
Genomic Transition to Pathogenicity in Chytrid Fungi
Understanding the molecular mechanisms of pathogen emergence is central to mitigating the impacts of novel infectious disease agents. The chytrid fungus Batrachochytrium dendrobatidis (Bd) is an emerging pathogen of amphibians that has been implicated in amphibian declines worldwide. Bd is the only member of its clade known to attack vertebrates. However, little is known about the molecular determinants of - or evolutionary transition to - pathogenicity in Bd. Here we sequence the genome of Bd's closest known relative - a non-pathogenic chytrid Homolaphlyctis polyrhiza (Hp). We first describe the genome of Hp, which is comparable to other chytrid genomes in size and number of predicted proteins. We then compare the genomes of Hp, Bd, and 19 additional fungal genomes to identify unique or recent evolutionary elements in the Bd genome. We identified 1,974 Bd-specific genes, a gene set that is enriched for protease, lipase, and microbial effector Gene Ontology terms. We describe significant lineage-specific expansions in three Bd protease families (metallo-, serine-type, and aspartyl proteases). We show that these protease gene family expansions occurred after the divergence of Bd and Hp from their common ancestor and thus are localized to the Bd branch. Finally, we demonstrate that the timing of the protease gene family expansions predates the emergence of Bd as a globally important amphibian pathogen.
Published in the journal:
. PLoS Pathog 7(11): e32767. doi:10.1371/journal.ppat.1002338
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002338
Summary
Understanding the molecular mechanisms of pathogen emergence is central to mitigating the impacts of novel infectious disease agents. The chytrid fungus Batrachochytrium dendrobatidis (Bd) is an emerging pathogen of amphibians that has been implicated in amphibian declines worldwide. Bd is the only member of its clade known to attack vertebrates. However, little is known about the molecular determinants of - or evolutionary transition to - pathogenicity in Bd. Here we sequence the genome of Bd's closest known relative - a non-pathogenic chytrid Homolaphlyctis polyrhiza (Hp). We first describe the genome of Hp, which is comparable to other chytrid genomes in size and number of predicted proteins. We then compare the genomes of Hp, Bd, and 19 additional fungal genomes to identify unique or recent evolutionary elements in the Bd genome. We identified 1,974 Bd-specific genes, a gene set that is enriched for protease, lipase, and microbial effector Gene Ontology terms. We describe significant lineage-specific expansions in three Bd protease families (metallo-, serine-type, and aspartyl proteases). We show that these protease gene family expansions occurred after the divergence of Bd and Hp from their common ancestor and thus are localized to the Bd branch. Finally, we demonstrate that the timing of the protease gene family expansions predates the emergence of Bd as a globally important amphibian pathogen.
Introduction
Understanding the emergence of novel pathogens is a central challenge in epidemiology, disease ecology, and evolutionary biology. Emerging pathogens of humans, wildlife, and agriculturally important crops generally have a dynamic recent evolutionary past. For example, many emerging pathogens have become adapted to new environmental conditions, shifted their host range, and/or evolved more virulent forms [1]–[3]. Identifying the genetic basis of these evolutionary shifts can lend insight into the mechanisms of pathogen emergence.
Studies of the amphibian-killing fungus Batrachochytrium dendrobatidis (Bd) provide an opportunity to better understand evolutionary transitions to pathogenicity. Bd is considered the leading cause of amphibian declines worldwide and is found on every continent where amphibians occur [4]–[5]. Bd infects amphibian skin and the resulting disease, chytridiomycosis, is responsible for population declines and extirpations in hundreds of amphibian species [6]–[7]. Bd is the only documented vertebrate pathogen in a diverse, early-branching lineage of fungi called the Chytridiomycota. Some chytrids are pathogens of plants, but most chytrids are primarily known to survive on decaying organic material as saprobes [8]. The question of how an amphibian-killing fungus evolved from an ancestor that was not a vertebrate pathogen is vital to understanding and mitigating the chytridiomycosis epidemic and will also shed light on the evolution of novel pathogens more broadly.
Investigating the transition to pathogenicity in chytrid fungi requires an explicitly evolutionary perspective. Specifically, identifying elements of the genome that have undergone recent evolution in the branch leading to Bd may help us determine how Bd attacks its amphibian hosts. Previously we identified several families of proteases that may be involved in Bd's ability to infect amphibian skin. Specifically, we found expanded gene families of metallo - and serine proteases in the Bd genome that exhibit life-stage specific gene expression patterns [9]. These proteases have been hypothesized to play a role in the ability of other fungal pathogens to invade and degrade host tissue [10]–[13]. However, previous studies could not resolve if these gene family expansions occurred along the branch leading to Bd because the fungal genomes available for comparison were only distantly related to Bd.
To determine what unique features of the Bd genome might relate to its ability to colonize amphibian skin, we compared genomes of Bd and its closest known relative, Homolaphlyctis polyrhiza (Hp) (this isolate has been described by Joyce Longcore (pers. comm.) and has been referred to as “JEL142” in previous publications [8]). Bd and Hp are in the same Rhizophydiales order [14], and Bd is the only member of this clade known to be a vertebrate pathogen [8]. We first confirmed that Hp cannot survive on amphibian skin alone. We then sequenced and characterized the genome of Hp using Roche-454 pyrosequencing. Finally, we used a comparative genomics approach to identify differences between Bd and Hp using additional fungal species as outgroups. Based on identified unique elements of the Bd genome, we develop hypotheses for the mechanisms and evolution of Bd pathogenicity.
Materials and Methods
Taxon Sampling
Our focal isolates were the JAM81 strain of Bd and the JEL142 strain of Hp. JAM81 was isolated from Rana muscosa in the Sierra Nevada Mountains in California, where Bd has caused catastrophic declines in R. muscosa populations [15]. Hp was collected from leaf litter in Maine and is a presumed saprobe. We also used the information from publically available genomes of an additional Bd isolate - JEL423 (http://www.broadinstitute.org/annotation/genome/batrachochytrium_dendrobatidis/MultiHome.html), andan additional chytrid, Spizellomyces punctatus, a terrestrial saprobe (Origins of Multicellularity Sequencing Project, Broad Institute of Harvard and MIT (http://www.broadinstitute.org/)). Finally we used the genome information from 17 additional publicly available fungal genomes (Table S1). We chose these outgroups to represent a broad phylogenetic survey of fungi that span four additional fungal phyla: Blastocladiomycota (Allomyces macrogynus), Zygomycota (Phycomyces blakesleeanus), Basidiomycota (Coprinopsis cinerea, Cryptococcus neoformans, Puccinia graminis f. sp. tritici, Ustilago maydis), and Ascomycota (Arthroderma benhamiae, Aspergillus nidulans, Blastomyces dermatitidis, Botrytis cinerea, Coccidioides immitis, Fusarium graminearum, Microsporum canis, Neurospora crassa, Pyrenophora tritici-repentis, Trichophyton rubrum, and Uncinocarpus reesii). Arthroderma benhamiae, M. canis, and T. rubrum were chosen in particular because they are dermatophytes (i.e. fungal pathogens that infect skin).
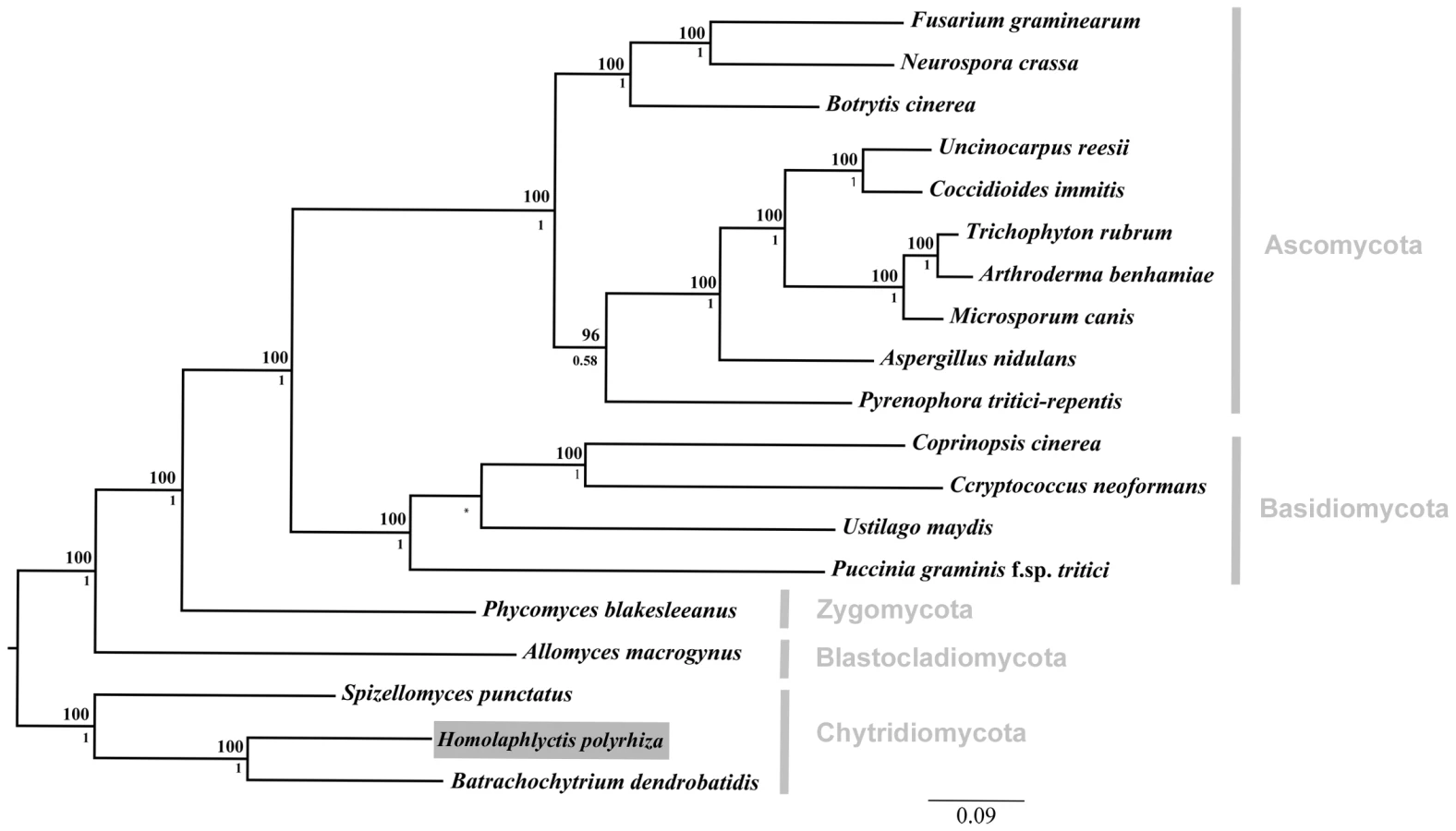
We reconstructed the phylogenetic relationships among the 19 taxa used in this study using Bayesian phylogenetic analyses of 51 single-copy genes. The alignment was comprised of 21182 total trimmed amino acid residues. The orthologous sequences were aligned with T-Coffee [16], concatenated, and trimmed with trimAl [17]. The Basidiomycota phylum was constrained by members Ustilago maydis and Puccinia graminis, and the tree rooted with the Chytridiomycota clade based on [8]. Bayesian posterior probabilities are shown below internal nodes and ML bootstrap values from 100 replicates above the nodes.
Growth of Bd and Hp on Amphibian Skin
We grew Bd (JAM81) and Hp on the standard growth medium PmTG (made from peptonized milk, tryptone and glucose) [18]. After one week of growth, we transferred 3.8× 106 zoospores from each isolate to 3 mL of two liquid growth conditions: standard growth media and amphibian skin. For standard growth media we used 1% liquid PmTG, and for amphibian skin we used 10% w/v pulverized and autoclaved cane-toad skin in water. We established six technical replicates of each isolate in each condition. Liquid cultures were gently shaken in 6-well tissue culture plates. To test how long Bd and Hp survived in each growth condition, we tested an aliquot from each culture every day for 14 days. Each day we removed 15 µL from each of the technical replicates, pooled aliquots for each isolate in each treatment group, and inoculated PmTG-agar growth plates. We inspected growth plates every day using 200× magnification to visualize whether active zoospores were produced.
Hp Genome Sequence, Assembly, and Annotation
We grew Hp at room temperature (23–25C) in liquid PmTG medium with gentle agitation for approximately 2 weeks. We extracted Hp DNA using a Zolan and Pukkila [19] protocol modified by the use of 2% sodium dodecyl sulphate as extraction buffer in place of CTAB. We sequenced the Hp genome using a Roche 454 Genome Sequencer FLX with Titanium chemistry and standard Roche protocol. We screened and trimmed 1,100,797 reads of vector sequences and assembled them with Roche's GS De Novo Assembler. We improved the assembly by synteny-based alignment to the JAM81 genome sequence with Mercator [20].
We annotated the Hp genome with predicted proteins using the MAKER annotation pipeline [21]. MAKER predicts proteins based on homology with protein-coding sequences of other species, and with the consensus of the ab initio gene prediction algorithms GeneMark, AUGUSTUS, and SNAP. GeneMark is self training so we simply applied it to determine ab initio parameters. We trained AUGUSTUS using parameters provided in the MAKER package and previously determined Bd training parameters. We trained SNAP by iteratively running MAKER with SNAP Bd models and then retraining on the most confident gene model parameters from the initial run. All parameters files are available in http://fungalgenomes.org/public/Hp_JEL142/. Because MAKER's final set of predicted proteins (referred to hereafter as “Hp_Maker”) is a conservative estimate that relies upon the consensus of different prediction algorithms, we also used the set of ab initio predicted proteins in MAKER by GeneMark-ES [22] as an upper limit (referred to hereafter as “Hp_GeneMark”). Hp_Maker is not a perfect subset of Hp_GeneMark, so we considered both datasets when characterizing the proteome of Hp. We annotated Hp protein models by comparison to the Pfam database of protein domains [23] using HMMER 3.0 (http://hmmer.org/).
We used two methods that rely on different algorithms to confirm that we successfully identified the majority of Hp proteins. First, we used the eukaryotic genome annotation pipeline CEGMA to predict the number of core eukaryotic genes in the Hp alignment [24]. Second we determined the number of “chytrid-specific” orthologous groups that were present in the Hp genome. We defined chytrid-specific orthologous groups as those groups shared between all available Chytridiomycota genomes: two Bd isolates (JAM81 and JEL423) and one Spizellomyces punctatus isolate (DAOM BR117) (Table S1). We identified chytrid-specific orthologous groups using BLASTP [25] and OrthoMCL [26], and determined how many of these were also found within either set of Hp predicted proteins (i.e., Hp_Maker and Hp_GeneMark).
Bd Unique Genomic Features
We also used BLASTP and OrthoMCL to determine orthologous groups for all sampled taxa. These orthologous groups were used to determine “Bd-specific” genes which we defined as those groups or genes that were present in both sequenced Bd genomes (JAM81 and JEL423) but absent from all other sampled fungi. [Note that the Bd-specific gene set is distinct from the more broadly defined chytrid-specific gene set discussed above]. We used GO::TermFinder [27] to determine if the Pfam annotations for the set of Bd-specific genes showed enrichment for particular GO terms.
Bd Gene Family Expansions
We identified several gene family expansions in Bd through inspection of the top ten largest Bd-specific orthologous groups and inspection of enriched GO categories. We found gene family expansions in families with genes containing M36, S41, and Asp (both Asp and Asp_protease) protease Pfam signature domains (see Table S2 for sequences and their Pfam domain delimitation). We conducted an exhaustive search in the focal genomes for M36, S41, and Asp domains using HMMER3 (http://hmmer.org/). For Hp we conducted the HMMER3 search in both the MAKER and GENEMARK datasets. For S41 and Asp, the predicted proteins from Maker were subsets of those from GeneMark, so we only report GeneMark names. For M36 there were several Maker predicted proteins that were not included in the GeneMark set, so we report both Maker and GeneMark names. We then aligned the sequences of the protein domains for all members in each expanded family for the three Chytridiomycota genomes (Bd, Hp, and Spizellomyces punctatus) and one Blastocladiomycota outgroup (Allomyces macrogynus). We generated these alignments using the iterative alignment program MUSCLE [28]. After inspecting the alignments, we found that 8 M36 and 13 Asp protein sequences were missing >50% of their domain sequences. These partial sequences were likely mis-annotation or pseudogenes so we excluded them from further analysis (see Table S2B for identities of excluded partial sequences). After aligning the protein domain sequences of the remaining proteins (see Figure S1 for alignments), we reconstructed gene trees for each family using the Maximum Likelihood method implemented in RAxML [29]. We used the rapid bootstrap algorithm (400 replicates) with the Jones-Taylor-Thornton substitution matrix assuming a gamma model of rate heterogeneity. We report the Maximum Likelihood trees with the highest log likelihood score and bootstrap support values.
We calculated synonymous and non-synonymous substitution rates (Ks and Ka, respectively) with the yn00 program implemented in the PAML package [30] using full length annotated coding sequences. For each expanded protease gene family (containing M36, S41, and Asp domains) we calculated Ks and Ka of putative orthologs between all focal taxa pairs [i.e., chytrids (Bd, Hp, and Spizellomyces punctatus) and between all focal taxa and the outgroup (Allomyces macrogynus)]. We identified putative orthologs based on a cross-species reciprocal best match between any species pairs [31]. In addition, we used a second, more stringent approach that required sequence distances between reciprocal best matches to follow the relationships between the four focal species. Because the rate distributions from these two approaches were similar, we only report results from the first approach. Because yn00 does not robustly correct for multiple substitutions [32], and because Ks values are large between our focal taxa, we use Ks values to make a general comparison (within versus between species) for rates of molecular evolution.
We made rough divergence time estimates for the duplication events in the three expanded protease gene families using “node-Ks” as a proxy of time. The node Ks is defined as follows: for each node N in the mid-point rooted phylogeny, its Ks is the averaged Ks values between all operational taxonomic unit pairs across the two lineages that originated from N. There are no empirical estimates of chytrid substitution rates, so we do not propose specific dates for the duplication events. However, we do use a rough approximation for a reasonable substitution rate (following previous molecular evolution studies in fungi [33]) to test whether the timing of gene duplications was likely coincident with the emergence of Bd as a deadly amphibian pathogen.
Results
Taxon Sampling
The phylogenetic relationship among all 19 taxa in this study can be seen in Figure 1. As described above, we sampled genomes from across the diversity of five fungal phyla (i.e., Chytridiomycota, Blastocladiomycota, Zygomycota, Basidiomycota, Ascomycota). Our sampling scheme allowed us to determine, in a phylogenetic context, which elements of Bd's genome are shared with Hp and other fungal taxa.
Growth of Bd and Hp on Amphibian Skin
Both Bd and Hp grew well in standard PmTG growth media and produced viable zoospores throughout the entire 14 day observation period. However, only Bd survived on frog skin alone. Bd produced viable zoospores in the cane-toad skin treatment throughout the entire observation period, and after 14 days of incubation the Bd - frog skin solution was cloudy with chytrid growth and degraded skin (Figure 2). Conversely, Hp did not survive and reproduce on cane-toad skin alone. We observed viable zoospores for Hp in the cane-toad skin treatment only for the first three days (these zoospores most likely persisted from the initial inoculation), and after 14 days of incubation the Hp - cane-toad skin solution remained clear of chytrid growth and the cane-toad skin remained intact and not further degraded (Figure 2). We did not observe the growth of any bacterial or fungal contaminants in any of the treatments.

Hp Genome Sequence, Assembly, and Annotation
We achieved a roughly 11.2× coverage of the Hp genome (total number of aligned bases divided by final genome length, assuming that most of the genome is represented in the aligned reads). We assembled 922,085 screened and trimmed sequencing reads into 16,311 contigs (N50 = 36,162). We inferred a haploid genome size for Hp of 26.7 Mb, comparable to other Chytridiomycota genomes [Bd (JAM81) = 24.3 Mb, and Spizellomyces punctatus = 24.1 Mb]. We have deposited the Hp 454 reads in GenBank through the NCBI Sequence Read Archives under the accession SRA037431.1), and we have deposited the Whole Genome Shotgun project at DDBJ/EMBL/GenBank under the accession AFSM00000000 (the version described here is the first version, AFSM01000000).
We generated 5,355 high confidence MAKER predictions and 11,857 GeneMark ab initio predictions for Hp's protein coding genes. The number of predicted Hp proteins falls within the range of other annotated chytrid genomes (8,732 predicted proteins in Bd (JAM81) and 8,804 in Spizellomyces punctatus). The difference in number of Hp predicted protein numbers between MAKER and GeneMark is due to MAKER's conservative approach, which relies upon homology with protein-coding sequences of other species, and with the consensus of multiple ab initio gene prediction algorithms. We did not directly validate the number of expressed genes in our predicted protein sets with EST or RNA sequencing. However, we did compare the Hp predicted protein set to gene content in other species, which provides confidence in the Hp annotation and assembly. We recovered 92% (228/249) of the core eukaryotic genes using CEGMA in the Hp_Maker dataset. Similarly, we identified 3,216 orthologous groups of “chytrid-specific” proteins shared among both Bd isolates and S. punctatus (Table S3). Of the predicted chytrid-protein set we recovered 90% (2,885/3,216) in one or both Hp predicted protein sets (2,271 in Hp_Maker and 2,817 in Hp_GeneMark). Together, these results indicate that our sequencing efforts recovered a large proportion of genes that are predicted to occur in the Hp genome.
Bd Unique Genomic Features
We identified Bd-specific genes using the genomes of Hp and 17 additional fungi. We considered genes to be Bd-specific if they were present in orthologous groups in both sequenced Bd genomes (JAM81 and JEL423) and absent from all other fungi including Hp. Using OrthoMCL clustered proteins we defined 6,556 orthologous groups in Bd (Table S4). Of the 6,556 orthologous groups in Bd, 1700 were Bd-specific by the above definition. The Bd-specific orthologous groups were comprised of 1,974 protein encoding genes, 417 (21%) of which could be functionally categorized by a Pfam domain (with an e-value<0.01) (Table S4). We did not find any orthologous groups uniquely shared between Bd and the dermatophytes to the exclusion of all other fungal outgroups (Table S4). Although we defined orthologous groups using the sequenced genomes of both Bd isolates (JAM81 and JEL423), below we report gene IDs from JAM81 for simplicity.
We conducted enrichment analyses using Gene Ontology (GO) terms from the set of 417 Bd-specific genes associated with a Pfam domain and found enrichment in all 3 GO structured vocabularies: Cellular Component, Biological Process, and Molecular Function. We present all significantly enriched GO terms (with a corrected P-value of < = 0.05) for the Bd-specific gene set in Table 1. Briefly, in the Biological Process ontology we found enrichment for genes involved in metabolic processes and regulation of carbohydrates, proteins, and transcription. In the Cellular Component ontology we found enrichment of genes located extracellularly, in the nucleus, and in membranes. In the Molecular Function ontology we found enrichment for genes involved in zinc-ion binding, protein dimerization, DNA-binding, hydrolase activity, and protease and triglyceride lipase activity.

Within the set of Bd-specific and GO-enriched genes were several functional groups of particular interest for their possible role in Bd pathogenesis. First, many Bd-specific genes were proteases and were found in expanded gene families (see below). Second, the Bd-specific gene set was enriched for genes containing the Lipase_3 Pfam domain found in triacylglyceride lipases (6 of 417 in the Bd-specific gene list, vs 20 of 8732 in the genome, p<0.03) (BATDEDRAFT 93190, BATDEDRAFT 26490, BATDEDRAFT_ 86691, BATDEDRAFT 93191, BATDEDRAFT_89307, BATDEDRAFT_26489). Third, we identified 62 genes from the Bd-specific gene set that encode Crinkler or CRN-like microbial effectors (CRN), a class of genes previously reported only in oomycetes and not found in any of the other fungi considered here (Figure 3 and Table S5).

Bd Gene Family Expansions
We conducted more detailed analyses for three protease gene families that were identified in the Bd-specific gene set and showed GO term enrichment: metallo-, serine-type, and aspartyl proteases (M36, S41, and Asp Pfam domains, respectively). The Bd genome contained 38 metalloproteases, 32 serine-type proteases, and 99 aspartyl proteases, in all cases at least 4 times as many family members as Hp (Figure 3). We found that expansions of metalloproteases, serine-type proteases and aspartyl proteases were largely Bd specific, having occurred after the split between the Bd and Hp lineages from their most recent common ancestor (Summary in Figure 4, gene-names available in tree, Figure S2). In all three families, Bd had a greater number of gene copies than any of the other focal taxa, and the Bd gene copies were generally clustered together to the exclusion of homologues from other taxa. This clustering is consistent with lineage-specific gene family expansions in Bd (Figure 4). We observed a large number of metalloprotease genes not only in Bd but also in Allomyces macrogynus (38 and 31 gene family members, respectively) (Figure 3). However, the gene tree indicates that the expansion of metalloprotease genes in Bd and A. macrogynus were independent with most duplication events occurring after the divergence of Bd and A. macrogynus from their common ancestor (Figure 4A).

In addition to identifying many lineage-specific duplicates of proteases in Bd, we demonstrate that these Bd duplication events likely occurred significantly more recently than the divergence time between the species analyzed (Figure 5). To assess the timing of expansion in each protease gene family, we calculated synonymous substitution rate, Ks, between homologs and based on the phylogeny, we calculated a node Ks value for each lineage-specific duplication node (Figure 5, left panel). The median Ks values for the metallo-, serine-type, and aspartyl – proteases derived from Bd-specific duplications were 0.37, 0.14 and 0.24, respectively (Figure 5, left panel). We note that in the metalloprotease family there were similar numbers of lineage-specific duplications in Bd (24) and A. macrogynus (28). However the Ks values of Bd-specific duplicates were significantly lower A. macrogynus duplicates (median Ks: 0.37 and 1.56, respectively; Kolmogorov-Smirnov tests, p<4.6e-5), indicating that Bd-specific M36 duplications took place much more recently than the A. macrogynus duplications.

We also examined Ks values of putative orthologs between Bd and Hp, Bd and S. punctatus, and Bd and A. macrogynus (Figure 5B, right panel). As expected from the phylogenetic relationships of these four species [8], the median Ks for all species was high, but the median Ks for Bd-Hp (3.40) was significantly lower than that of Bd-S. punctatus (3.94) and Bd-A. macrogynus (4.03) (Kolmogorov-Smirnov tests, p<2.2e-16). Importantly, the median Bd-Hp orthologous Ks values were ∼9–24 fold higher than the median Ks of Bd lineage-specific duplicates. Therefore, the Bd-specific duplications occurred substantially more recently than the divergence of Bd and Hp. Previous molecular evolution studies in fungi have used a synonymous nucleotide substitution rate of 8.1e-9 substitutions per site per year to estimate the timing of molecular events [33]. If this substitution rate is reasonable for chytrid fungi, the duplication events leading to the metallo-, serine-type, and aspartyl protease gene family expansions in Bd would be millions of years old (Table S6). Even if the true substitution rate differs by several orders of magnitude, it is important to recognize that these protease duplication events occurred long before Bd emerged as a global pathogen of amphibians.
Discussion
To investigate the genomic changes that accompanied the evolution of pathogenicity, we compared Bd, the deadly chytrid pathogen of amphibians, with Hp, a closely related chytrid that is not a known pathogen of vertebrates. We confirm that Bd and Hp have different nutritional modes (Figure 2); unlike Hp, Bd is capable of growing on amphibian skin alone. Given the most chytrids are saprobes like Hp, Bd's ability to infect vertebrate skin likely arose after the divergence of Bd and Hp from their common ancestor. Fungal growth on vertebrate skin requires the expression of enzymes that break-down host epidermal tissue [10]–[13]. Because Bd causes chytridiomycosis by infecting frog skin [34]–[35] we were particularly interested in elements of the Bd genome whose evolution might have allowed Bd to colonize and degrade amphibian skin.
We compared the genomes of Bd and Hp in a broad taxonomic context of 18 diverse fungal genomes to identify genomic factors that make Bd unique. The Bd and Hp genomes are similar in size and number of predicted genes but show important differences in gene content. Therefore we could identify Bd-specific genes (i.e., genes that were found in Bd but not in Hp or other fungal outgroups). Bd-specific genes are enriched for GO terms related to extracellular and enzymatic activity. Many Bd-specific genes are members of recently expanded gene families (i.e., gene families with significantly more members than other fungal species). Below we discuss Bd-specific genes with particular emphasis on understanding how Bd may interact with its amphibian hosts.
Proteases are the most dramatically enriched class of Bd-specific genes. The Bd genome contains expanded gene families of metalloproteases, serine-type proteases, and aspartyl proteases. Each of these Bd gene families contains more than 30 family members and contains 4–10 times as many family members as found in Hp (Figure 3). Extracellular fungal proteases have been implicated in the adherence to, invasion of, and degradation of host cells by other fungal pathogens [10]–[13]. In particular, protease gene family expansions have been suggested as a link to pathogenesis in other fungal pathogens. Several fungal pathogens of vertebrates (e.g., Arthroderma benhamiae, Coccidioides spp, and Trichophyton spp.) exhibit gene family expansions specifically for metalloproteases and serine-type proteases [10], [36]–[37].
Here we strengthen the evidence implicating proteases in Bd pathogenesis in several ways. First and most importantly we demonstrate that protease gene families are not expanded in Hp and thus polarize the expansion events to a much shorter phylogenetic branch leading to Bd. Second, we present an additional protease gene family expansion. We previously reported the Bd gene family expansions for metallo - and serine proteases [9], and we now describe a dramatic expansion of aspartyl proteases in the Bd genome (Figure 3). Aspartyl proteases are of particular interest because they have been implicated in the adherence to and invasion of human host tissue by fungal pathogens (Candida spp.) [38]–[39]. Many genes in the expanded metallo -, serine - and aspartyl - protease gene families are highly expressed, and in some cases differentially expressed between Bd life stages [9]. Finally, we more rigorously document the dynamics of protease gene family expansions. Calculations using a range of reasonable substitution rates show that the three protease gene family expansions occurred substantially more recently than the divergence of Bd and Hp from their common ancestor.
It is important to caution that our comparative genomics results do not conclusively demonstrate a role for proteases as pathogenicity factors. First, we lack a specific mechanism by which proteases mediate Bd host invasion. Understanding the functional consequences of protease gene family expansions will require molecular assays to determine how specific enzymes contribute to host substrate metabolism. Second, protease gene family expansions are not always obviously correlated with fungal pathogenicity. For example we observed a large number of metalloprotease genes in Allomyces macrogynus and a variable number of aspartyl proteases in several of our outgroup taxa (Figure 4). These are independent expansion events relative to the Bd gene duplications and are not associated with a specific shift in substrate metabolism. Third, the estimated timing of the Bd protease gene duplications does not unambiguously link particular genes to the recent emergence of Bd as a global frog pathogen. Although the gene duplication events are relatively recent, most still likely occurred millions of years ago. More ancient duplication of protease genes may have set the stage for Bd's ability to infect frogs, but finer scale intraspecific data will be required to determine whether particular paralogs exhibit molecular signatures of recent selection.
While proteases may play the most obvious role in pathogen invasion and metabolism of host tissue, we also observed an enrichment of genes with triglyceride lipase activity in the Bd-specific gene set (Table 1). These enzymes are known to play a role in fungal-plant interactions [40], and have been hypothesized to play a role in at least one fungal-vertebrate interaction - between Malassezia furfur and the skin of its human host [41]. M. furfur incorporates host lipids into its own cell wall; this is thought to assist M. furfur in adhering to the host and evading the host's immune system. The extent to which Bd can utilize the products of triglyceride lipase activity for nutrition or adhesion remains to be tested. However, the enrichment of triglyceride lipase genes in the Bd-specific gene set suggests considering whether lipases could play a role in Bd's invasion of host tissue.
In addition to the genes that may be involved in host tissue metabolism we observed a large number of Bd-specific genes with similarity to microbial proteins known as Crinklers and Crinkler-like effectors (CRN). Microbial effectors in general act within the host cytoplasm to suppress host defenses and alter normal host cell metabolism [42]–[43]. It is unusual that a fungus contains CRN effectors as these proteins have so far only been reported from oomycetes, a group of important plant and fish pathogens within the kingdom Chromista [44]. CRN effectors are modular proteins consisting of a signal-peptide, a downstream translocator domain that allows CRN proteins to gain entry into host cells, and a C-terminus domain that interacts with host proteins [42]. While 62 unique Bd proteins show similarity to CRN effectors at the protein level, only one is predicted to be secreted (BATDEDRAFT_ 23205). Therefore while the function of putative CRN effectors in Bd remains to be determined, the possibility that they function as microbial effectors and interact with host elements merits further investigation.
We have sequenced the genome of Bd's closest known relative to develop hypotheses for genomic determinants of Bd's ability to infect and kill amphibians. However, the divergence between Bd and Hp is still substantial [8]. Recent research indicates that chytrids may be more ubiquitous than previously appreciated in both aquatic and terrestrial environments [45], and much chytrid diversity remains to be characterized. The discovery of additional taxa more closely related to Bd than Hp would help further localize genomic changes to the Bd lineage. Interspecific comparisons such as the one presented here can be complemented by intraspecific comparisons among Bd isolates to understand the evolutionary dynamics of genes hypothesized to play a role in Bd pathogenicity. However, robust hypothesis testing will require functional characterization of genes that may be important to Bd's ability to infect frogs. Bd currently lacks a transformation system in which to study gene function, but heterologous expression systems could potentially be used to determine specific gene functions. Additionally, understanding expression patterns of candidate genes under different nutrient conditions and during different stages of host invasion are likely to yield important insights. Ultimately, identifying the molecular mechanisms of host-pathogen interactions will provide new avenues for mitigating the devastating effects of chytridiomycosis.
Supporting Information
Zdroje
1. SmithKFGueganJF 2010 Changing Geographic Distributions of Human Pathogens. Annual Review of Ecology, Evolution, and Systematics, Vol 41 Palo Alto Annual Reviews 231 250
2. WoolhouseMGauntE 2007 Ecological origins of novel human pathogens. Crit Rev Microbiol 33 231 242
3. HoskissonPATrevorsJT 2010 Shifting trends in pathogen dynamics on a changing planet. Antonie Van Leeuwenhoek 98 423 427
4. BergerLSpeareRDaszakPGreenDECunninghamAA 1998 Chytridiomycosis causes amphibian mortality associated with population declines in the rain forests of Australia and Central America. Proc Natl Acad Sci U S A 95 9031 9036
5. LipsKRBremFBrenesRReeveJDAlfordRA 2006 Emerging infectious disease and the loss of biodiversity in a Neotropical amphibian community. Proc Natl Acad Sci U S A 103 3165 3170
6. LöttersSLa MarcaEStuartSGagliardoRVeithM 2004 A new dimension of current biodiversity loss. Herpetotropicos 1 29 31
7. SkerrattLFBergerLSpeareRCashinsSMcDonaldKR 2007 Spread of chytridiomycosis has caused the rapid global decline and extinction of frogs. EcoHealth 4 125 134
8. JamesTYLetcherPMLongcoreJEMozley-StandridgeSEPorterD 2006 A molecular phylogeny of the flagellated fungi (Chytridiomycota) and description of a new phylum (Blastocladiomycota). Mycologia 98 860 871
9. RosenblumEBStajichJEMaddoxNEisenMB 2008 Global gene expression profiles for life stages of the deadly amphibian pathogen Batrachochytrium dendrobatidis. Proc Natl Acad Sci U S A 105 17034 17039
10. BurmesterAShelestEGlöcknerGHeddergottCSchindlerS 2011 Comparative and functional genomics provide insights into the pathogenicity of dermatophytic fungi. Genome Biol 12 R7
11. MonodM 2008 Secreted Proteases from Dermatophytes. Mycopathologia 166 285 294
12. MonodMCapocciaSLéchenneBZauggCHoldomM 2002 Secreted proteases from pathogenic fungi. Int J Med Microbiol 292 405 419
13. da SilvaBAdos SantosALSBarreto-BergterEPintoMR 2006 Extracellular peptidase in the fungal pathogen Pseudallescheria boydii. Curr Microbiol 53 18 22
14. LetcherPMPowellMJChurchillPFChambersJG 2006 Ultrastructural and molecular phylogenetic delineation of a new order, the Rhizophydiales (Chytridiomycota). Mycol Res 110 898 915
15. RachowiczLJKnappRAMorganJATSticeMJVredenburgVT 2006 Emerging infectious disease as a proximate cause of amphibian mass mortality. Ecology 87 1671 1683
16. NotredameCHigginsDGHeringaJ 2000 T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol 302 205 217
17. Capella-GutiérrezSSilla-MartínezJMGabaldónT 2009 trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25 1972 1973
18. BarrDJS 1986 Allochytridium expandens rediscovered - morphology, physiology and zoospore ultrastructure. Mycologia 78 439 448
19. ZolanMEPukkilaPJ 1986 Inheritance of DNA methylation in Coprinus cinereus. Mol Cell Biol 6 195 200
20. DeweyC 2007 Aligning multiple whole genomes with Mercator and MAVID. Methods Mol Biol 395 221 236
21. CantarelBLKorfIRobbSMCParraGRossE 2008 MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res 18 188 196
22. Ter-HovhannisyanVLomsadzeAChernoffYOBorodovskyM 2008 Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res 18 1979 1990
23. FinnRDMistryJTateJCoggillPHegerA 2010 The Pfam protein families database. Nucleic Acids Res 38 D211 D222
24. ParraGBradnamKKorfI 2007 CEGMA: a pipeline to accurately annotate core genes in eukaryotic genornes. Bioinformatics 23 1061 1067
25. AltschulSFGishWMillerWMyersEWLipmanDJ 1990 Basic local alignment search tool. J Mol Biol 215 403 410
26. LiLStoeckertCJRoosDS 2003 OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res 13 2178 2189
27. BoyleEIWengSAGollubJJinHBotsteinD 2004 GO::TermFinder - open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 20 3710 3715
28. EdgarRC 2004 MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32 1792 1797
29. StamatakisALudwigTMeierH 2005 RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21 456 463
30. YangZH 2007 PAML 4: Phylogenetic analysis by maximum likelihood. Mol Biol Evol 24 1586 1591
31. HanadaKZouCLehti-ShiuMDShinozakiKShiuSH 2008 Importance of lineage-specific expansion of plant tandem duplicates in the adaptive response to environmental stimuli. Plant Physiol 148 993 1003
32. YangZHBielawskiJP 2000 Statistical methods for detecting molecular adaptation. Trends Ecol Evol 15 496 503
33. LynchMConeryJS 2000 The evolutionary fate and consequences of duplicate genes. Science 290 1151 1155
34. LongcoreJEPessierAPNicholsDK 1999 Batrachochytrium dendrobatidis gen et sp nov, a chytrid pathogenic to amphibians. Mycologia 91 219 227
35. VoylesJYoungSBergerLCampbellCVoylesWF 2009 Pathogenesis of Chytridiomycosis, a Cause of Catastrophic Amphibian Declines. Science 326 582 585
36. JoussonOLéchenneBBontemsOCapocciaSMignonB 2004 Multiplication of an ancestral gene encoding secreted fungalysin preceded species differentiation in the dermatophytes Trichophyton and Microsporum. Microbiology 150 301 310
37. SharptonTJStajichJERounsleySDGardnerMJWortmanJR 2009 Comparative genomic analyses of the human fungal pathogens Coccidioides and their relatives. Genome Res 19 1722 1731
38. KaurRMaBCormackBP 2007 A family of glycosylphosphatidylinositol-linked aspartyl proteases is required for virulence of Candida glabrata. Proc Natl Acad Sci U S A 104 7628 7633
39. MonodMBorg-von ZepelinM 2002 Secreted aspartic proteases as virulence factors of Candida species. Biol Chem 383 1087 1093
40. GaillardinC 2010 Lipases as Pathogenicity Factors of Fungi. TimmisKN Handbook of Hydrocarbon and Lipid Microbiology Springer 3259 3268
41. BrunkeSHubeB 2006 MfLIP1, a gene encoding an extracellular lipase of the lipid-dependent fungus Malassezia furfur. Microbiology 152 547 554
42. HaasBJKamounSZodyMCJiangRHYHandsakerRE 2009 Genome sequence and analysis of the Irish potato famine pathogen Phytophthora infestans. Nature 461 393 398
43. KamounS 2006 A catalogue of the effector secretome of plant pathogenic oomycetes. Annu Rev Phytopathol 44 41 60
44. Cavalier-SmithTChaoEEY 2006 Phylogeny and megasystematics of phagotrophic heterokonts (kingdom Chromista). J Mol Evol 62 388 420
45. FreemanKRMartinAPKarkiDLynchRCMitterMS 2009 Evidence that chytrids dominate fungal communities in high-elevation soils. Proc Natl Acad Sci U S A 106 18315 18320
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 11
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Multiple Candidate Effectors from the Oomycete Pathogen Suppress Host Plant Immunity
- The Splicing Factor Proline-Glutamine Rich (SFPQ/PSF) Is Involved in Influenza Virus Transcription
- A TNF-Regulated Recombinatorial Macrophage Immune Receptor Implicated in Granuloma Formation in Tuberculosis
- SH3 Domain-Mediated Recruitment of Host Cell Amphiphysins by Alphavirus nsP3 Promotes Viral RNA Replication
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy