
Evolution of Multidrug Resistance during Infection Involves Mutation of the Essential Two Component Regulator WalKR
Antimicrobial resistance in Staphylococcus aureus is a major public health threat, compounded by emergence of strains with resistance to vancomycin and daptomycin, both last line antimicrobials. Here we have performed high throughput DNA sequencing and comparative genomics for five clinical pairs of vancomycin-susceptible (VSSA) and vancomycin-intermediate ST239 S. aureus (VISA); each pair isolated before and after vancomycin treatment failure. These comparisons revealed a frequent pattern of mutation among the VISA strains within the essential walKR two-component regulatory locus involved in control of cell wall metabolism. We then conducted bi-directional allelic exchange experiments in our clinical VSSA and VISA strains and showed that single nucleotide substitutions within either walK or walR lead to co-resistance to vancomycin and daptomycin, and caused the typical cell wall thickening observed in resistant clinical isolates. Ion Torrent genome sequencing confirmed no additional regulatory mutations had been introduced into either the walR or walK VISA mutants during the allelic exchange process. However, two potential compensatory mutations were detected within putative transport genes for the walK mutant. The minimal genetic changes in either walK or walR also attenuated virulence, reduced biofilm formation, and led to consistent transcriptional changes that suggest an important role for this regulator in control of central metabolism. This study highlights the dramatic impacts of single mutations that arise during persistent S. aureus infections and demonstrates the role played by walKR to increase drug resistance, control metabolism and alter the virulence potential of this pathogen.
Published in the journal:
. PLoS Pathog 7(11): e32767. doi:10.1371/journal.ppat.1002359
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002359
Summary
Antimicrobial resistance in Staphylococcus aureus is a major public health threat, compounded by emergence of strains with resistance to vancomycin and daptomycin, both last line antimicrobials. Here we have performed high throughput DNA sequencing and comparative genomics for five clinical pairs of vancomycin-susceptible (VSSA) and vancomycin-intermediate ST239 S. aureus (VISA); each pair isolated before and after vancomycin treatment failure. These comparisons revealed a frequent pattern of mutation among the VISA strains within the essential walKR two-component regulatory locus involved in control of cell wall metabolism. We then conducted bi-directional allelic exchange experiments in our clinical VSSA and VISA strains and showed that single nucleotide substitutions within either walK or walR lead to co-resistance to vancomycin and daptomycin, and caused the typical cell wall thickening observed in resistant clinical isolates. Ion Torrent genome sequencing confirmed no additional regulatory mutations had been introduced into either the walR or walK VISA mutants during the allelic exchange process. However, two potential compensatory mutations were detected within putative transport genes for the walK mutant. The minimal genetic changes in either walK or walR also attenuated virulence, reduced biofilm formation, and led to consistent transcriptional changes that suggest an important role for this regulator in control of central metabolism. This study highlights the dramatic impacts of single mutations that arise during persistent S. aureus infections and demonstrates the role played by walKR to increase drug resistance, control metabolism and alter the virulence potential of this pathogen.
Introduction
In hospitals worldwide infections with methicillin-resistant S. aureus (MRSA) remain a significant cause of morbidity and mortality, with a small number of clones accounting for a large number of hospital acquired infections. In Australasia, multi-locus sequence type (MLST) 239 (ST239) is the major hospital acquired clone of MRSA, and has been present in the region for over 30 years. This clone is resistant to almost all antibiotic classes; therefore the mainstay of therapy for serious MRSA infections has been the glycopeptide antibiotic vancomycin. However, resistant strains have recently emerged [1], and although the level of resistance is low there is an impact on treatment outcome [2]. These vancomycin-intermediate S. aureus (VISA, vancomycin MIC 4–8 µg/ml) and heterogenous-VISA (hVISA, vancomycin MIC ≤2 µg/ml with a “resistant subpopulation”) strains are increasingly common, however the genetics of resistance are incompletely defined [3]. While the emergence of VISA in Australia has been in strains of the ST239 clone [4], the first VISA strain Mu50 was reported from Japan in 1997 [5], and a number of other reported VISA strains belong to the same clonal complex as Mu50 (CC5) [6]–[8].
In many cases VISA emerge from fully-vancomycin susceptible S. aureus (VSSA) parental strains during persistent infection [6], [8], [9], and in some cases this has been associated with the evolution of daptomycin non-susceptibility despite the absence of exposure to daptomycin [10]. VISA strains appear to arise via sequential point mutations in key staphylococcal regulatory genes [11]–[13], however the breadth of mutations that can contribute to resistance are poorly defined. In addition, it is not clear if there are differences in resistance mechanisms and pathways to VISA in different clones of S. aureus. Commonly described phenotypic changes in VISA compared to VSSA include increased cell wall thickness and reduced autolytic activity [7], [14], [15], in addition to other significant phenotypic changes that are predicted to impact the virulence of the organism. These include a reduction in biofilm formation, reduced activity of the agr quorum sensing system, and enhanced capsule production [3], [4], [15], [16]. The link(s) between development of antimicrobial resistance and the regulation of these virulence factors is unknown.
A number of studies have used comparative genomics of paired S. aureus isolates to detect mutations that occur in the resistant strain compared to the parent strain, including a landmark study by Mwangi et al where increasing vancomycin resistance in sequential clinical isolates of S. aureus were linked to accumulated mutations in the increasingly resistant strain [11]. However, the genetic loci where mutations in clinical S. aureus strains have been experimentally confirmed using allelic replacement experiments to contribute to VISA are limited to vraSR, graRS, and more recently rpoB [13], [17], [18]. We have previously used functional genomics to show that a point mutation in graS can lead to reduced vancomycin susceptibility in one clinical pair of ST239 VSSA (JKD6009) and VISA (JKD6008) [18]. However this mutation, while leading to a reduction in vancomycin susceptibility, did not restore the full VISA resistance profile. It is worth noting that all these studies have focussed on a total of three clinical and laboratory induced VISA isolates, and screening of additional ST239 VISA strains has failed to demonstrate that mutations in these loci are common to other VISA [4], suggesting that there are mutations in as yet undefined loci contributing to VISA in other clinical isolates.
Daptomycin is an antibiotic that exerts its effect at the cell membrane, and while a link between VISA and daptomycin non-susceptibility has been demonstrated [10], [19], the genetics of this relationship are undefined. While mutations in mprF, rpoB and rpoC are thought to be the genetic basis for daptomycin non-susceptibility in S. aureus, mutations have also been detected in walK [20]. The contribution of the walK mutations to daptomycin non-susceptibility has not been defined as these strains also harboured mprF mutations.
To expand understanding of the mechanisms of VISA and identify other loci contributing to vancomycin resistance we fully sequenced four additional clinical pairs of VSSA and VISA, and then compared them to the fully assembled and annotated genome of our previously described VISA strain JKD6008 [21]. We also re-analyzed our original sequenced pair (JKD6008 and JKD6009) after fully assembling the genome sequence of the VISA strain JKD6008. Using this approach, followed by allelic replacement experiments, we show that WalKR (also known as YycGF and VicKR) plays a major role during the in vivo evolution of extensive drug resistance in clinical S. aureus. Our findings highlight an unexpected degree of plasticity within this essential two-component regulator and are particularly pertinent, with inhibitors of WalK recently proposed as novel anti-staphylococcal agents [22]–[24].
Results
Isolate Selection and Characteristics
To identify the genetic mechanisms leading to VISA five clinical pairs of VSSA and VISA were selected, where the resistant strain evolved from the susceptible parent strain during failed vancomycin therapy [15], [18]. In addition, we also examined eight global non-paired hVISA and VISA (Table 1). The clinical pairs were selected from patients that had been treated with vancomycin, and not daptomycin. This meant that any changes in daptomycin susceptibility linked to vancomycin exposure and increasing vancomycin resistance could be specifically assessed. The duration of in vivo vancomycin exposure ranged from 8 to 42 days in these clinical isolate pairs and the majority of isolates fulfilled the criteria for VISA (vancomycin broth MIC 4–8 µg per ml). For all VISA strains in the isolate pairs an increase in daptomycin MIC was seen compared to the parental VSSA strain, and five of the 13 hVISA/VISA isolates were daptomycin non-susceptible (Table 1). All of the clinical strains were typed using the StaphyType96 DNA Array (CLONDIAG, Jena, Germany) to predict the sequence type, mec type, and agr type (Table 1). All of the clinical pairs were from Australia and New Zealand, and were ST239 MRSA, the dominant hospital clone of MRSA in the region, while the additional isolates represented a range of staphylococcal sequence types within clonal complexes 5 and 8.
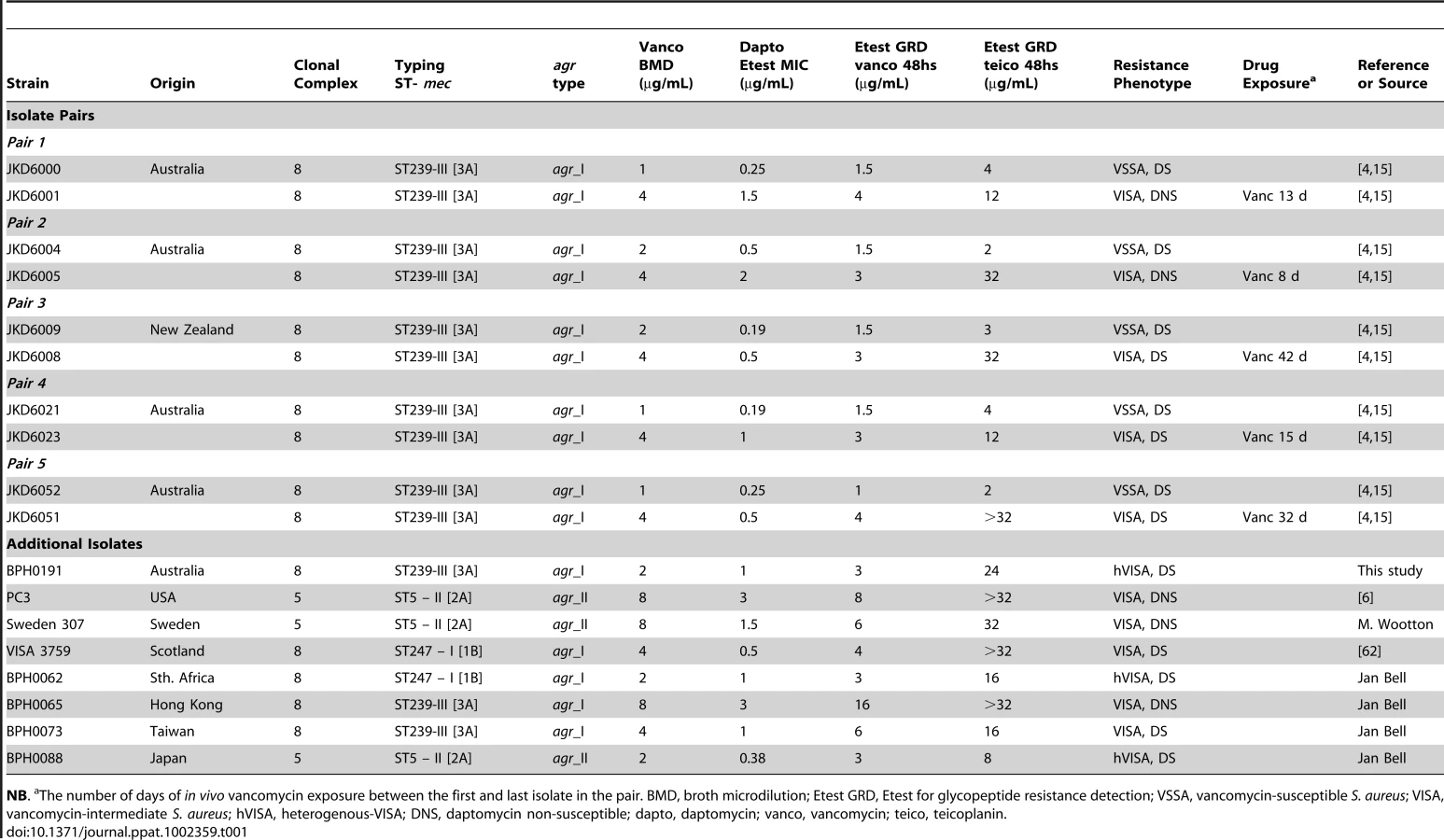
Genome Comparisons Highlight Mutations in walKR Associated with Reduced Vancomycin and Daptomycin Susceptibility in S. aureus
In an earlier study of the VSSA/VISA pair JKD6009/JKD6008 we compared partially assembled 454 GS20 sequences and found six nucleotide substitutions in JKD6008 [18]. We then showed that a mutation occurring in the sensor region of the graS gene partly explained the reduced vancomycin susceptibility of this strain. To now comprehensively address the question of mutations that contribute to VISA we used our recently completed JKD6008 reference genome [21] which we have shown is closely related to other Australasian ST239 strains (unpublished data), and is therefore an appropriate reference genome for analysis, and used our read-mapping technique to re-examine the genetic differences between JKD6008 and JKD6009 as well as comparing four other clinical VSSA/VISA pairs (Table 1, strain pairs 1, 2, 4 and 5). Using either SOLiD or Illumina technologies, high coverage short-read sequences were obtained for the clinical pairs with high mean fold coverage (JKD6009 [SOLiD] mean fold coverage 338.7x; other strains [Illumina] mean fold coverage 85.5 to 230.6x). The list of differences between JKD6009 and JKD6008 increased from six to 10, but each pair presented a limited list of mutations in the VISA strain compared to its VSSA parent (Table 2). The most interesting observation was the presence of a previously undetected SNP in the walK gene of JKD6008. Strikingly, three of the four other clinical pairs also had single mutations within the walKR locus (Table 2). In one pair (JKD6004/JKD6005), the only mutation identified was a single SNP in walR of VISA strain JKD6005. JKD6051 was the only VISA strain among the five sequenced pairs without a walKR mutation. Mutations of potential interest in this strain included a mutation in the regulator SarR (A68T), and a mutation in RpoB (H481Y).

Frequency and Role of walKR Mutations in Reduced Vancomycin and Daptomycin Susceptibility in S. aureus
Given the previous reports of diverse genetic pathways involved in VISA we were surprised to find a single locus that was mutated in four of our five pairs of sequenced strains. To extend this analysis the walKR locus was sequenced from 8 additional, unpaired global isolates of hVISA/VISA that were available for study (Table 3). This demonstrated that, in addition to the four VISA strains in the genome sequencing analysis, 6 of the 8 additional resistant strains also had a mutation in the walKR locus. The downstream genes yycHIJ are involved in repression of walR in B. subtilis [25], therefore we sequenced yycHIJ for the two strains where a mutation in walKR was not detected but no mutations were found (Table 3).

Analysis of the positions of the mutations within the walKR genes in this study indicates that they are not limited to a specific domain or region (Figure 1A). Indeed, the mutations occur across the spectrum of the domains that contribute to two-component regulator function. Each change therefore presumably exerts its effect via distinct mechanisms. For example the WalR A96T mutation occurs in a conserved region that is important for phosphorylation-mediated protein conformational changes [26] (Figure 1B), while the K208R mutation occurs in a highly conserved α3 DNA recognition helix region (Figure 1B). Recently, the crystal structure of DNA-binding domain of S. aureus WalR protein has been solved, and the interaction of the protein with target DNA examined [27]. Modelling the WalR DNA-binding domain with the K208R mutation from the VISA strain JKD6005 highlights its proximal location to the critical α3–β5 DNA binding loop (Figure 1C).

Within WalK, amino acid substitutions were detected in a range of functional domains (Figure 1), including the G223D mutation in JKD6008 at a highly conserved residue required for a sharp reverse turn between the α1 helix and the connector region of the HAMP domain [28], the 3-amino acid deletion Δ337–340 in BPH0191 in the β scaffold of the PAS domain, which is also known to be involved in its structural integrity [29], and the N382S mutation in WalK from BPH0073 in the phosphor-acceptor domain, only 3 amino acids from the conserved histidine that undergoes autophosphorylation and is essential for autokinase activity (Figure 1B).
Generation and Whole Genome Sequencing of walKR Mutants
To measure the impact of these single nucleotide changes in walKR, bi-directional allelic replacement experiments were performed using two of the clinical pairs (Table 2, pairs 2 and 3). The walK mutation from VISA strain JKD6008 was introduced into the parent VSSA JKD6009, generating TPS3130 (Table 4). We have previously generated a GraS T136I mutation in JKD6009 (JKD6208) [18], therefore to measure the impact of the walK/graS double mutation, the walK mutation from VISA JKD6008 was also introduced into the previously produced graS mutant (JKD6208), generating TPS3128 (Table 4). The walR allele from the VISA strain JKD6005 was used to replace the walR allele in the VSSA parent JKD6004 generating TPS3190, and the walR allele from VSSA parent (JKD6004) was used to replace the walR allele from VISA strain (JKD6005) generating the vancomycin-susceptible strain TPS3124 (Table 4).
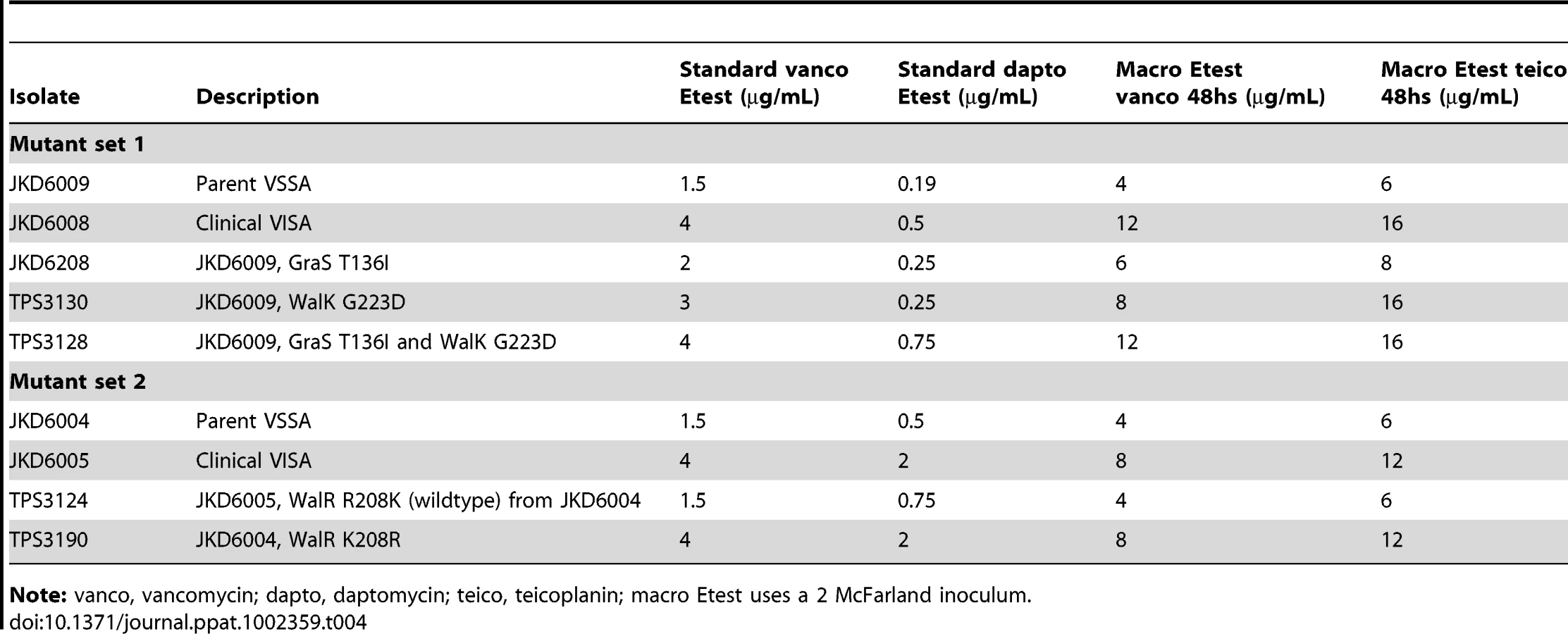
We next sequenced the genomes of S. aureus mutants TPS3130 and TPS3190 to determine if unintended mutations had been introduced during the pKOR1-mediated allelic exchange process, particularly in other regulatory loci such as agr. Coverage of the reference strain JKD6008 was 99.7% and 96.9% for TPS3130 and TPS3190, respectively. For strain TPS3190, Ion Torrent sequencing confirmed the expected walR mutation at position 25010 and the presence of a single silent mutation in walK at position 26026, but no other changes. The unintended change at nucleotide 26026 was a PCR-induced error, introduced during cloning in pKOR1. The situation with strain TPS3130 was more complex. TPS3130, which is VSSA strain JKD6009 modified by replacing its walK gene with the allele from VISA strain JKD6008 (conferring the G223D amino acid change) had the predicted SNP at position 25769 (Figure S1) but also carried an additional four SNPs not present in JKD6009 (Table 5). Two of these four SNPs were the same as changes found in VISA strains JKD6008 and we propose that these might be compensatory mutations linked to the walK mutation. The probability of these changes occurring by chance at exactly these positions in JKD6008 and TPS3190 is small (p<2.3E13) and one can exclude the possibility of a strain mix-up, as the remaining two SNPs were specific to TPS3130 (Table 5). PCR and Sanger sequencing confirmed that the sequences at all five of these altered loci in JKD6009, JKD6008 and TPS3130 were correct.


Impact of walKR Mutations on Antimicrobial Resistance
Vancomycin susceptibility significantly decreased when either the walK allele from JKD6008 (VISA) or the walR allele from JKD6005 (VISA) was swapped into its respective VSSA parent (Table 4 and Figure 2A and B). While the single G223D mutation in WalK converted the VSSA parent strain JKD6009 to VISA, the combination of the G223D mutation in WalK and the T136I mutation in GraS was required to convert JKD6009 to the full intermediate resistance of JKD6008. In comparison, the single K208R mutation in WalR from VISA strain JKD6005 was sufficient to convert JKD6004 (VSSA parent) to intermediate vancomycin resistance.

The in vivo evolution of VISA occurred without daptomycin exposure, however an increase in daptomycin MIC was detected in the majority of VISA strains in this study. Examination of daptomycin susceptibility in the mutants (Table 4) showed that these mutations in walKR promoted daptomycin co-resistance. The single WalR K208R substitution increased the daptomycin MIC of the parental VSSA strain JKD6004 from 0.5 µg per ml to 2 µg per ml generating a daptomycin non-susceptible strain (TPS3190), while the double walK/graS mutant (TPS3128) had a distinct increase in daptomycin MIC compared to the parental strain, although it remained within the defined susceptible range.
The Impact of walKR Mutations on Other Recognized VISA Phenotypes
We next measured autolytic activity and cell wall thickness in the walKR allele-swapped strains compared to their VSSA and VISA parents. The impacts of the single substitutions in either WalR or WalK were dramatic, with significant reductions in autolytic activity and increases in cell wall thickness linked to the introduction of the walR or walk allele from the VISA strain into the VSSA parent (Figure 2C and 3). Conversely, the phenotypes were reversed when the VSSA walR allele from JKD6004 replaced walR in VISA strain JKD6005 (Figure 2C and 3).

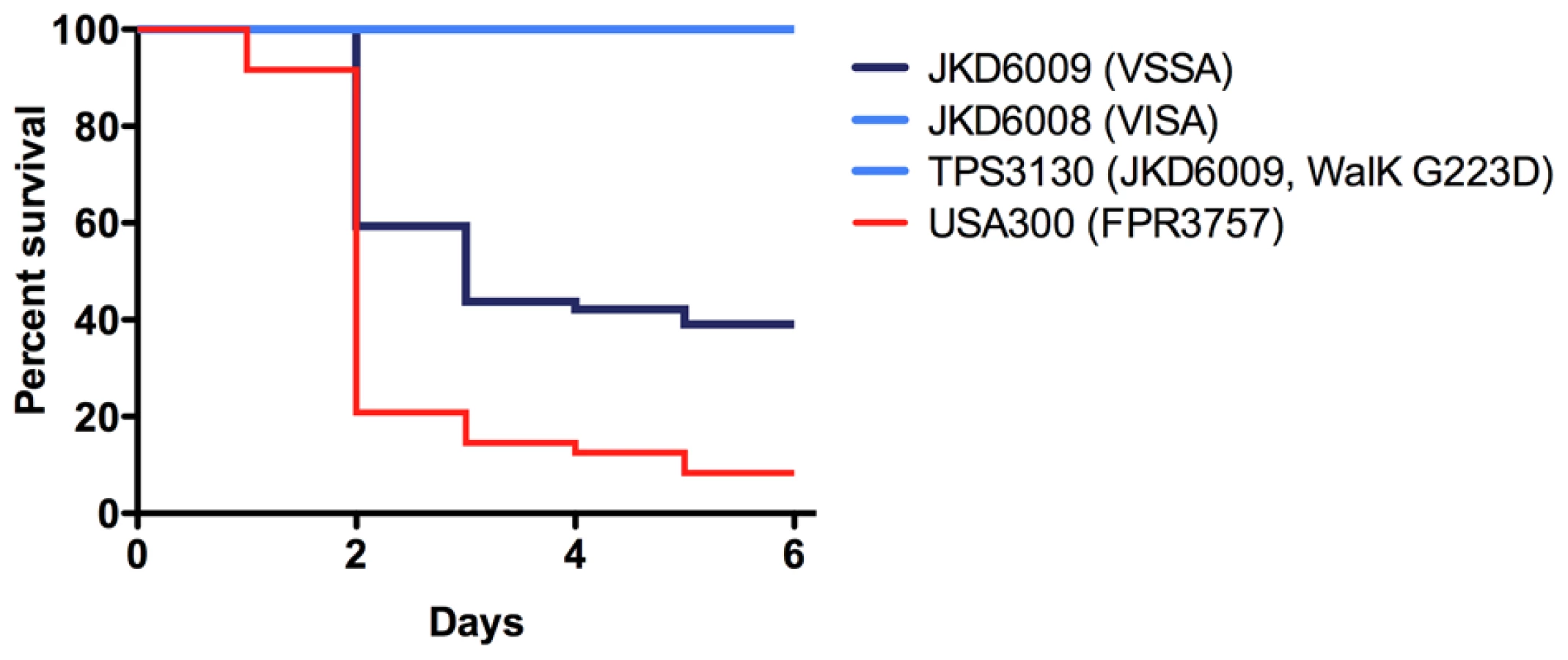
In our previous studies using these VSSA and VISA clinical pairs we had detected significant changes in a number of key staphylococcal virulence mechanisms in VISA [4]. Most strikingly, the VISA strains had marked reduction in agr activation despite the absence of mutations in the agr locus. We measured expression of RNAIII, the effector molecule of the agr locus, in the walKR mutants during exponential growth phase (Figure 2F) and showed that the mutations in either walK or walR had dramatic effects on agr expression with minimal RNAIII expression in VISA strains or the mutants harbouring the VISA walKR alleles compared to the VSSA parents. Additionally, it has previously been demonstrated using the Galleria mellonella model that clinical VISA has reduced virulence compared to parent strains [30], however the genetic basis for this has not been defined. Using this model the clinical VISA strain JKD6008, and the walK mutant TPS3130 demonstrated significantly reduced virulence compared to the parent strain JKD6009 (Figure 4), indicating that WalKR also plays an important role in control of virulence. The parent VSSA strain JKD6004 was relatively avirulent in this model, therefore significant changes in the clinical VISA (JKD6005) and the mutant strain (TPS3190) were not observed.
While biofilm formation is reduced in some VISA strains, the mechanisms underlying this have been unclear. We found that mutations in walK and walR led to significant reduction in bioflm formation that mimicked the changes seen in the clinical VISA strains (Figure 2E). There was no significant difference in growth rates of mutant and clinical strains to explain these findings.
Transcriptional Effects of WalK and WalR Mutations, and Implications for WalKR Control of Central Metabolism
We next used microarray analysis to explore the global regulatory effects of the walK (TPS3130) and walR (TPS3190) mutants compared to the parent strains (Figure 5A, B; Table S1). Any gene that demonstrated a fold change ≥1.5 accompanied by an adjusted p-value of <0.05 was included in the comparisons, and this included 507 genes in the TPS3130 vs JKD6009 analysis and 334 genes in the TPS3190 vs JKD6004 analysis. An analysis of genes that were significantly differentially regulated in the two experiments, in the same direction, revealed 90 genes that were down-regulated, and 73 genes that were up-regulated in WalKR mutants compared to parent strains (Table S1 and Figure 5).

Three genes in S. aureus have been previously shown to have upstream walR biding sites (orthologs in JKD6008 are SAA6008_02607, isaA; SAA6008_02335, ssaA_1; SAA6008_00250, lytM) [31], and six other genes involved in cell wall metabolism and virulence have predicted upstream walR binding sequences, and have been shown to have increased expression upon walKR induction in a mutant with inducible walKR activity [32]. Unexpectedly, only two of these genes were differentially regulated in both experiments. SAA6008_02607 (isaA) was up-regulated in both experiments, 2.3-fold in TPS3190 compared to parent and 6.2-fold in TPS3130 compared to parent, while SAA6008_1007 (atl) was down-regulated in both experiments, 1.8-fold (Table S1). Among 24 genes with predicted upstream WalR binding sequences [31], only two were differentially regulated in both our microarray experiments. These genes included SA6008_00455 (ndhF) encoding an NADH dehydrogenase subunit, which was up-regulated 2.2 to 2.4-fold, and SAA6008_02705 (sasF) encoding an LPXTG cell wall surface anchor protein, which was down-regulated 2.8 to 3.7-fold.
While WalKR has been identified as a master regulator of cell wall metabolism in S. aureus [25], the transcriptional profile of the two mutant strains in this analysis painted a different picture of WalKR control within the cell. The most dramatic and consistent transcriptional changes were found in genes involved in central metabolism. Using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis (www.genome.jp) [33], the major pathways where differential transcription occurred in both mutants were genes whose products are involved in amino acid metabolism (in particular alanine, aspartate, glutamate metabolism [up-regulation of ald1, argG, argH; down-regulation of gltB, gltD, SA6008_02661]; arginine and proline metabolism [up-regulation of agrG, agrH, and down-regulation of aldA, ureA, ureB, ureC]; valine, leucine and isoleucine metabolism [down-regulation of ilvB, ilvC, ilvD]), in addition to significant up-regulation of genes whose products are involved in purine and pyrimidine metabolism (purF, purN, purM, purH, pyrC, pyrB, pyrAA, pyrR, pdp, tdk). In contrast, only five out of 27 genes that have been consistently grouped as part of the staphylococcal “cell wall stimulon” (sbtB, vraS, sgtB, SA6008_01906, and SA6008_02595) [34]–[38] were differentially regulated in the two experiments. All five of these genes were down-regulated (1.6 to 3.6-fold) in the walKR mutant compared to the parent strain.
The veracity of the microarray data was checked by quantitative RT-PCR analysis of six genes from different pathways, including argH, purF, pyrA, ureA, atl, and gltB. The qRT-PCR data was consistent with the microarray results (Figure S2).
Discussion
Methicillin-resistance in S. aureus remains a significant public health issue that is compounded by the evolution of strains with low-level resistance to last-line agents such as vancomycin and daptomycin. While the first VISA strain was reported in 1997 [5], our understanding of the genetic determinants of VISA remains incomplete. To address this issue we took a comparative genomics approach that included whole genome sequencing of 10 clinical S. aureus isolates, and showed that mutations within the essential S. aureus regulatory locus walKR are important mediators of intermediate vancomycin resistance. The contribution of both walK and walR mutations to vancomycin resistance was experimentally confirmed using bi-directional allelic exchange experiments. By swapping walK and walR alleles between VSSA and VISA strains we could make susceptible strains resistant and then revert resistant strains to susceptible. In addition, consistently observed features of VISA strains worldwide include cell wall thickening and reduced autolytic activity [6]-[8], [14], [15], and analysis of our walKR mutants showed the key role played by this locus in VISA, as single nucleotide changes in either walK or walR led to strains exhibiting these well-described VISA phenotypes.
Daptomycin is another cell wall active antibiotic with a different mode-of-action to vancomycin but there is increasing data demonstrating a link between VISA and daptomycin non-susceptibility in S. aureus [10], [19]. The clinical pairs of VSSA and VISA used in this study were selected from patients that had been treated with vancomycin, and not daptomycin. The vancomycin induced walKR mutations led to an increase in daptomycin MIC in clinical S. aureus, in some cases generating full daptomycin non-susceptibility. While walK mutations have been previously associated with daptomycin non-susceptibility in association with mprF mutations [20], no genetic manipulation of the walKR locus has been performed to confirm the contribution of these mutations to daptomycin resistance. Here we have shown for the first time the contribution of mutations in walK or walR (without mprF mutations) to reduced daptomycin susceptibility. Interestingly, the combination of the graS (T136I) and walK (G223D) mutation in JKD6008 was associated with a higher daptomycin MIC than either mutation alone (Table 4), suggesting an additive effect of these two mutations to increase both vancomycin and daptomycin resistance.
WalKR is highly conserved and specific to low GC gram-positive pathogens [25], and is the only known essential two component regulatory system in S. aureus [39], [40]. It has been best studied in B. subtilis, S. aureus and S. pneumoniae where cell wall metabolism genes dominate the predicted regulon of WalR [25]. A high frequency of walKR mutations was detected in this study, with mutations in eight of the 10 VISA, and two of the three hVISA strains tested, suggesting that mutations within this locus are a previously under-appreciated mediator of VISA. Our findings are supported by recent data from Shoji et al who found walK mutations were common in an international collection of VISA strains, although no walR mutations were reported [41]. While much attention has been focussed on the role of vraRS and graRS mutations in VISA [12], [13], [18], [41] none of the five sequenced strains in our study contained a vraRS mutation, and only one strain (JKD6008) contained a previously recognized graS mutation [18], suggesting these regulators are not the dominant mediators of VISA. It is unclear if there is a clone-specific pathway to VISA, such that we may have uncovered a frequent pattern of walKR mutation because we sequenced ST239 strains of S. aureus, the dominant hospital clone from our region. We attempted to address this potential bias by including a global collection of VISA isolates that were available to us for testing, and included non-ST239 strains. It is notable then that all three ST5 VISA strains tested also contained a walK mutation, however the ST247 strains and one of the ST239 strains did not contain walKR mutations, nor mutations in the downstream negative regulators of walKR, yycHIJ. Mutations of potential interest in the strain JKD6051 that is the only fully sequenced strain not to contain a walKR mutation included a mutation in the regulator sarR (A68T), and a mutation in rpoB (H481Y). There has been recent interest in the role of rpoB mutations in VISA, with the demonstration that an A621E amino acid change in RpoB leads to the hVISA phenotype [17], and more recently a link demonstrated between rifampicin resistance in S. aureus (due to rpoB mutations) and hVISA [42]. Therefore it is possible that the rpoB mutation detected in JKD6051 contributed to the VISA phenotype, however this was not specifically tested in this study.
Whole genome analysis of the allelic exchange VISA mutant TPS3190 using Ion Torrent sequencing confirmed the absence of any significant additional mutations in this laboratory derived strain, in particular an absence of agr mutations that might otherwise explain some of the described phenotypes. The sequencing of TPS3130 revealed a more complex story, with the detection of four additional SNPs, two of which were also present in the clinical VISA strain JKD6008 (Table 5). WalKR is thought to control cell-wall metabolism through sensing the lipid-II intermediate during peptidoglycan biosynthesis and regulating autolysin gene expression in this organism [25], however the basis for its essentiality is not well understood but appears to be at least partly related to WalKR control of cell wall metabolism [40]. It is therefore likely that S. aureus is very sensitive to changes in walKR because of its essential nature. We propose that the modification of walK in JKD6008 (and TPS3130) has been accompanied by at least two compensatory mutations. Both of these SNPs have occurred in genes linked to substrate transport. A deeper understanding of the role of WalKR will be required before we can say how the changes we observed in JKD6008 and TPS3130 might compensate for a mutation in walK. The two additional TPS3130-specific changes are equally intriguing and may also be compensatory, with a nucleotide deletion that corrects a frame-shift mutation in a gene encoding a component of the trehalose group translocation phosphate transporter and a predicted amino acid change in a hypothetical gene with a putative lipid kinase domain (Table 5).
Two key questions arising are how do mutations in walKR influence function of the regulator, and then cause VISA. Mwangi et al reported a mutation in the downstream negative regulator of WalKR, yycH that was associated with an increase in vancomycin resistance in a series of clinical isolates. It was predicted that this mutation would increase WalKR activity by reducing negative feedback to the regulator [11]. In another study, an IS256 insertion into the walKR promoter region was associated with a change in vancomycin resistance, and the authors predicted that a hybrid, overactive promoter was responsible for increased walKR expression and resistance [43]. In the present study, however, we found a range of SNPs across all the functional domains of WalKR in VISA strains, leading us to propose that decreased WalKR performance is probably the more likely consequence of these changes and that the VISA phenotype is initiated by a reduction in WalKR activity. WalKR has also been shown to positively regulate genes encoding enzymes with cell wall lytic activity. The reduction in autolytic activity and attenuated biofilm formation of the walKR mutants in this study also suggest that the mutations are restricting WalKR function, as these phenotypes have been previously demonstrated to occur in a conditional mutant with reduced WalKR activity [32]. We have also recently described a similar phenomenon where a point mutation in another essential S. aureus gene (relA) contributed to a reduction in function of the enzyme, but not complete absence of activity [44].
To further investigate the contribution of the walK and walR mutations to resistance we performed a global transcriptional profile of the mutant strains compared to parent strains (Figure 5). We were surprised to find a relative paucity of cell wall metabolism genes represented in the transcription expression data. We intentionally used a low fold cut off of 1.5 for inclusion of differentially expressed genes in the analysis, and despite this we only found four genes that were differentially expressed in both experiments that have been previously predicted to be part of the WalR regulon [31], [32]. While down-regulation of major autolysin expression (atl) was detected (1.8-fold in both mutants) and this could potentially explain the changes in autolytic activity detected in WalK and WalR mutants (Figure 2), the dominant theme of the transcriptional profiles was a change in transcription of genes encoding products involved in selected aspects of central metabolism. For example, pathway analysis based on our microarray data suggests WalKR interacts directly or indirectly with several components of the urea cycle. So while cellular metabolism in the mutants has been shifted away from anabolism of branched chain amino acids (valine, leucine and isoleucine), metabolites within the urea cycle, including glutamine, arginine and aspartate, have gained more prominence, with up-regulation of genes encoding enzymes that could increase yields of substrates that feed purine and pyrimidine biosynthesis. An increase in pyrimidine metabolism would tie in neatly with the observed increased cell wall thickness and decreased capsular polysaccharide in the walKR mutant strains. Two distinct pathways are used by S. aureus to synthesise UDP N-acetylglucosamine (UDP-GlcNAc), a key intermediate for both peptidoglycan and capsular polysaccharide biosynthesis. Pools of UDP-GlcNAc for peptidoglycan biosynthesis are produced by glycolysis and pyrimidine metabolism while UDP-GlcNAc for capsule biosynthesis is linked to gluconeogenesis and aminosugar metabolism [45]. Therefore an increase in pyrimidine metabolism might be predicted to increase pools of UDP-GlcNAc available for peptidoglycan synthesis, with a concomitant decrease in the availability of the same metabolite for capsule production.
Finally, this study has demonstrated a link between walKR mutations and a number of VISA associated phenotypes. While biofilm formation is reduced in some VISA strains, including those previously characterized strains used in this study [15], the mechanisms underlying this have been unclear. We found that mutations in walK and walR led to significant reduction in biofilm formation that mimicked the changes seen in the VISA strains (Figure 2E). Reduced walKR activity in a conditional mutant was previously associated with reduced biofilm production [32], and is consistent with the changes in biofilm formation seen with S. aureus mutants deficient in autolysis [46], [47]. Extracellular DNA (eDNA) that is released by autolysis is an important initial step in S. aureus biofilm formation [48]. We propose that there may be an absence of eDNA in VISA and that this may explain the biofilm defect observed in VISA strains with walKR mutations. The agr locus is a quorum sensing toxin regulatory system that is critical to S. aureus virulence [49]. Using real time RNAIII PCR and microarray transcriptional analysis we have demonstrated that mutation of walKR is associated with reduced agr activation during exponential growth, however it is unclear if this is a direct or indirect relationship. Little is known about the role WalKR plays in S. aureus virulence. While virulence studies have been performed using streptococcal species, the impact of walK mutants in these experiments has been conflicting [50], [51]. Using the invertebrate model Galleria mellonella a significant attenuation of virulence of the walK mutant strain TPS3130 was demonstrated (Figure 4). However, because there was no difference in the virulence of the clinical pair JKD6004/JKD6005, no impact of the walR mutation in the parental strain was discernable.
In conclusion, this study highlights the potential for single nucleotide changes in S. aureus to dramatically affect bacterial behaviour and antimicrobial resistance. Complete bacterial genome sequencing of carefully selected strains has revealed mutations in walKR as a common mechanism for in vivo evolution of multi-drug resistance in this pathogen, leading to daptomycin cross-resistance, and impacting virulence mechanisms of the organism. Additionally, transcription profiling of strains with single base substitutions in walK or walR indicate that this regulatory locus not only controls autolysis, but more generally impacts metabolic activities within the cell. Efforts to design therapeutic strategies based on inhibiting walKR should be aware of the potential impact on the organism of inducing mutations in this locus.
Materials and Methods
Strains and Growth Conditions
Bacterial strains and a description of their antibiotic susceptibility, geographical origin, and plasmids used in the study are listed in Tables 1 and 6. Staphylococcal strains were stored in glycerol broth at −80°C and subcultured twice onto Horse Blood Agar (Oxoid) before being used for any experiment. Unless otherwise indicated all S. aureus isolates were grown in BHIB (Oxoid), and E. coli grown in LB broth (Oxoid). When required media was supplemented with the following antibiotics at the indicated concentrations: for E. coli, ampicillin 100 µg/ml; for S. aureus RN4220, chloramphenicol 10 µg/ml; for S. aureus clinical isolates, chloramphenicol 25 µg/ml.
Antibiotic Susceptibility and Molecular Typing
Vancomycin MICs were determined by microbroth MIC according to CLSI criteria [52], and VISA defined as a strain with a vancomycin broth MIC of 4–8 µg per ml [52]. Detection of heterogenous-VISA (hVISA) was performed by Etest GRD according to recommendations of the manufacturer, while clinical and mutant strain pairs were tested by Macro method Etest using a 2 McFarland inoculum [3]. Daptomycin MICs were performed by Etest (AB Biodisk), according to manufacturer’s instructions, and isolates were defined as daptomycin non-susceptible according to CLSI criteria (daptomycin MIC >1 µg per ml) [52]. Clinical isolates underwent molecular characterisation using the DNA microarray StaphyType96 (CLONDIAG, Jena, Germany). DNA extraction was performed using the DNeasy Tissue Kit (Qiagen, Hilden, Germany), and the microarray and data analysis were performed as previously described [53]. The DNA microarray assigns strains to clonal complexes (CC) using reference strains previously defined by multilocus sequence typing (MLST) and spa typing. Pulsed-field gel electrophoresis using SmaI enzyme restriction was performed using the CHEF DR III system (BioRad, Berkeley, California) to confirm clonal group if necessary [54].
DNA Methods, Molecular Techniques and Construction of Mutants
Standard procedures were used for DNA manipulation, molecular techniques, PCR, sequencing and plasmid extraction. To generate walK and walR mutants, allelic replacement experiments were performed using the vector pKOR1, as described previously [18]. The locus containing the walKR mutation was amplified from JKD6008, JKD6004, and JKD6005 for exchange into the respective parental strains using primers 1901 and 1908 (Table S2). The amplified product was cloned into the attB sites of pKOR1 and then transformed into E. coli DH5alpha. After transformation into E. coli, pKOR1 with the integrated walKR locus was extracted and sequenced to confirm the correct sequence, prior to performing allelic exchange in RN4220 intermediate and the clinical S. aureus strain. To confirm that no other significant mutations were introduced during the homologous recombination, the whole walKR locus was sequenced from the mutant strains using oligonucleotides covering the whole replaced sequence and its flanking ends (Table S2).
High Throughput DNA Sequencing
Genome sequences for four ST239 VSSA and VISA clinical pairs (VSSA: JKD6000, JKD6004, JKD6021 and JKD6052; VISA: JKD6001, JKD6005, JKD6023, and JKD6051) were obtained from an Illumina Genome Analyzer II using 36-cycle paired-end chemistry. SOLiDv2 26 bp mate-pair sequencing was also performed on the clinical VSSA strain JKD6009, for which previous 454 GS20 shotgun sequence data was available. Single-end genome sequencing of the two laboratory-induced mutants TPS3130 and TPS3190 was performed using Ion Torrent sequencing as described [55]. TPS3130 yielded 88.7 Mbp from four 314 chips and TPS3190 yielded 56.9 Mbp from two 314 chips.
Comparative Genomics
A read mapping approach was used to compare the sequences from the four VSSA/VISA clinical pairs described above, the previously described clinical pair JKD6009/JKD6008 (NCBI Genbank accession numbers NZ_ABSA00000000 and CP002120) and the Ion Torrent sequences from TPS3130 and TPS3190. The reads from all genomes were aligned to the JKD6008 reference using SHRiMP 2.0 [56]. SNPs were identified using Nesoni v0.52, which uses the aligned reads of each genome to the reference to construct a tally of putative differences at each position, including substitutions, insertions, and deletions [www.bioinformatics.net.au]. This tally was input to a Bayesian model to decide whether a base (or deletion) could be called for the position, and if so, whether it differed from the reference. Each position is treated as possibly containing a mixture of bases. A particular base is called if the likelihood that it makes up more than 50% of the mixture exceeds a threshold, set to 0.99 by default. This likelihood is calculated by updating a prior distribution over all possible mixtures, in the form of a Dirichlet distribution, as bases are observed. A similar procedure is used to call the presence or absence of insertions between positions in the reference. Using the whole genome sequence of JKD6008 as a reference a global SNP analysis was performed, and allelic variability at any nucleotide position was tallied to generate a global SNP analysis for every genome compared to JKD6008. Protein modelling was performed using the crystal structure of the WalR DNA-binding domain of S. aureus (PDB structure: 2ZXJ) using ModBase [27], [57].
Phenotypic Assays
Biofilm and autolytic assays were performed as described previously [15]. Cell wall thickness was measured by taking 100 readings of wall thickness per strain from multiple cells after preparation of electron microscopic images as described previously [15].
Galleria mellonella Virulence Assay
This invertebrate S. aureus infection model was used to study the virulence of clinical and mutant strains as previously described [44]. Briefly, a HPLC syringe was used to inject 10 µL of bacterial suspension (approx 1.0×106 CFU) into the last left proleg of each caterpillar. Bacterial colony counts were performed to confirm consistency of inoculum, and caterpillars monitored daily for 6 days. Multiple biological replicates were performed for each strain.
Microarray Transcriptional Analysis
Microarray transcriptional analysis was performed with TIGR version 9 S. aureus arrays, as previously described [4]. For preparation of total RNA shaking flasks (50 mL BHI broth in 250 mL flasks) were inoculated with 500 µL overnight BHI broth culture and incubated on a 225rpm shaker at 37°C. Optical density was closely monitored, and one millilitre of sample was collected at exponential growth phase (optical density at 600 nm of 0.5) and 0.5 mL RNA stabilization reagent (RNA later, Qiagen) was added and mixed immediately. The mixture was allowed to stand in room temperature for 10 minutes before total RNA was extracted using the RNeasy micro kit (Qiagen). RNA extractions and hybridisations were performed on four different occasions, and the dye swapped with each biological replicate. The images were combined and quantified and then imported into BASE and analyzed using Bioconductor and Limma [58], [59]. The fold ratio of gene expression for the mutant strains TPS3130 and TPS3190 relative to the parental MRSA strain JKD6009 and JKD6004, respectively, were calculated. Using a modified t-test p values were calculated and adjusted for multiple testing using false discovery rate (FDR) correction. A ≥ 1.5-fold change with p<0.05 was considered significant.
Quantitative RT-PCR
To investigate activity of the agr locus (RNAIII) and confirm the microarray transcriptional results qRT-PCR was performed for RNAIII, argH, purF, pyrA, ureA, atl, and gltB, using oligos in Table S2. RNA was prepared from exponential phase cultures as previously described [15]. Two on-column DNase I digestion steps were performed and cDNA synthesis using SuperScript II RNase H reverse transcriptase (Invitrogen) included a SuperScript II negative control to confirm the absence of genomic DNA. Relative expression was determined as previously described [15], and was normalised against gyrB as previously described [60]. Results were obtained from 3 biological replicates each performed in triplicate.
Statistical Analysis
Statistical analyses of mutant strains were performed using the two-tailed Mann Whitney U test, with a p<0.05 set for statistical significance. Kaplan Meier plots of G. mellonella killing results were analyzed using the log rank test. All analyses were performed using Prism for Macintosh ver 5.0 (GraphPad Software Inc., CA, USA)
Accession Numbers
For the sequenced clinical strains JKD6000, JKD6001, JKD6004, JKD6005, JKD6009, JKD6021, JKD6023, JKD6051, JKD6052 the reads have been deposited in the NCBI Sequence Read Archive under study accession number SRA027352. The Ion Torrent reads for the mutant strains TPS3130 and TPS3190 have been deposited in the NCBI Sequence Read Archive under study accession number SRA044879.2. Microarray data has been submitted to GEO with accession number GSE29157. Protein modelling was performed using the crystal structure of the WalR DNA-binding domain of S. aureus (PDB structure: 2ZXJ).
Supporting Information
{kind=link}
{kind=link}
Zdroje
1. WardPBJohnsonPDGrabschEAMayallBCGraysonML 2001 Treatment failure due to methicillin-resistant Staphylococcus aureus (MRSA) with reduced susceptibility to vancomycin. Med J Aust 175 480 483
2. HowdenBPWardPBCharlesPGKormanTMFullerA 2004 Treatment outcomes for serious infections caused by methicillin-resistant Staphylococcus aureus with reduced vancomycin susceptibility. Clin Infect Dis 38 521 528
3. HowdenBPDaviesJKJohnsonPDStinearTPGraysonML 2010 Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin Microbiol Rev 23 99 139
4. HowdenBPSmithDJMansellAJohnsonPDWardPB 2008 Different bacterial gene expression patterns and attenuated host immune responses are associated with the evolution of low-level vancomycin resistance during persistent methicillin-resistant Staphylococcus aureus bacteraemia. BMC Microbiol 8 39
5. HiramatsuKAritakaNHanakiHKawasakiSHosodaY 1997 Dissemination in Japanese hospitals of strains of Staphylococcus aureus heterogeneously resistant to vancomycin. Lancet 350 1670 1673
6. SieradzkiKRobertsRBHaberSWTomaszA 1999 The development of vancomycin resistance in a patient with methicillin-resistant Staphylococcus aureus infection. N Engl J Med 340 517 523
7. SieradzkiKTomaszA 2003 Alterations of cell wall structure and metabolism accompany reduced susceptibility to vancomycin in an isogenic series of clinical isolates of Staphylococcus aureus. J Bacteriol 185 7103 7110
8. SmithTLPearsonMLWilcoxKRCruzCLancasterMV 1999 Emergence of vancomycin resistance in Staphylococcus aureus. Glycopeptide-Intermediate Staphylococcus aureus Working Group. N Engl J Med 340 493 501
9. MooreMRPerdreau-RemingtonFChambersHF 2003 Vancomycin treatment failure associated with heterogeneous vancomycin-intermediate Staphylococcus aureus in a patient with endocarditis and in the rabbit model of endocarditis. Antimicrob Agents Chemother 47 1262 1266
10. KelleyPGGaoWWardPBHowdenBP 2011 Daptomycin non-susceptibility in vancomycin-intermediate Staphylococcus aureus (VISA) and heterogeneous-VISA (hVISA): implications for therapy after vancomycin treatment failure. J Antimicrob Chemother 66 1057 1060
11. MwangiMMWuSWZhouYSieradzkiKde LencastreH 2007 Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc Natl Acad Sci U S A 104 9451 9456
12. NeohHMCuiLYuzawaHTakeuchiFMatsuoM 2008 Mutated response regulator graR is responsible for phenotypic conversion of Staphylococcus aureus from heterogeneous vancomycin-intermediate resistance to vancomycin-intermediate resistance. Antimicrob Agents Chemother 52 45 53
13. CuiLNeohHMShojiMHiramatsuK 2009 Contribution of vraSR and graSR point mutations to vancomycin resistance in vancomycin-intermediate Staphylococcus aureus. Antimicrob Agents Chemother 53 1231 1234
14. CuiLMurakamiHKuwahara-AraiKHanakiHHiramatsuK 2000 Contribution of a thickened cell wall and its glutamine nonamidated component to the vancomycin resistance expressed by Staphylococcus aureus Mu50. Antimicrob Agents Chemother 44 2276 2285
15. HowdenBPJohnsonPDWardPBStinearTPDaviesJK 2006 Isolates with low-level vancomycin resistance associated with persistent methicillin-resistant Staphylococcus aureus bacteremia. Antimicrob Agents Chemother 50 3039 3047
16. RenzoniABarrasCFrancoisPCharbonnierYHugglerE 2006 Transcriptomic and functional analysis of an autolysis-deficient, teicoplanin-resistant derivative of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 50 3048 3061
17. CuiLIsiiTFukudaMOchiaiTNeohHM 2010 An RpoB mutation confers dual heteroresistance to daptomycin and vancomycin in Staphylococcus aureus. Antimicrob Agents Chemother 54 5222 5233
18. HowdenBPStinearTPAllenDLJohnsonPDWardPB 2008 Genomic analysis reveals a point mutation in the two-component sensor gene graS that leads to intermediate vancomycin resistance in clinical Staphylococcus aureus. Antimicrob Agents Chemother 52 3755 3762
19. MoisePANorthDSteenbergenJNSakoulasG 2009 Susceptibility relationship between vancomycin and daptomycin in Staphylococcus aureus: facts and assumptions. Lancet Infect Dis 9 617 624
20. FriedmanLAlderJDSilvermanJA 2006 Genetic changes that correlate with reduced susceptibility to daptomycin in Staphylococcus aureus. Antimicrob Agents Chemother 50 2137 2145
21. HowdenBPSeemannTHarrisonPFMcEvoyCRStantonJA 2010 Complete genome sequence of Staphylococcus aureus strain JKD6008, an ST239 clone of methicillin-resistant Staphylococcus aureus with intermediate-level vancomycin resistance. J Bacteriol 192 5848 5849
22. OkadaAIgarashiMOkajimaTKinoshitaNUmekitaM 2010 Walkmycin B targets WalK (YycG), a histidine kinase essential for bacterial cell growth. J Antibiot (Tokyo) 63 89 94
23. QinZLeeBYangLZhangJYangX 2007 Antimicrobial activities of YycG histidine kinase inhibitors against Staphylococcus epidermidis biofilms. FEMS Microbiol Lett 273 149 156
24. GotohYDoiAFurutaEDubracSIshizakiY 2010 Novel antibacterial compounds specifically targeting the essential WalR response regulator. J Antibiot (Tokyo) 63 127 134
25. DubracSBisicchiaPDevineKMMsadekT 2008 A matter of life and death: cell wall homeostasis and the WalKR (YycGF) essential signal transduction pathway. Mol Microbiol 70 1307 1322
26. BourretRB 2010 Receiver domain structure and function in response regulator proteins. Curr Opin Microbiol 13 142 149
27. DoiAOkajimaTGotohYTanizawaKUtsumiR 2010 X-ray crystal structure of the DNA-binding domain of response regulator WalR essential to the cell viability of Staphylococcus aureus and interaction with target DNA. Biosci Biotechnol Biochem 74 1901 1907
28. HulkoMBerndtFGruberMLinderJUTruffaultV 2006 The HAMP domain structure implies helix rotation in transmembrane signaling. Cell 126 929 940
29. RepikARebbapragadaAJohnsonMSHaznedarJOZhulinIB 2000 PAS domain residues involved in signal transduction by the Aer redox sensor of Escherichia coli. Mol Microbiol 36 806 816
30. PelegAYMongaDPillaiSMylonakisEMoelleringRCJr 2009 Reduced susceptibility to vancomycin influences pathogenicity in Staphylococcus aureus infection. J Infect Dis 199 532 536
31. DubracSMsadekT 2004 Identification of genes controlled by the essential YycG/YycF two-component system of Staphylococcus aureus. J Bacteriol 186 1175 1181
32. DubracSBonecaIGPoupelOMsadekT 2007 New insights into the WalK/WalR (YycG/YycF) essential signal transduction pathway reveal a major role in controlling cell wall metabolism and biofilm formation in Staphylococcus aureus. J Bacteriol 189 8257 8269
33. KanehisaM 2002 The KEGG database. Novartis Found Symp 247 : 91-101; discussion 101-103, 119-128, 244-152
34. KurodaMKurodaHOshimaTTakeuchiFMoriH 2003 Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol Microbiol 49 807 821
35. McAleeseFWuSWSieradzkiKDunmanPMurphyE 2006 Overexpression of genes of the cell wall stimulon in clinical isolates of Staphylococcus aureus exhibiting vancomycin-intermediate - S. aureus-type resistance to vancomycin. J Bacteriol 188 1120 1133
36. McCallumNSpeharGBischoffMBerger-BachiB 2006 Strain dependence of the cell wall-damage induced stimulon in Staphylococcus aureus. Biochim Biophys Acta 1760 1475 1481
37. SobralRGJonesAEDes EtagesSGDoughertyTJPeitzschRM 2007 Extensive and genome-wide changes in the transcription profile of Staphylococcus aureus induced by modulating the transcription of the cell wall synthesis gene murF. J Bacteriol 189 2376 2391
38. UtaidaSDunmanPMMacapagalDMurphyEProjanSJ 2003 Genome-wide transcriptional profiling of the response of Staphylococcus aureus to cell-wall-active antibiotics reveals a cell-wall-stress stimulon. Microbiology 149 2719 2732
39. MartinPKLiTSunDBiekDPSchmidMB 1999 Role in cell permeability of an essential two-component system in Staphylococcus aureus. J Bacteriol 181 3666 3673
40. DelauneAPoupelOMalletACoicYMMsadekT 2011 Peptidoglycan crosslinking relaxation plays an important role in Staphylococcus aureus WalKR-dependent cell viability. PLoS One 6 e17054
41. ShojiMCuiLIizukaRKomotoANeohHM 2011 walK and clpP Mutations Confer Reduced Vancomycin Susceptibility in Staphylococcus aureus. Antimicrob Agents Chemother 55 3870 3881
42. WatanabeYCuiLKatayamaYKozueKHirakawaH 2011 Impact of rpoB mutations on reduced vancomycin susceptibility in Staphylococcus aureus. J Clin Microbiol 49 2680 2684
43. JansenATurckMSzekatCNagelMCleverI 2007 Role of insertion elements and yycFG in the development of decreased susceptibility to vancomycin in Staphylococcus aureus. Int J Med Microbiol 297 205 215
44. GaoWChuaKDaviesJKNewtonHJSeemannT 2010 Two novel point mutations in clinical Staphylococcus aureus reduce linezolid susceptibility and switch on the stringent response to promote persistent infection. PLoS Pathog 6 e1000944
45. SadykovMRMattesTALuongTTZhuYDaySR 2010 Tricarboxylic acid cycle-dependent synthesis of Staphylococcus aureus Type 5 and 8 capsular polysaccharides. J Bacteriol 192 1459 1462
46. BiswasRVogguLSimonUKHentschelPThummG 2006 Activity of the major staphylococcal autolysin Atl. FEMS Microbiol Lett 259 260 268
47. BolesBRThoendelMRothAJHorswillAR 2010 Identification of genes involved in polysaccharide-independent Staphylococcus aureus biofilm formation. PLoS One 5 e10146
48. MannEERiceKCBolesBREndresJLRanjitD 2009 Modulation of eDNA release and degradation affects Staphylococcus aureus biofilm maturation. PLoS One 4 e5822
49. NovickRP 2003 Autoinduction and signal transduction in the regulation of staphylococcal virulence. Mol Microbiol 48 1429 1449
50. WagnerCSaizieu AdASchonfeldHJKamberMLangeR 2002 Genetic analysis and functional characterization of the Streptococcus pneumoniae vic operon. Infect Immun 70 6121 6128
51. ThroupJPKoretkeKKBryantAPIngrahamKAChalkerAF 2000 A genomic analysis of two-component signal transduction in Streptococcus pneumoniae. Mol Microbiol 35 566 576
52. CLSI 2010 Performance Standards for Antimicrobial Susceptibility Testing. Approved Standard. CLSI document M100-S20 Wayne, , Pennsylvania CLSI; 2010
53. MoneckeSKanigHRudolphWMullerECoombsG 2010 Characterisation of Australian MRSA strains ST75 - and ST883-MRSA-IV and analysis of their accessory gene regulator locus. PLoS One 5 e14025
54. O'BrienFGUdoEEGrubbWB 2006 Contour-clamped homogeneous electric field electrophoresis of Staphylococcus aureus. Nat Protoc 1 3028 3033
55. RothbergJMHinzWRearickTMSchultzJMileskiW 2011 An integrated semiconductor device enabling non-optical genome sequencing. Nature 475 348 352
56. RumbleSMLacroutePDalcaAVFiumeMSidowA 2009 SHRiMP: accurate mapping of short color-space reads. PLoS Comput Biol 5 e1000386
57. PieperUEswarNWebbBMEramianDKellyL 2009 MODBASE, a database of annotated comparative protein structure models and associated resources. Nucleic Acids Res 37 D347 354
58. SaalLHTroeinCVallon-ChristerssonJGruvbergerSBorgA 2002 BioArray Software Environment (BASE): a platform for comprehensive management and analysis of microarray data. Genome Biol 15 8
59. SmythGK 2004 Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3 Article 3
60. BurianMRautenbergMKohlerTFritzMKrismerB 2010 Temporal expression of adhesion factors and activity of global regulators during establishment of Staphylococcus aureus nasal colonization. J Infect Dis 201 1414 1421
61. TatusovRLFedorovaNDJacksonJDJacobsARKiryutinB 2003 The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4 41
62. PatronRLClimoMWGoldsteinBPArcherGL 1999 Lysostaphin treatment of experimental aortic valve endocarditis caused by a Staphylococcus aureus isolate with reduced susceptibility to vancomycin. Antimicrob Agents Chemother 43 1754 1755
63. KreiswirthBNLofdahlSBetleyMJO'ReillyMSchlievertPM 1983 The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305 709 712
64. DiepBAGillSRChangRFPhanTHChenJH 2006 Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 367 731 739
65. BaeTSchneewindO 2006 Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55 58 63
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 11
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Multiple Candidate Effectors from the Oomycete Pathogen Suppress Host Plant Immunity
- The Splicing Factor Proline-Glutamine Rich (SFPQ/PSF) Is Involved in Influenza Virus Transcription
- A TNF-Regulated Recombinatorial Macrophage Immune Receptor Implicated in Granuloma Formation in Tuberculosis
- SH3 Domain-Mediated Recruitment of Host Cell Amphiphysins by Alphavirus nsP3 Promotes Viral RNA Replication
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy