have not been yet exposed to IFNα/β [29]. Accordingly, this signaling does not require the ligand and hence can impair the ability of a naïve cell to respond to its future encounters with IFNα/β. Such a pathway can be further stimulated by the inducers of the unfolded protein response (UPR). UPR is a complex of signaling pathways that are essential for protein quality control in cells [30], [31] and can be induced by various stress stimuli such as thapsigargin (TG) as well as by viral infection [32]. Inducers of UPR (including RNA-containing viruses such as vesicular stomatitis virus, VSV, and hepatitis C virus, HCV) were shown to promote IFNAR1 degradation via a pathway that required the activation of a PKR-like ER kinase (PERK, [33]). Subsequent delineation of the ligand-independent pathway revealed an important role for casein kinase 1α (CK1α) in the direct phosphorylation of S535/S526 within the IFNAR1 degron [34]. The ability of constitutively active CK1α to phosphorylate the IFNAR1 degron was augmented in cells treated with UPR inducers or infected with VSV via priming phosphorylation of another serine – S532 (S523 in mouse IFNAR1). The latter phosphorylation was mediated by a yet to be identified kinase that functioned downstream of PERK [35].
Here we report that the DNA-containing herpes simplex virus (HSV) also induced the ligand/TYK2-independent phosphorylation and downregulation of IFNAR1. Remarkably, these effects required neither viral protein synthesis nor PERK activity but relied on the activation of the PRR signaling pathways that center around the activation of the p38 protein kinase. This kinase was required for the phosphorylation of the IFNAR1 priming site leading to an ensuing phosphorylation of the IFNAR1 degron on Ser535/526, acceleration of IFNAR1 degradation, and attenuation of IFNα/β signaling. These events appear to be important for the protection of DCs from autocrine/paracrine Type I IFN.
Results
Herpes Simplex Virus downregulates IFNAR1 via the ligand independent pathway
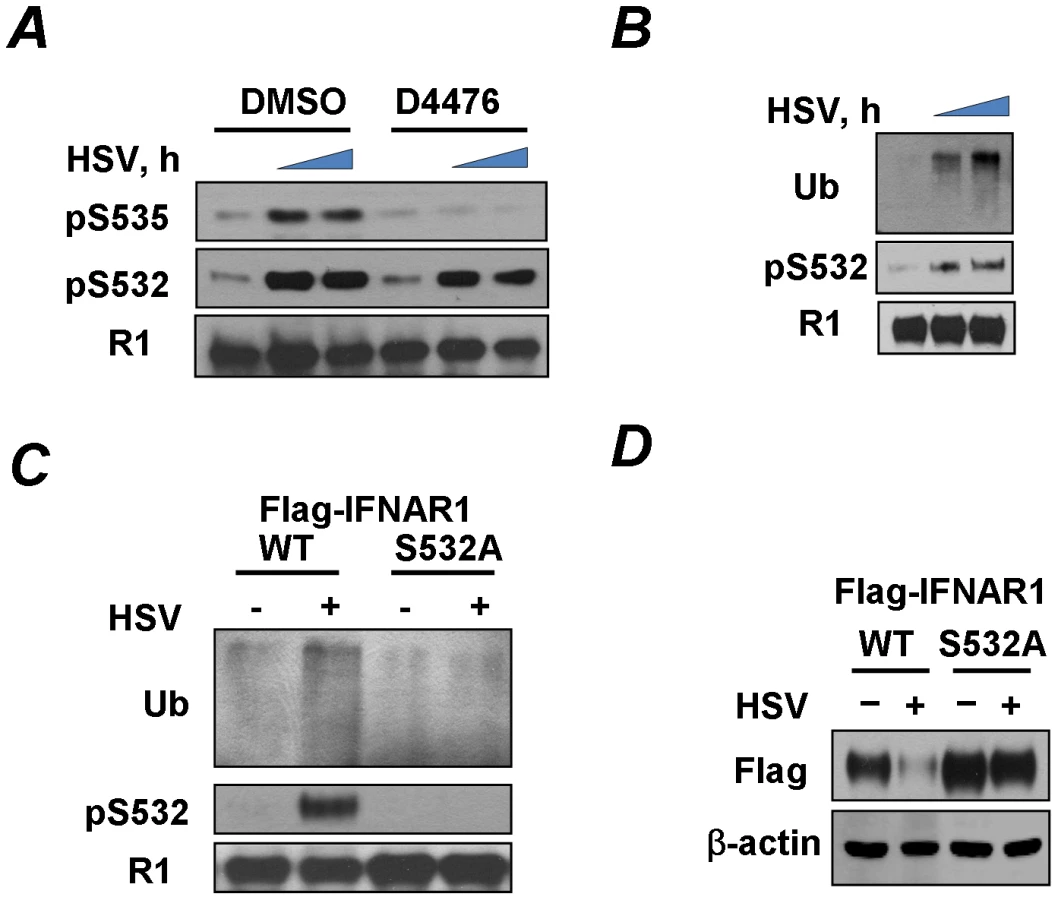
RNA-containing viruses (HCV and VSV) can downregulate IFNAR1 in human KR-2 cells that harbor a catalytically inactive TYK2 and that are deficient in IFNα-stimulated Ser535 phosphorylation of IFNAR1 and the degradation of this receptor chain [27], [33]. We sought to investigate whether a DNA-containing virus such as the herpes simplex virus (HSV) is also capable of such activity. We observed that the infection of KR-2 cells with HSV stimulates degron phosphorylation of endogenous human IFNAR1 and robustly downregulates the levels of this receptor (Figure 1A). A marked decrease in cell surface levels of murine IFNAR1 in response to HSV infection was also seen in mouse embryo fibroblasts (MEFs, Figure 1B).

To determine whether downregulation of IFNAR1 by HSV depends on IFNAR1 degron phosphorylation, we have generated a knock-in mouse that expresses a phosphorylation-deficient IFNAR1 mutant. To this end, mouse ES cells, in which one wild type Ifnar1 allele was replaced with a mutant allele that lacks Ser526 (a serine residue homologous to Ser535 within human IFNAR1; cells were described in [33]) were rid of the Neo cassette. They were then used to generate knock-in mice that express the IFNAR1S526A mutant (“SA”, Figure S1). Importantly, MEFs derived from these mice were noticeably resistant to an HSV-induced downregulation of IFNAR1 levels on the cell surface (Figure 1B). This result suggests that HSV downregulates IFNAR1 in a degron phosphorylation-dependent manner.
Given that HSV has been known to induce the production of both IFNα and IFNβ [36], we next sought to delineate the mechanisms by which HSV infection stimulates the phosphorylation of IFNAR1 degron. We chose to analyze endogenous IFNAR1 in human fibrosarcoma-derived cell lines harvested at the earlier time points (20–22 hr) of HSV infection at low MOI (0.1). Under these conditions, the total levels of IFNAR1 were not yet dramatically downregulated, enabling a better detection of Ser535 phosphorylation. HSV infection of WT-5 cells that express wild type TYK2 [37], [38] induced a robust S535 phosphorylation of IFNAR1 (Figure 1C). Comparable levels of IFNAR1 degron phosphorylation were seen in the isogenic KR-2, which express kinase-dead TYK2 and are responsive to IFNβ [38] but not to IFNα [27], [37] suggesting that the activity of TYK2 is not required for the effects of the virus. On the contrary, the phosphorylation of IFNAR1 Ser535 in response to treatment with recombinant IFNβ was much less evident in KR-2 cells that displayed only a rather modest STAT1 phosphorylation in response to this cytokine (Figure 1C). Furthermore, in either WT-5 or KR-2 cells, infection with HSV hardly induced any STAT1 phosphorylation indicating that an increase in IFNAR1 degron phosphorylation may not rely on HSV-induced production of Type I IFN. Finally, in the isogenic fibrosarcoma U5A, cells that lack the IFNAR2 chain of the Type I IFN receptor and are insensitive to any Type I IFN [39], infection with HSV robustly induced Ser535 phosphorylation of IFNAR1 (Figure 1C). These results suggest that the effects of HSV on phosphorylation of IFNAR1 are ligand-independent.
These isogenic WT-5, KR-2 and U5A cell lines were infected with HSV for longer periods of time to determine the role of TYK2 kinase activity and endogenous Type I IFN in downregulation. A comparable decrease of IFNAR1 levels in response to infection was seen in WT-5 and KR-2 cells. Furthermore, HSV robustly decreased levels of IFNAR1 in U5A cells (Figure 1D). These results indicate that neither production of the endogenous ligands nor catalytic activities of TYK2 are required for IFNAR1 downregulation in cells infected with HSV.
Given the latter results we sought to determine whether, similarly to VSV, the effects of HSV infection on IFNAR1 might be mediated by the constitutively active kinase CK1α [34] whose ability to phosphorylate IFNAR1 degron is augmented by a priming phosphorylation of IFNAR1 on S532 [35]. Phosphorylation of the degron (S535) in HSV-infected KR-2 cells was indeed attenuated by treating the cells with a CK1 inhibitor (Figure 2A). Furthermore, HSV infection induced the priming phosphorylation of IFNAR1 on S532 (Figures 1C and 2A–C) and IFNAR1 ubiquitination (Figures 2B–C). The IFNAR1S532A mutant lacking the priming site was noticeably more resistant to the HSV-stimulated ubiquitination and downregulation than the wild type receptor (Figures 2C–D). Together, these results suggest that, similar to VSV, HSV stimulates the ligand/TYK2-independent pathway of IFNAR1 phosphorylation-dependent ubiquitination and downregulation that requires the priming phosphorylation.
PERK is not required for phosphorylation and downregulation of IFNAR1 in response to HSV
The experimental settings of all these experiments included infection at low doses (MOI 0.1) that did not induce changes in the status of IFNAR1 (data not shown) until later periods of the infection (24–30 h post infection) which were chosen for the maximal expression of viral proteins to induce UPR. Under these conditions, HCV and VSV required PERK activity to promote IFNAR1 phosphorylation and degradation [33]. Although KR-2 cells display a noticeable increase in the phosphorylation of translational regulator eIF2α (a major PERK substrate) in response to thapsigargin or VSV [33], [35], stimulation of this signaling event by HSV infection was modest at best (Figure 2A). These results are consistent with previous reports that attributed low levels of eIF2α phosphorylation either to the stimulation of eIF2α de-phosphorylation by the HSV protein γ134.5 under conditions where PERK is activated [40] or to the suppression of PERK activation by the viral glycoprotein B [41]. Under the conditions used in our experiments in KR-2 cells to influence the phosphorylation of IFNAR1 degron, an induction of PERK phosphorylation (indicative of its activation) was observed in response to thapsigargin but not to infection with HSV (Figures 3B–C). Both thapsigargin and HSV infection stimulated the priming phosphorylation of IFNAR1 on Ser532 in KR-2 cells. Whereas the knockdown of PERK noticeably attenuated the effects of thapsigargin, priming phosphorylation of IFNAR1 stimulated by, HSV infection was impervious to the modulations of PERK expression (Figure 3C). This result suggests that PERK is dispensable for HSV-stimulated IFNAR1 phosphorylation in human cells.

We further tested the requirement of PERK by using MEFs from mice harboring a conditional knockout allele of PERK (PERKfl/fl) where PERK is acutely excised upon transduction with a retrovirus encoding the Cre recombinase [42]. Consistent with a previous report [33], the acute deletion of PERK prevented the downregulation of IFNAR1 upon VSV infection. However, the downregulation of IFNAR1 stimulated by HSV was not affected by the status of PERK (Figure 3D). These data suggest that HSV is capable of stimulating the ligand-independent pathway in a manner that differs from viral protein synthesis-induced UPR and activation of PERK described for HCV and VSV.
We next tested a possibility that a requirement for prolonged infection and ensuing HSV replication needed to observe the effects of low doses of HSV (MOI 0.1) might be foregone if more viruses are used initially. To this end, we compared the effects of HSV (at MOI 5.0) that was either sham treated or irradiated with UV for inactivation. The latter procedure decreased the titer of this viral preparation from 7×107 to 3 pfu. Remarkably, the treatment of KR-2 cells with a high dose of either active or inactive HSV sufficed for inducing the priming phosphorylation of IFNAR1 within 60–90 minutes (Figure 4A). Furthermore, treatment with either active or inactive HSV comparably decreased the levels of IFNAR1 (Figure 4B) indicating that the downregulation of IFNAR1 can be stimulated by HSV in a manner that does not require virus replication.

The downregulation of IFNAR1 can plausibly occur through diverse mechanisms including an increase in protein degradation and decrease in protein synthesis mediated by translational or pre-translational events (e.g., a decrease in mRNA levels). To determine the role of IFNAR1 proteolytic turnover in this process we used a standard approach of blocking the protein synthesis by treating the cells with cycloheximide. Under these conditions, the treatment of cells with inactivated HSV markedly accelerated the rate of degradation of either endogenous IFNAR1 (Figure 4C) or exogenously expressed Flag-tagged IFNAR1 (Figure 4D). Importantly, inactivated HSV did not increase the rate of proteolytic turnover for the priming site deficient IFNAR1S532A mutant (Figure 4D). Together, these results suggest that HSV rapidly initiates a specific PERK-independent signaling pathway that leads to IFNAR1 priming phosphorylation and degradation.
Pathogen receptor recognition signaling induces IFNAR1 phosphorylation and degradation
One possibility is that such a signaling pathway could be initiated by the recognition of pathogen patterns within inactivated HSV. HSV was reported to activate Toll like receptors (TLR), including TLR9, via viral genomic DNA [43], [44] as well as TLR2 via an unidentified molecular structure on the virion [45]. While we failed to detect an increase in priming or degron phosphorylation of IFNAR1 in KR-2 cells upon treatment with an activator of TLR2 muramyl dipeptide (MDP, data not shown), such phosphorylation was readily observed when KR-2 cells were treated with HSV or with TLR9 inducer CpG (Figure 5A). Whereas these results do not prove or disprove the participation of specific TLR in IFNAR1 phosphorylation mediated by HSV, they indicate a possibility that the stimulation of PRR signaling in general might lead to the same result.

To test this possibility, we aimed to determine whether other known inducers of PRR signaling were capable of stimulating priming phosphorylation of IFNAR1. To this end, we switched from KR-2 fibrosarcoma cells to the types of cells that actually function to present foreign antigens, and, accordingly, express numerous types of pathogen recognition receptors. The treatment of human monocytic U937 cells with inducers of TLR9 (CpG) or TLR4 (lipopolysaccharide, LPS) led to a robust phosphorylation of IFNAR1 on its priming site (Figures 5B, C). Furthermore, an increase in Ser532 phosphorylation of IFNAR1 in U937 cells was also seen in response to other inducers of PRR such as the TLR3 ligand poly I:C and NOD2/TLR2/TLR4 ligand MDP, and in response to high doses of inactivated VSV (Figure 5D). It is plausible that our previous studies, designed to test the effects of VSV using infection at low MOI and analyzed at the late time point of infection when the induction of UPR is at its maximum, [33] had missed this early effect.
Role of p38 kinase in pathogen receptor signaling-induced IFNAR1 phosphorylation and degradation
Signaling pathways triggered by the activation of PRR are known to induce a number of important regulatory kinases such as Jun N-terminal kinases (JNK), IκB kinases (IKK), stress-activated p38 protein kinases, and mitogen-activated Erk kinases (reviewed in [6]). The pre-treatment of U937 cells with a pharmacologic inhibitor of p38 kinase (SB203580) prevented an increase in priming phosphorylation of IFNAR1 in response to LPS. Such an effect was not observed when the JNK inhibitor SP600125 was used (Figure 5C). The inhibition of p38 kinase by SB203580 decreased the phosphorylation of S532 in response to all tested inducers of PRR signaling (Figure 5D). These results collectively implicate p38 protein kinase in mediating the priming phosphorylation of IFNAR1 in response to PRR signaling.
To further investigate the contribution of the p38 kinase, we used an in vitro assay in which S532 phosphorylation of bacterially-produced GST-IFNAR1 by cell lysates (as a source of kinase activity) was assessed by immunoblotting using a phosho-S532-specific antibody (as in [35]). Under these conditions, lysates from cells treated with UV-inactivated HSV exhibited a greater ability to phosphorylate GST-IFNAR1 on S532 in vitro than lysates from untreated cells. This activity could be tempered by adding p38 inhibitors (SB203580 or VX702) but not by adding the JNK inhibitor SP6000125 (Figure 6A). Furthermore, recombinant p38 kinase was capable of incorporating radiolabeled phosphate groups into the wild type GST-IFNAR1 protein whereas this incorporation was lower when the GST-IFNAR1S532A mutant was used as a substrate (Figure 6B). Finally, the Flag-tagged p38α kinase immunopurified from KR-2 cells was capable of phosphorylating GST-IFNAR1 on S532 in an immunokinase reaction (Figure 6C). This activity was increased when the kinase was purified from cells pre-treated with inactivated HSV. Importantly, no activity was observed when either the catalytically inactive p38AGF mutant was used as a source of kinase or when the phosphorylation-deficient GST-IFNAR1S532A mutant was used as a substrate (Figure 6C). Given that the knock-down of endogenous p38α in U937 cells by shRNA also noticeably decreased the extent of S532 phosphorylation of endogenous IFNAR1 (Figure 6D), these results collectively suggest that the p38 kinase activated by PRR signaling mediates the phosphorylation of the priming site on IFNAR1. Whereas these data indicate that p38 kinase is capable of phosphorylating Ser532, our results do not exclude the possibility that another kinase that associates with p38 and depends on p38 activation could function as a direct priming kinase.

Consistent with the importance of priming phosphorylation in the ligand-independent pathway, the treatment of U937 cells with LPS activated p38 kinase and also stimulated the phosphorylation of S535 within the degron of IFNAR1 (Figure 6E). This phosphorylation was compromised by pre-treating the cells with a p38 kinase inhibitor SB203580. An important observation to note here is that this compound did not affect the Ser535 phosphorylation stimulated by IFNα (Figure 6E). Given that IFNα is a poor inducer of priming phosphorylation and of p38 activation and is capable of stimulating the degron phosphorylation independently of the priming site ([35] and Figure 6E), it appears that the role of p38 kinase in IFNAR1 phosphorylation is largely limited to the ligand-independent pathway. Furthermore, these results (together with a rather poor induction of STAT1 phosphorylation in cells treated with LPS under conditions reported in Figure 6E) render unlikely the possibility that phosphorylation of the IFNAR1 degron in response to PRR signaling is indirectly mediated by autocrine IFNα/β.
We then investigated the effects of PRR signaling on the degradation of IFNAR1. Consistent with previous reports in other cell lines [27], [29], treatment of U937 cells with IFNα markedly increased the rate of IFNAR1 turnover (Figure 6F); a similar effect observed upon the LPS treatment. Importantly, the effects of LPS (but not of IFNα) were blocked by pre-treating the cells with a p38 kinase inhibitor (Figure 6F). These results indicate that PRR signaling specifically accelerates the degradation of IFNAR1 through the activation of p38 kinase whereas this kinase is dispensable for the stimulation of IFNAR1 proteolytic turnover by IFNα.
Pathogen recognition receptor signaling impedes cellular responses to Type I IFN
We next sought to investigate whether the acceleration of IFNAR1 degradation by PRR signaling may attenuate the cellular responses to Type I IFN. We focused on the activation of STAT1 (assessed by its tyrosine phosphorylation) in mouse bone marrow macrophages that robustly responded to exogenous murine IFNβ, and where such a response could be readily abolished if neutralizing antibodies against IFNα and IFNβ were added to the reaction (Figure 7A, lanes 1–2 and 13–14). These settings were then used to examine the effect of PRR activators on the extent of IFN signaling. To this end, we pre-treated cells with various combinations of PRR activator LPS, p38 kinase inhibitor SB203580, and IFNα/β neutralizing antibodies. We then washed off the pre-treatment agents and proceeded to treat the cells with exogenous IFNβ and detect STAT1 phosphorylation.

The pre-treatment of macrophages with LPS alone noticeably increased the basal levels of STAT1 phosphorylation (Figure 7A, lane 3 vs 1). A similar result was observed when pre-treatment with LPS also included anti-IFNα/β neutralizing antibodies (lane 9 vs. 3). This outcome suggests that the effects of LPS on STAT1 phosphorylation should not be attributed to production of endogenous Type I IFN and were likely mediated by other cytokines (for example, IFNλ that is known to be produced upon PRR stimulation [46], [47], [48]). However, the activation of STAT1 in response to the subsequent addition of exogenous IFNβ was noticeably impaired in cells pre-treated with LPS (Figure 7A, compare lanes 2 and 4). This result suggests that the activation of PRR in cells prior to their encounter with Type I IFN may temper future responses of a cell to these cytokines.
Remarkably, the suppressive effect of LPS on IFNβ signaling was partially alleviated when pre-treatment with LPS was carried out in the presence of the p38 kinase inhibitor SB203580 (Figure 7A, lane 8 vs. lane 4). Given that the effects of the p38 inhibitor were largely unaffected by adding the neutralizing antibodies to the pre-treatment combination of LPS and SB203580 (Figure 7A, lane 12 vs. lane 8), it is unlikely that the inhibitor acted through stimulating an additional production of endogenous Type I IFN. Collectively, these results suggest that PRR signaling-induced activation of p38 kinase in cells may attenuate their responses to a subsequent exposure to Type I IFN.
To further determine whether the suppression of IFN signaling by PRR inducers requires IFNAR1 phosphorylation, we used the knock-in mice harboring the IFNAR1S526A mutant, which is insensitive to downregulation in response to HSV infection (Figure 1B). Whereas pre-treatment with LPS dramatically inhibited IFNβ-induced STAT1 phosphorylation in bone marrow macrophages from wild type mice, this inhibition was not seen in cells that express the non-degradable IFNAR1 mutant (Figure 7B). These data collectively suggest that p38 kinase activity-dependent phosphorylation and the downregulation of IFNAR1 in response to PRR signaling decrease the extent of cellular responses to Type I IFN.
Intriguingly, LPS was reported to promote the maturation of human DCs of monocytic origin, a process during which the downregulation of IFNAR1 and the entire Type I IFN receptor had been previously reported [4], [49], [50]. Autocrine/paracrine Type I IFN produced by DCs not only plays an important role in their function but also exerts pro-apoptotic effects on DCs themselves [12], [16]. Given that suppression of cell viability elicited by some of Type I IFN species are most prominent in cells that express high levels of receptor chains [51], it is plausible that IFNAR1 downregulation may help DCs to survive under exposure to their own IFNα/β. To test this hypothesis, we assessed the viability of mouse bone marrow-derived DCs activated by LPS. Treatment with a p38 kinase inhibitor significantly decreased the viability of these cells (Figures 7C). Remarkably, this effect was less evident in DCs that were either derived from IFNAR1-null mice (Figure 7C) or treated with the IFNAR1-neutralizing antibody (Figure 7D) indicating that the activation of p38 kinase may be important for protecting DCs from detrimental effects of Type I IFN. Furthermore, whereas the percent of viable LPS-activated BMDC from the IFNAR1S526A knock-in mice was relatively low (10.7±2.0%), it could be noticeably increased by incubating these cells with the IFNAR1-neutralizing antibody (20.3±3.4%, p<0.01). Similar data were obtained when Annexin V-negative CD11c-expressing cells were analyzed (Figure S2). Taken together, these data suggest that PRR-stimulated p38 kinase-dependent degradation of IFNAR1 results in protection of activated DCs from the detrimental effects of autocrine/paracrine IFNα/β.
Discussion
We have previously reported that the induction of UPR activates a ligand/JAK-independent signaling pathway that leads to phosphorylation, ubiquitination, and degradation of IFNAR1. This signaling involves stimulation of PERK-dependent priming phosphorylation of IFNAR1 followed by its degron phosphorylation by CK1α. This pathway, which can be activated in response to VSV or HCV infection, plays an important role in regulating the levels of IFNAR1 in naïve cells and in determining the sensitivity of cells to the future exposure to Type I IFN [29], [33], [34], [35].
In the present study, we investigated whether HSV infection also negatively affects IFNAR1 stability and signaling. We found that ligand/TYK2-independent phosphorylation and downregulation of IFNAR1 can indeed be observed in cells infected with HSV (Figure 1). Similar to VSV [35], HSV infection induced the priming phosphorylation of IFNAR1; and this phosphorylation was required for IFNAR1 ubiquitination and downregulation (Figure 2). However, when we next investigated the role of PERK in these processes, the differences between HSV and VSV became apparent. Unlike VSV, HSV infection caused little (if any) increase in the phosphorylation of the major PERK substrate, translational regulator eIF2α (Figure 3A). Although available literature suggests that some of this effect could be attributed to the action of phosphatases directed by the HSV protein γ134.5 [40], our studies together with another report [41] indicated a deficient activation of PERK in cells infected by HSV (Figures 3B–C). Furthermore, genetic experiments using PERK knockdown in human cells and PERK knockout in mouse cells clearly demonstrated that PERK is dispensable for IFNAR1 phosphorylation and downregulation by HSV (Figure 3C–D). Given that high doses of inactivated HSV also stimulated IFNAR1 phosphorylation, downregulation, and degradation (Figure 4), we proposed that there is a novel branch of the ligand-independent pathway. We hypothesized that this signaling branch could be induced by pathogen recognition receptors.
Once this initial hypothesis received support from experiments that used canonical selective activators of PRR signaling (Figure 5), we changed the focus of our study to follow the effects of this signaling. Accordingly, we modified the experimental design and switched to using these canonical activators (due to their commercial availability and better reproducibility over preparations of inactivated virus) and to cell models that specifically reflected the function of pathogenic patterns recognition and ensuing reactions of innate immunity. Our subsequent studies demonstrated that, even in the absence of viral infection, the activation of PRR signaling robustly induces priming phosphorylation and the degradation of IFNAR1 in a manner that requires the activation of p38 kinase (Figures 5–6). Furthermore, p38 kinase-dependent phosphorylation of IFNAR1 leads to IFNAR1 degradation and, accordingly, tempers the responses of cells to their future encounters to IFNα/β (Figures 6–7).
The role of the p38 kinase in these processes is intriguing. On one hand, the results of pharmacologic (Figure 5D) and genetic (Figure 6D) studies implicate p38 kinase in the PRR-induced downregulation of IFNAR1. Furthermore, our biochemical data suggest that p38 kinase is capable of directly phosphorylating the priming site on IFNAR1 in vitro (Figure 4A–C). However, given a known preference of this kinase for proline-directed Ser and Thr residues as phospho-acceptor sites [52] and the fact that the priming site on IFNAR1 does not conform to these criteria, it is plausible that the direct phosphorylation of Ser532 in cells might be carried out by a SB203580-sensitive kinase that associates with p38 kinase and depends on p38 activation.
On another hand, p38 kinase can be also activated by Type I IFN in several types of cells [53], [54], [55]. In fact, within cells that have already encountered it, IFNα/β, p38 kinase activity is proven to contribute to the maximal extent of the IFN-induced transcriptional program [10], [56], [57]. Yet it appears that the ligand-induced phosphorylation and degradation of IFNAR1 does not depend on p38 kinase activity (Figure 6E–F). Indeed, our recent study identified protein kinase D2 as a key TYK2-dependent IFN-inducible kinase that mediates the ligand-stimulated IFNAR1 phosphorylation, ubiquitination, endocytosis and degradation [28]. Furthermore, it appears that the activation of p38 kinase in cells that have not been yet exposed to IFNα/β may temper future sensitivity to these cytokines through an elimination of the receptor (Figure 7E).
Collectively, these studies describe a novel link between an activation of innate immune responses that usually govern production of Type I IFN, with modulation of the extent of cellular responses elicited by these cytokines (Figure 7E). It is plausible that the temporal downregulation of IFNAR1 that precedes or coincides with the peak of IFNα/β synthesis could be important for various aspects of the host defenses. These aspects may include the maintenance of the viability of IFN-producing cells, limiting the extent of IFNα/β pathway, and affecting the sensitivity of the host to the secondary infection.
Among cell types capable of producing IFNα/β, the dendritic cells (DCs) are distinguished with an advanced ability to recognize a variety of pathogenic patterns and, upon this activation, synthesize and secrete Type I IFNs and other cytokines that participate in shaping the immune responses [6], [58]. Activated DCs that produce IFNα/β have to be protected from the detrimental effects of autocrine IFN [4], [12], [16]. Indeed, it has been shown that activated DCs are prone to apoptosis, the extent of which is decreased in cells from IFNAR1-null animals [12], [16]. It has also been demonstrated that, upon their maturation, DCs downregulate Type I IFN receptor [49], [50] though the mechanism underlying this downregulation or its role in DC maturation and survival remain unclear. In this study, we demonstrated that PRR-stimulated p38 kinase-dependent degradation of IFNAR1 leads to an attenuation of Type I IFN signaling and ameliorates its negative effects in DCs (Figure 7). These data suggest that the activation of the signaling pathway that involves phosphorylation and degradation of IFNAR1 may render DCs (and perhaps other IFNα/β-producing cells) refractory to their own Type I IFN.
As indicated by our data (presented in Figure 7), such refractoriness may improve the viability of DCs. In addition, the mechanism described here may plausibly contribute to discontinuing an otherwise potentially harmful cycle of DCs being activated by Type I IFN followed by the production of more of these cytokines and a subsequent greater activation of DCs. Given the well documented role of IFNα/β in the pathogenesis of autoimmune disorders (including systemic lupus erythematosus, psoriasis, and Type 1 diabetes mellitus [17], [18], [19], [20]), temporal downregulation of IFNAR1 might play an important role in protecting the host from such autoimmune reactions.
Whereas the latter outcomes of IFNAR1 degradation stimulated by signaling induced by pathogenic patterns may benefit the host, it could also decrease the ability of some cell types exposed to PRR inducers to mount an appropriate anti-viral response. It remains to be seen whether the suppression of Type I IFN signaling by chronic exposure to other pathogens in other cell types may play a role in the development of secondary viral infections, which have been linked to insufficient IFNα/β function [59]. Future studies in vivo aimed at a further delineation of the mechanisms of IFNAR1 degradation and its role in sensitivity to secondary infections and autoimmune disorders are consequently warranted.
Materials and Methods
Plasmids and reagents
Recombinant GST-p38α was purchased (Cell Signaling). Vectors for bacterial expression of GST-IFNAR1 and mammalian expression of human and murine Flag-IFNAR1 were described previously [25], [33], [35]. The plasmids for expression of Flag-p38 (wild type or catalytically inactive AGF) were a generous gift from R. J. Davis. ShRNA constructs for knocking down p38α kinase were from Sigma. Constructs for knock down of PERK were previously described [33]. Recombinant human IFNα2 (Roferon) was from Roche. Recombinant murine IFNβ and mouse IFNα/β neutralizing antibodies were from PBL. LPS, poly IC, MDP and CpG were from InVivogen. Neutralizing antibody against mouse IFNAR1 (MAR) was from Leinco. P38 inhibitor SB203580 and JNK inhibitor SP600125 were from EMD Biosciences. P38 inhibitor VX-702 was from ChemTek. CK1 inhibitor D4476 was from Tocris. All other reagents were from Sigma.
Viruses
VSV (Indiana serotype, a gift from R. Harty) and HSV-1 (KOS strain, a gift from G. Cohen and R. Eisenberg) were propagated in HeLa cells. These cells were also used to determine the viral titers in serially diluted stocks using methylcellulose method. In experiments requiring inactivated virus, virus suspension was placed in Petri dishes and exposed either to UV-C light (254 nm) for 5 min to achieve the total dose of 1500 J/m2 or sham treated for the same time period.
Cells and gene delivery
Human monocytic U937 and HeLa cells were from ATTC. Replication-deficient lentiviral particles encoding shRNA against GFP (shCON), PERK, p38, or the empty virus control were prepared via co-transfecting 293T cells with three other helper vectors as described previously [33], [35]. Viral supernatants were concentrated by PEG8000 precipitation and were used to infect U937 cells in the presence of polybrene (4 µg/mL, Sigma). Cells were selected and maintained in the presence of 1 µg/mL of puromycin. Human fibrosarcoma 2fTGH-derived IFNAR2-null U5A [39] cells were kindly provided by G. Stark; the isogenic derivatives of TYK2-null 11.1 cells including the KR-2 cells that harbor catalytically inactive TYK2 or WT-5 cells that re-express wild type TYK2 [27] were a generous gift from S. Pellegrini.
Ethics statement and animals
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Pennsylvania (protocol # 800992). Every effort was made to minimize animal suffering. IFNAR1-null mice were kindly provided by D.E. Zhang (UCSD). Bone marrow-derived macrophages (BMM) and bone marrow-derived dendritic cells (BMDC) were produced as described previously [12].
Immunotechniques and cell viability
These assays were carried out as described previously [34]. Monoclonal antibodies against human IFNAR1 that were used for immunoprecipitation (EA12) or immunoblotting (GB8) were described in detail elsewhere [60]. Antibodies against PERK [33] were kindly provided by J.A. Diehl. Antibodies against p-STAT1, p-p38 (Cell Signaling), phospho-Ser532, phospho-Ser535 (or phospho-Ser523, phospho-Ser526, respectively, in the murine receptor) [26], [35], murine IFNAR1 (LeinCo), STAT1, p38, phospho-PERK (Santa Cruz), Flag, β-actin (Sigma) and ubiquitin (clone FK2, Biomol) were used for immunoprecipitation (IP) and immunoblotting (IB) as described previously [26], [35]. Cell viability assays were analyzed by FACS (BD Calibur) to calculate CD11c positive and propidium iodide negative cell population as described previously [12], [33].
In vitro kinase assays
Kinase assays were carried out as described previously [29], [34]. In brief, p38 was immunoprecipitated and the immunoprecipitates were incubated with 1 µg of substrates (bacterially-expressed and purified wild type or S532A GST-IFNAR1 mutant) in kinase buffer (25 mM Tris-HCl pH 7.4, 10 mM MgCl2, 1 mM NaF, 1 mM NaVO3) and ATP (1 mM) at 30°C for 30 min. Samples were then separated by 10% SDS-PAGE and analyzed by immunoblotting with phospho-specific antibodies.
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. O'NeillLABowieAG 2010 Sensing and signaling in antiviral innate immunity. Curr Biol 20 R328 333
2. BlasiusALBeutlerB 2010 Intracellular toll-like receptors. Immunity 32 305 315
3. TakeuchiOAkiraS 2009 Innate immunity to virus infection. Immunol Rev 227 75 86
4. CocciaEM 2008 IFN regulation and functions in myeloid dendritic cells. Cytokine Growth Factor Rev 19 21 32
5. IwasakiAMedzhitovR 2010 Regulation of adaptive immunity by the innate immune system. Science 327 291 295
6. BrodskyIEMedzhitovR 2009 Targeting of immune signalling networks by bacterial pathogens. Nat Cell Biol 11 521 526
7. PalmNWMedzhitovR 2009 Pattern recognition receptors and control of adaptive immunity. Immunol Rev 227 221 233
8. StarkGR 2007 How cells respond to interferons revisited: from early history to current complexity. Cytokine Growth Factor Rev 18 419 423
9. AaronsonDSHorvathCM 2002 A road map for those who don't know JAK-STAT. Science 296 1653 1655
10. PlataniasLC 2005 Mechanisms of type-I - and type-II-interferon-mediated signalling. Nat Rev Immunol 5 375 386
11. HasanUACauxCPerrotIDoffinACMenetrier-CauxC 2007 Cell proliferation and survival induced by Toll-like receptors is antagonized by type I IFNs. Proc Natl Acad Sci U S A 104 8047 8052
12. YenJHGaneaD 2009 Interferon beta induces mature dendritic cell apoptosis through caspase-11/caspase-3 activation. Blood 114 1344 1354
13. LonghiMPTrumpfhellerCIdoyagaJCaskeyMMatosI 2009 Dendritic cells require a systemic type I interferon response to mature and induce CD4+ Th1 immunity with poly IC as adjuvant. J Exp Med 206 1589 1602
14. LongmanRSBraunDPellegriniSRiceCMDarnellRB 2007 Dendritic-cell maturation alters intracellular signaling networks, enabling differential effects of IFN-alpha/beta on antigen cross-presentation. Blood 109 1113 1122
15. GaoYMajchrzak-KitaBFishENGommermanJL 2009 Dynamic accumulation of plasmacytoid dendritic cells in lymph nodes is regulated by interferon-beta. Blood 114 2623 2631
16. MatteiFBracciLToughDFBelardelliFSchiavoniG 2009 Type I IFN regulate DC turnover in vivo. Eur J Immunol 39 1807 1818
17. Delgado-VegaAMAlarcon-RiquelmeMEKozyrevSV 2010 Genetic associations in type I interferon related pathways with autoimmunity. Arthritis Res Ther 12 Suppl 1 S2
18. HallJCRosenA 2010 Type I interferons: crucial participants in disease amplification in autoimmunity. Nat Rev Rheumatol 6 40 49
19. KunzMIbrahimSM 2009 Cytokines and cytokine profiles in human autoimmune diseases and animal models of autoimmunity. Mediators Inflamm 2009 979258
20. FinkeDElorantaMLRonnblomL 2009 Endogenous type I interferon inducers in autoimmune diseases. Autoimmunity 42 349 352
21. UzeGSchreiberGPiehlerJPellegriniS 2007 The receptor of the type I interferon family. Curr Top Microbiol Immunol 316 71 95
22. CocciaEMUzeGPellegriniS 2006 Negative regulation of type I interferon signaling: facts and mechanisms. Cell Mol Biol (Noisy-le-grand) 52 77 87
23. HuangfuWCFuchsSY 2010 Ubiquitination-dependent regulation of signaling receptors in cancer. Genes Cancer 1 725 734
24. KumarKGBarriereHCarboneCJLiuJSwaminathanG 2007 Site-specific ubiquitination exposes a linear motif to promote interferon-alpha receptor endocytosis. J Cell Biol 179 935 950
25. KumarKGTangWRavindranathAKClarkWACrozeE 2003 SCF(HOS) ubiquitin ligase mediates the ligand-induced down-regulation of the interferon-alpha receptor. EMBO J 22 5480 5490
26. KumarKGKrolewskiJJFuchsSY 2004 Phosphorylation and specific ubiquitin acceptor sites are required for ubiquitination and degradation of the IFNAR1 subunit of type I interferon receptor. J Biol Chem 279 46614 46620
27. MarijanovicZRagimbeauJKumarKGFuchsSYPellegriniS 2006 TYK2 activity promotes ligand-induced IFNAR1 proteolysis. Biochem J 397 31 38
28. ZhengHQianJVargheseBBakerDPFuchsS 2011 Ligand-stimulated downregulation of the alpha interferon receptor: role of protein kinase d2. Mol Cell Biol 31 710 720
29. LiuJPlotnikovABanerjeeASuresh KumarKGRagimbeauJ 2008 Ligand-independent pathway that controls stability of interferon alpha receptor. Biochem Biophys Res Commun 367 388 393
30. WekRCCavenerDR 2007 Translational control and the unfolded protein response. Antioxid Redox Signal 9 2357 2371
31. RonDWalterP 2007 Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8 519 529
32. HeB 2006 Viruses, endoplasmic reticulum stress, and interferon responses. Cell Death Differ 13 393 403
33. LiuJHuangFuWCKumarKGQianJCaseyJP 2009 Virus-induced unfolded protein response attenuates antiviral defenses via phosphorylation-dependent degradation of the type I interferon receptor. Cell Host Microbe 5 72 83
34. LiuJCarvalhoLPBhattacharyaSCarboneCJKumarKG 2009 Mammalian casein kinase 1alpha and its leishmanial ortholog regulate stability of IFNAR1 and type I interferon signaling. Mol Cell Biol 29 6401 6412
35. BhattacharyaSHuangFuWCLiuJVeerankiSBakerDP 2010 Inducible priming phosphorylation promotes ligand-independent degradation of the IFNAR1 chain of type I interferon receptor. J Biol Chem 285 2318 2325
36. KirchnerHEnglerHSchroderCHZawatzkyRStorchE 1983 Herpes simplex virus type 1-induced interferon production and activation of natural killer cells in mice. J Gen Virol 64 Pt 2 437 441
37. GauzziMCVelazquezLMcKendryRMogensenKEFellousM 1996 Interferon-alpha-dependent activation of Tyk2 requires phosphorylation of positive regulatory tyrosines by another kinase. J Biol Chem 271 20494 20500
38. RaniMRGauzziCPellegriniSFishENWeiT 1999 Induction of beta-R1/I-TAC by interferon-beta requires catalytically active TYK2. J Biol Chem 274 1891 1897
39. LutfallaGHollandSJCinatoEMonneronDReboulJ 1995 Mutant U5A cells are complemented by an interferon-alpha beta receptor subunit generated by alternative processing of a new member of a cytokine receptor gene cluster. EMBO J 14 5100 5108
40. ChengGFengZHeB 2005 Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2alpha dephosphorylation by the gamma(1)34.5 protein. J Virol 79 1379 1388
41. MulveyMAriasCMohrI 2007 Maintenance of endoplasmic reticulum (ER) homeostasis in herpes simplex virus type 1-infected cells through the association of a viral glycoprotein with PERK, a cellular ER stress sensor. J Virol 81 3377 3390
42. ZhangPMcGrathBLiSFrankAZambitoF 2002 The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol 22 3864 3874
43. LundJSatoAAkiraSMedzhitovRIwasakiA 2003 Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med 198 513 520
44. KrugALukerGDBarchetWLeibDAAkiraS 2004 Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 103 1433 1437
45. Kurt-JonesEAChanMZhouSWangJReedG 2004 Herpes simplex virus 1 interaction with Toll-like receptor 2 contributes to lethal encephalitis. Proc Natl Acad Sci U S A 101 1315 1320
46. YangKPuelAZhangSEidenschenkCKuCL 2005 Human TLR-7-, -8-, and -9-mediated induction of IFN-alpha/beta and -lambda Is IRAK-4 dependent and redundant for protective immunity to viruses. Immunity 23 465 478
47. OsterlundPIPietilaTEVeckmanVKotenkoSVJulkunenI 2007 IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-lambda) genes. J Immunol 179 3434 3442
48. CocciaEMSeveraMGiacominiEMonneronDRemoliME 2004 Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol 34 796 805
49. SeveraMRemoliMEGiacominiERagimbeauJLandeR 2006 Differential responsiveness to IFN-alpha and IFN-beta of human mature DC through modulation of IFNAR expression. J Leukoc Biol 79 1286 1294
50. GauzziMCCaniniIEidPBelardelliFGessaniS 2002 Loss of type I IFN receptors and impaired IFN responsiveness during terminal maturation of monocyte-derived human dendritic cells. J Immunol 169 3038 3045
51. MoragaIHarariDSchreiberGUzeGPellegriniS 2009 Receptor density is key to the alpha2/beta interferon differential activities. Mol Cell Biol 29 4778 4787
52. RouxPPBlenisJ 2004 ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68 320 344
53. KatsoulidisELiYMearsHPlataniasLC 2005 The p38 mitogen-activated protein kinase pathway in interferon signal transduction. J Interferon Cytokine Res 25 749 756
54. PlataniasLC 2003 The p38 mitogen-activated protein kinase pathway and its role in interferon signaling. Pharmacol Ther 98 129 142
55. UddinSMajchrzakBWoodsonJArunkumarPAlsayedY 1999 Activation of the p38 mitogen-activated protein kinase by type I interferons. J Biol Chem 274 30127 30131
56. VermaADebDKSassanoAKambhampatiSWickremaA 2002 Cutting edge: activation of the p38 mitogen-activated protein kinase signaling pathway mediates cytokine-induced hemopoietic suppression in aplastic anemia. J Immunol 168 5984 5988
57. UddinSLekmineFSharmaNMajchrzakBMayerI 2000 The Rac1/p38 mitogen-activated protein kinase pathway is required for interferon alpha-dependent transcriptional activation but not serine phosphorylation of Stat proteins. J Biol Chem 275 27634 27640
58. SoloffACBarratt-BoyesSM 2010 Enemy at the gates: dendritic cells and immunity to mucosal pathogens. Cell Res 20 872 885
59. AlsharifiMMullbacherARegnerM 2008 Interferon type I responses in primary and secondary infections. Immunol Cell Biol 86 239 245
60. GoldmanLAZafariMCutroneECDangABrickelmeierM 1999 Characterization of antihuman IFNAR-1 monoclonal antibodies: epitope localization and functional analysis. J Interferon Cytokine Res 19 15 26
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 6
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- High Affinity Nanobodies against the VSG Are Potent Trypanolytic Agents that Block Endocytosis
- Structural and Mechanistic Studies of Measles Virus Illuminate Paramyxovirus Entry
- Sporangiospore Size Dimorphism Is Linked to Virulence of
- The Binding of Triclosan to SmeT, the Repressor of the Multidrug Efflux Pump SmeDEF, Induces Antibiotic Resistance in
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy