
Protease-Resistant Prions Selectively Decrease Shadoo Protein
The central event in prion diseases is the conformational conversion of the cellular prion protein (PrPC) into PrPSc, a partially protease-resistant and infectious conformer. However, the mechanism by which PrPSc causes neuronal dysfunction remains poorly understood. Levels of Shadoo (Sho), a protein that resembles the flexibly disordered N-terminal domain of PrPC, were found to be reduced in the brains of mice infected with the RML strain of prions [1], implying that Sho levels may reflect the presence of PrPSc in the brain. To test this hypothesis, we examined levels of Sho during prion infection using a variety of experimental systems. Sho protein levels were decreased in the brains of mice, hamsters, voles, and sheep infected with different natural and experimental prion strains. Furthermore, Sho levels were decreased in the brains of prion-infected, transgenic mice overexpressing Sho and in infected neuroblastoma cells. Time-course experiments revealed that Sho levels were inversely proportional to levels of protease-resistant PrPSc. Membrane anchoring and the N-terminal domain of PrP both influenced the inverse relationship between Sho and PrPSc. Although increased Sho levels had no discernible effect on prion replication in mice, we conclude that Sho is the first non-PrP marker specific for prion disease. Additional studies using this paradigm may provide insight into the cellular pathways and systems subverted by PrPSc during prion disease.
Published in the journal:
. PLoS Pathog 7(11): e32767. doi:10.1371/journal.ppat.1002382
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002382
Summary
The central event in prion diseases is the conformational conversion of the cellular prion protein (PrPC) into PrPSc, a partially protease-resistant and infectious conformer. However, the mechanism by which PrPSc causes neuronal dysfunction remains poorly understood. Levels of Shadoo (Sho), a protein that resembles the flexibly disordered N-terminal domain of PrPC, were found to be reduced in the brains of mice infected with the RML strain of prions [1], implying that Sho levels may reflect the presence of PrPSc in the brain. To test this hypothesis, we examined levels of Sho during prion infection using a variety of experimental systems. Sho protein levels were decreased in the brains of mice, hamsters, voles, and sheep infected with different natural and experimental prion strains. Furthermore, Sho levels were decreased in the brains of prion-infected, transgenic mice overexpressing Sho and in infected neuroblastoma cells. Time-course experiments revealed that Sho levels were inversely proportional to levels of protease-resistant PrPSc. Membrane anchoring and the N-terminal domain of PrP both influenced the inverse relationship between Sho and PrPSc. Although increased Sho levels had no discernible effect on prion replication in mice, we conclude that Sho is the first non-PrP marker specific for prion disease. Additional studies using this paradigm may provide insight into the cellular pathways and systems subverted by PrPSc during prion disease.
Introduction
Prion diseases, such as Creutzfeldt-Jakob disease (CJD) in humans, bovine spongiform encephalopathy (BSE), and chronic wasting disease (CWD) in cervids, are invariably fatal neurodegenerative disorders caused by the accumulation of misprocessed prion protein (PrPSc) in the brain. The central pathognomonic event in prion diseases is the post-translational refolding of the cellular prion protein (PrPC) into PrPSc, a partially protease-resistant and β-sheet-enriched conformation that is infectious [2], [3]. Mice lacking PrPC fail to develop prion disease and do not propagate infectious prions in their brains, indicating that PrPC expression is required for prion replication [4], [5]. Despite a clear involvement in pathogenesis, the mechanism by which PrPSc causes neuronal dysfunction during prion disease remains obscure. Although PrPC is known to interact with or reside in close spatial proximity to numerous other proteins in the cell membrane [6], [7], [8], none of these identified proteins has been shown to be associated with prion disease pathogenesis or prion replication.
The mammalian prion protein family consists of three members: PrPC; Doppel (Dpl), a testes-specific protein involved in the proper functioning of the male reproductive system [9], [10]; and Shadoo (Sho), a recently identified neuronal paralog of PrPC encoded by the Sprn gene [1], [11]. Unlike Dpl, which resembles the alpha-helical C-terminal domain of PrPC [12], Sho is reminiscent of the flexibly disordered N-terminal domain of PrP. In particular, the similarity between PrP and Sho is striking within the alanine/glycine-rich hydrophobic tract. This region of PrP is of particular interest because (i) it is the most-conserved region among PrP ortholog sequences; (ii) it is conformationally altered in PrPSc [13]; (iii) its deletion renders PrP toxic to cerebellar neurons [14], [15]; and (iv) deletions within this region result in a loss of PrPC-associated neuroprotective activity [1]. Like PrPC, Sho is an N-glycosylated GPI-anchored protein that is expressed in the brain and exhibits neuroprotective properties in response to various neurotoxic stimuli in cells [1], [16]. Both PrPC and Sho undergo endoproteolytic cleavage just N-terminal to the hydrophobic tract to generate a C-terminal fragment termed C1 [1],[17]. PrPC is also cleaved in the vicinity of residue 88 to generate a distinct C-terminal fragment termed C2 [17]. Production of the C2 fragment is greatly increased during prion disease, likely due to the inability of the cell to clear aggregated PrPSc via lysosomal degradation [18], [19]. Although the biological role of Sho in the brain is currently unknown, knockdown of Sprn in mouse embryos lacking PrP expression results in a lethal phenotype [20], arguing for an overlapping function with PrPC. However, Sho levels are unchanged in the brains of adult mice lacking PrPC [1], indicating that cross-regulation of protein expression does not occur between the two proteins.
The influence, if any, of Sho on prion replication and pathogenesis remains to be evaluated. Analogously to Prnp encoding the prion protein, polymorphisms have been identified in the human, ovine, and murine Sprn genes; whether these are linked to prion disease incubation time or susceptibility is not completely understood [21], [22], [23], [24]. Recently, it has been shown that Sho protein levels are reduced in the brains of clinically ill mice infected with the RML strain of prions [1]. This observation suggests that Sho protein levels may be inherently linked to prion replication or reflect the presence of PrPSc in the brain. Indeed, Sho is known to reside in spatial proximity to PrPC within the cell membrane as demonstrated by cross-linking experiments [8]. Furthermore, nonsense mutations in the SPRN gene were found in two patients with variant CJD, but not in control patients [21]. These results argue that a thorough evaluation of the effect of Sho on prion disease is warranted.
Here we report that levels of Sho and PrPSc were inversely correlated in the brains of prion-infected rodents and sheep. This association was observed for 14 different prion strains and required the presence of the N-terminal domain of PrP. Furthermore, Sho overexpression did not influence the kinetics of prion replication in mice. Additional studies of the relationship between Sho and PrP may help to reveal neurotoxic mechanisms utilized by PrPSc during prion disease.
Results
Decreased Sho levels in the brains of prion-infected rodents and sheep
Previously, it was shown that Sho protein levels were reduced in the brains of clinically ill, wild-type (wt) mice infected with the RML strain of prions [1]. To investigate whether this phenomenon occurs with other prion strains and other animal species, we examined Sho levels in the brains of wt CD-1 mice, meadow voles, and sheep infected with prions. Consistent with previous findings, Sho levels were notably reduced in the brains of wt CD-1 mice and meadow voles infected with RML prions at the onset of neurological symptoms (Figure 1A). In addition, we observed diminished Sho levels in the brains of three sheep with natural (non-experimental) scrapie and in a sheep inoculated with the CH1641 scrapie strain [25] (Figure 1B). Sho levels also were decreased in RML-infected Tg(NSE-MoPrP) mice, which selectively express PrPC in neurons (Figure 1C).

Despite the reduction in Sho protein levels in prion-infected mice, Sprn mRNA levels did not decrease in two different lines of wt mice (FVB and C57BL/6) infected with RML prions relative to mice inoculated with uninfected brain homogenate (Figure 1D; data from [26]), consistent with findings from others [22], [27]. Thus, depletion of Sho during prion disease occurs via a post-transcriptional process.
A recent paper demonstrated that recombinant Sho readily converts to amyloid under physiological conditions [28]. We therefore tested if sequestration of Sho in large SDS-insoluble aggregates within prion-infected brains hinders the detection of Sho by Western blotting. We solubilized prion-infected hamster brains with 6 M guanidine hydrochloride but found no increase in the Sho signal (Figure S1), arguing that Sho does not form insoluble aggregates in the brains of prion-infected rodents.
Inverse relationship between Sho and PrPSc levels during prion disease
To investigate the relationship between Sho and PrPSc levels, we examined Sho levels in wt FVB mice at different time points following inoculation with RML prions. At 74 days postinoculation (dpi), Sho levels in the brain began to decrease as protease-resistant PrPSc first became visible by Western blotting (Figure 2A). As PrPSc levels continued to increase until the mice reached the clinical phase of prion disease at 133 dpi, Sho protein levels also decreased progressively. Relative quantification of Sho and PrPSc levels in the brains of RML-infected mice revealed that the inflection points for Sho depletion and protease-resistant PrPSc accumulation were at ∼70 dpi (Figure 2B), although small amounts of protease-resistant PrPSc were apparent by 60 dpi. Statistical analysis revealed an inverse correlation between Sho and protease-resistant PrPSc levels (Figure 2C).

Decreased Sho levels in diverse Tg models of prion disease
We next investigated whether Sho levels decrease upon prion infection with other strains and in other animal species. In wt FVB mice (which express the PrP-A allotype), we examined 3 additional prion strains: 22L, Me7, and 301V (Figure 3A). Both 22L and Me7 prions originated from sheep with scrapie, like RML prions, and were passaged in wt mice [29], [30]. The 301V strain was derived from passage of brain homogenate from a cow with BSE to VM mice [31], and then passaged in B6.I mice, which express the PrP-B allotype, or in FVB mice (Figure 3A). Another mouse-passaged scrapie strain, 87V [32], was also passaged in B6.I mice. In all cases, Sho levels in the brain, examined at the onset of neurological symptoms, were depleted in response to prion infection.

Similarly, a near-complete reduction in Sho levels was observed in hamsters infected with the Sc237, 139H, HY, or DY strains of prions (Figure 3B). Sc237 prions originated from sheep with scrapie, then passaged in Syrian hamsters; 139H was also isolated in scrapie-infected sheep, passaged first in mice then in Syrian hamsters [33]. HY and DY were isolated by passage of transmissible mink encephalopathy (TME) prions into Syrian hamsters [34]. The incubation periods for all prion strain-host combinations examined are shown in Table S1.
We also tested whether Sho levels were diminished in response to infection with naturally occurring prion strains. SSBP/1 scrapie prions were injected into transgenic (Tg) mice expressing ovine PrP, and elk CWD prions were inoculated in Tg mice expressing elk PrP (Figure 3C). In addition, Tg mice expressing human PrP with either the M129 or V129 polymorphism were infected with sporadic CJD prions of subtype MM1 and VV2, respectively (Figure 3D). All inoculated Tg mice showed decreased Sho levels as PrPSc accumulated during prion disease. Taken together, these results demonstrate that Sho depletion in the brain occurs in different animal species in response to a variety of prion strains.
Decreased Sho in prion-infected cultured cells
Next, we assessed whether Sho levels were decreased in cultured cells replicating prions. Because N2a neuroblastoma cells express very low levels of endogenous Sho that are not detectable by Western blotting [8], we generated an N2a cell line that stably overexpresses Sho; these cells are denoted N2a-Sho (Figure 4A). Western blot analysis revealed that the Sho protein expressed in N2a-Sho cells exhibited similar biochemical properties to Sho in mouse brains [1], including N-glycosylation and endoproteolytic processing to generate a C-terminal (ShoC1) fragment (Figure 4A).

Similar to ScN2a cells that stably propagate RML prions [35], N2a-Sho cells were infected with RML prions, and denoted ScN2a-Sho cells. Following extensive passage to remove all traces of the inoculum, Sho levels were assessed in infected ScN2a-Sho cells. For comparison, Sho levels were also determined in uninfected N2a-Sho cells. No consistent decrease in Sho levels was observed in infected ScN2a-Sho cells despite the presence of protease-resistant PrP (Figure 4B, C). Because N2a cells show heterogeneous potential for infection with prions, even in clonal populations of cells [36], we performed further subcloning of the ScN2a-Sho cells in order to isolate subclones that were more uniformly infected. Two such subclones with the highest levels of protease-resistant PrPSc (referred to as ScN2a-Sho-1 and ScN2a-Sho-2 cells) were selected for further analysis. In these lines, Sho levels were decreased by 40–45%, a significant reduction compared to Sho levels in uninfected N2a-Sho cells (Figure 4B, C). These results demonstrate that Sho reduction can also occur in cultured cells as a result of prion infection. In contrast, we did not observe any change in Sho following amplification of hamster Sc237 prions in vitro by protein misfolding cyclic amplification (PMCA), a cell-free system for studying prion replication [37] (Figure S2).
Decreased Sho is specific for protease-resistant PrPSc
To address the specificity of Sho depletion to prion disease, we examined levels of Sho in the brains of mice with other neurodegenerative illnesses not associated with the accumulation of protease-resistant PrPSc. These mice include two Tg mouse models of Alzheimer's disease (AD); Tg(MoDpl)/Prnp0/0 mice that show cerebellar degeneration as a result of Dpl expression [38]; a Tg mouse model of Parkinson's disease; and a Tg mouse model of frontotemporal dementia. In the AD mouse models, Tg(APP23) and Tg(CRND8) mice [39], [40], expression of mutant amyloid precursor protein results in the progressive accumulation of Aβ. Despite high levels of cerebral Aβ accumulation in aged Tg(APP23) and Tg(CRND8) mice, Sho levels were similar to those in wt mice and younger control mice (Figure 5A). Similarly, Sho levels in the brains of Tg(MoDpl)/Prnp0/0 mice did not diminish as a result of Dpl-induced degeneration (Figure S3). In Tg(SNCA,A53T) mice that express mutant human alpha-synuclein associated with Parkinson's disease, Sho levels were also unaltered (Figure S4). Tg(MAPT,P301S) mice do not show decreased Sho levels in their brains despite expressing mutant human tau causing frontotemporal dementia (Figure S4). Together, these observations argue that Sho depletion in the brain is a specific indicator of protease-resistant PrPSc accumulation in prion disease.

Next, we investigated Sho levels in Tg mice with a neurodegenerative illness characterized by the accumulation of protease-sensitive PrPSc in the brain. Tg mice expressing mutant MoPrP(P101L), analogous to the P102L mutation causing Gerstmann-Sträussler-Scheinker (GSS) disease in humans, show spongiform degeneration and astrocytosis reminiscent of prion disease but do not harbor protease-resistant PrPSc in their brains [41], [42]. However, the disease was transmissible to Tg mice expressing lower levels of mutant PrP(P101L), arguing for the presence of protease-sensitive PrPSc [43], [44]. Only a small decrease in Sho levels was observed in the brains of clinically ill Tg(MoPrP,P101L) animals compared to the reduction observed in wt mice infected with RML prions (Figure 5B, C). PrP in the brains of these animals was precipitable by PTA but sensitive to PK digestion, arguing for the presence of protease-sensitive PrPSc. Sho levels were also not substantially decreased in mice infected with MoSP2 prions (Figure 5D), a protease-sensitive synthetic prion strain generated from recombinant PrP amyloid [45]. The presence of protease-sensitive PrPSc in the brains of MoSP2-infected mice was confirmed by their ability to seed the polymerization of recombinant PrP in the real-time quaking-induced conversion (RT-QuIC) assay [46], which is derived from the amyloid seeding assay [47]. Cumulatively, these results argue that Sho is a specific indicator of protease-resistant PrPSc conformations in the brains of animals with prion disease.
Interaction between Sho and PrP in prion-infected cells
To investigate whether Sho and PrPSc directly interact with each other, we performed coimmunoprecipitation analyses. When Sho was immunoprecipitated from ScN2a-Sho-1 cells, coprecipitation of PrP was observed (Figure 6). In contrast, no copurification of PrP was observed when the immunoprecipitation was performed on uninfected N2a-Sho cells, suggesting that Sho binds to misfolded PrP but does not interact with PrPC. As a control for the non-specific binding of aggregated forms of PrPSc to the immunoprecipitation matrix, we performed immunoprecipitations on ScN2a-Sho cells in which the anti-Sho antibody was omitted. No copurification of PrP was observed under these conditions. Furthermore, only minute amounts of PrP were observed after performing Sho immunoprecipitations on ScN2a cells, which do not express detectable levels of Sho (Figure 6). These results indicate that Sho possesses an intrinsic ability to bind misfolded PrP, providing a potential mechanism for the PrPSc-correlated depletion of Sho during prion disease.

Sho overexpression does not influence incubation times
Having shown that Sho levels are linked to protease-resistant PrPSc levels in the brains of prion-infected mice, we next asked whether Sho levels have any effect on prion replication. We therefore generated Tg mice overexpressing murine or human Sho under the control of the hamster Prnp promoter, denoted Tg(MoSho) and Tg(HuSho) mice, respectively. Two independent Tg(MoSho) lines were obtained, which express Sho in the brain at approximately 12 - and 20-fold the levels present in the brains of wt mice (Table 1). Two distinct Tg(HuSho) lines were also generated. Both Tg(MoSho) lines remained free of neurological symptoms up to 500 days of age. At older ages, a proportion of Tg(MoSho) mice began to exhibit signs of neurological illness including circling, ataxia, and dysmetria (Figure S5). No Thioflavin S–reactive deposits were observed in the brains of aged Tg(MoSho) mice, indicating that overexpression of Sho did not lead to the formation of amyloid in the brain. The most prominent neuropathological finding in aged Tg(MoSho) mice was mild vacuolation accompanied by astrocytic gliosis, predominantly in white matter tracts (Figure S5). However, these changes are consistent with normal aging in mice, indicating that prolonged overexpression of Sho likely has minimal consequences for normal brain homeostasis.

As determined by Western blotting, Sho protein in the brains from both lines of Tg(MoSho) mice exhibited N-glycosylation and was subject to endoproteolytic trimming to generate the ShoC1 fragment (Figure 7A). Levels of PrPC were unchanged in uninfected Tg(MoSho) and Tg(HuSho) mice compared to wt mice (Figure 7B), confirming the absence of cross-regulation of protein expression between the two proteins. Tg(MoSho) mice were then inoculated with three different mouse-adapted prion strains: RML, Me7, and 301V. Incubation periods for the three inocula were not substantially different between wt mice and Tg(MoSho) mice (Table 1). For RML prions, no significant difference (P>0.05 by the Log-rank test) was observed between the survival curves for wt mice and either of the Tg(MoSho) lines (Figure 7C), indicating that Sho levels do not influence the incubation period in mice. Infection of either line of Tg(HuSho) mice with RML prions also failed to alter the incubation period compared to that of wt mice (Table 1). The banding pattern and level of PK-resistant PrPSc in the brains of RML-infected, Tg(MoSho) mice was similar to that of RML-infected, wt mice (Figure 7D). Neuropathological signs of prion disease, including spongiform degeneration and PrP deposition, were similar in prion-infected Tg(MoSho) mice and infected wt FVB mice, regardless of the prion strain used (Figure 7E–J, S6). In contrast to a recent report [48], we found no evidence of Thioflavin S–reactive amyloid deposits in the brains of prion-infected Tg(MoSho) or Tg(HuSho) mice. Collectively, these results argue that increased Sho levels in the brain do not modulate prion disease in mice.

We next examined Sho levels in the brains of prion-infected Tg(MoSho) mice. By Western blotting, Sho levels were clearly decreased in the brains of RML-inoculated Tg(MoSho) mice sacrificed at the onset of clinical signs of prion disease, compared to age-matched, uninfected controls (Figure 8A). Levels of full-length Sho and the ShoC1 fragment decreased proportionately in prion-infected Tg(MoSho) mice (Figure 8B). Quantification of Sho levels in wt and Tg(MoSho) mice at the onset of clinical prion disease revealed that Sho levels decreased ∼70% in each line, regardless of the initial Sho expression level (Figure 8C). This observation may indicate that there are two distinct pools of Sho in the brain: one that can be eliminated by the presence of protease-resistant PrPSc and one that is refractory to this phenomenon. Furthermore, it suggests that Sho levels are substoichiometric to PrPSc levels in the brain since the same proportional decrease in Sho was observed in RML-infected Tg(MoSho) mice despite much higher levels of Sho expression. In two lines of RML-infected Tg(HuSho) mice, Sho levels were also reduced compared to uninfected controls (Figure 8D). When taken together, these results argue that although Sho and PrPSc levels are inversely correlated, Sho levels do not affect the onset of prion disease in mice.

The N-terminus and GPI anchor in PrP influence Sho reduction
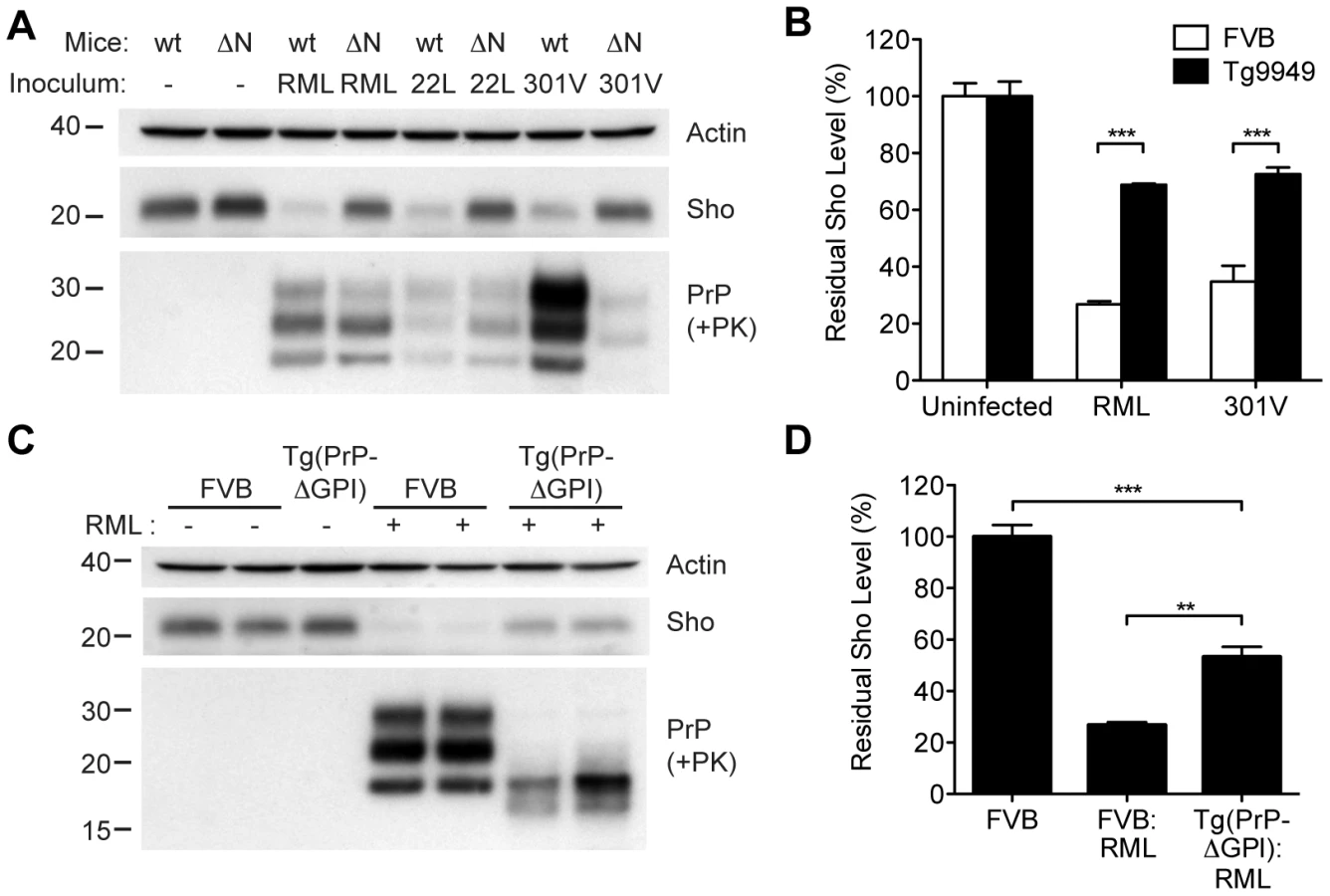
In order to gain mechanistic insight into the diminution of Sho protein during prion disease, we examined Sho levels in prion-infected transgenic mice that express various PrP constructs. Tg9949 mice express N-terminally truncated PrP lacking residues 23–88 at ∼16× the PrP levels found in wt mice. These mice are susceptible to prion disease, albeit with longer-than-expected incubation periods [49]. We inoculated Tg9949 mice with three different mouse-passaged prion strains: RML, 22L, and 301V. Tg9949 mice were susceptible to all three strains with mean incubation periods between 104 and 161 days (Table S1). Sho levels in their brains were evaluated when the mice developed clinical signs of prion disease. Surprisingly, Sho levels in prion-infected Tg9949 mice were decreased by ∼30% (Figure 9A), compared to the ∼70% diminution observed in prion-infected, wt mice (Figure 9B). Despite these significant differences in Sho levels, amounts of protease-resistant PrPSc were similar in Tg9949 mice and wt mice after inoculation with either RML or 22L prions (Figure 9A). Similar results were found in Tg mice expressing full-length PrP lacking its GPI anchor [Tg(PrP-ΔGPI) mice; 50]. Following infection with RML prions, levels of Sho decreased by only ∼45% in Tg(PrP-ΔGPI) mice compared to ∼75% in wt mice (Figure 9C, D). These results indicate that the presence of both the N-terminus and GPI anchor of PrP significantly influence the strong inverse relationship between PrPSc and Sho during prion disease.
Correlation of Sho depletion and PrPSc C2 fragment
Although Sho levels are clearly correlated with protease-resistant PrPSc levels, the relative ratios of Sho and PrPSc may differ by prion strain. To investigate this issue, we challenged meadow voles with three distinct prion strains (Table S1); voles are known to be susceptible to a variety of prion strains in a PrP sequence–independent manner [51]. In meadow voles, Sho levels were decreased by ∼90% in RML-infected animals compared to ∼80% in Sc237 - or 301V-infected animals (Figure 10A). However, based on the examination of four animals per strain, similar or higher levels of PK-resistant PrPSc were consistently found in the Sc237-infected brains compared to the RML-infected brains (Figure 10B), indicating that prion strain-specific differences in the extent of Sho reduction cannot be explained by the relative amount of protease-resistant PrPSc in the brain. To test if a different PrPSc species may correlate better with the extent of Sho reduction, we digested the prion-infected meadow vole brain homogenates with thermolysin (TL), which is a bacterial protease that completely digests PrPC but leaves PrPSc intact. Notably, unlike PK, TL can be used to isolate full-length PrPSc due to an absence of preferred cleavage sites in the N-terminal domain of PrP 52. When brain homogenates were digested with TL and then deglycosylated with PNGaseF to reveal the C2 proteolytic fragment of PrPSc, which corresponds to the generation of “endogenous” protease-resistant PrP in prion-infected cells due to intracellular proteolysis [19], [53], a significant inverse relationship between Sho and PrPSc C2 fragment levels was found for the three prion strains (Figure 10B, C). For example, brains from meadow voles infected with RML prions exhibited the lowest Sho level and the highest PrPSc C2 fragment level. No C2 fragment was observed in uninfected voles following TL digestion, indicating that the extent of Sho reduction is correlated with the PrPSc C2 fragment, not the PrPC C2 fragment. A similar relationship was observed in the brains of prion-infected Tg(MoSho) mice. Tg24474 mice infected with RML or Me7 prions exhibited the largest reduction in Sho levels and had higher levels of the PrPSc C2 fragment compared to mice infected with 301V prions, which had higher residual Sho levels and lower levels of the PrPSc C2 fragment (Figure 10D). Thus, prion strain-specific differences in the extent of Sho depletion can be explained by the relative amount of PrPSc C2 fragment produced for a given strain.

Discussion
We describe here a quantitative relationship between Sho and protease-resistant PrPSc levels in the brains of prion-infected animals. Brain Sho levels were reduced in response to numerous natural and experimental prion strains, but not in response to the accumulation of protease-sensitive PrPSc, Aβ, alpha-synuclein, or tau aggregates, indicating that Sho depletion is a specific indicator of protease-resistant PrPSc in the brain. Thus, our experiments, as well as similar results presented in this issue by Westaway et al. 54 indicate that Sho is not a mere bystander during prion disease even though Sho levels did not modulate the kinetics of prion replication in mice.
Sho as an indicator of PrPSc in the brain
The quantitative inverse relationship between Sho and protease-resistant PrPSc levels in the brain suggests that the relative levels of these two proteins are mechanistically linked. Sho depletion occurred not only in experimentally infected rodents (Figure 1A), but also in sheep naturally infected with scrapie (Figure 1B), eliminating the possibility that this phenomenon results from an artifact of intracerebral inoculation. Furthermore, Sho reduction is not a general indicator of neuronal dysfunction or protein aggregation because Sho levels were unaltered in the brains of Tg mice with large quantities of either Aβ, alpha-synuclein, or tau deposits (Figure 5, S4). Because reduction of human Sho levels occurred in response to mouse PrPSc accumulation (Figure 8D) and reduction of mouse Sho occurred in response to human PrPSc (Figure 3D), Sho depletion is unlikely to require species-specific contacts between Sho and protease-resistant PrPSc, in contrast to the species-specific interactions between PrPC and PrPSc necessary for efficient prion replication. Because Sho expression appears to be restricted to neurons in the brain [1], [55] and neuron-specific expression of PrPC is sufficient to elicit Sho depletion in prion-infected mice (Figure 1C), it is likely that interactions between Sho and PrPSc in neurons are responsible for reductions in the Sho protein. Coimmunostaining of PrPSc deposits and Sho in prion-infected animals may more precisely pinpoint the location of Sho depletion in the brain.
A recent paper has also described Sho depletion in response to various mouse-adapted prion strains, including strain-specific effects on Sho levels [56]. In our study, infection with all prion strains characterized by protease-resistant PrPSc resulted in substantial depletion of Sho although strain-specific responses were also observed. For a given prion strain, Sho levels correlated well with levels of protease-resistant PrPSc in the brain (Figure 2). However, differences in the extent of Sho depletion between different prion strains were not correlated with absolute levels of protease-resistant PrPSc in the brain at the onset of clinical disease but with relative amounts of the PrPSc C2 fragment (Figure 10B–D). This finding is consistent with the much higher extent of Sho reduction in mice infected with mouse-adapated kuru compared to mice challenged with mouse-adapted variant CJD, which appears to have much less endogenous truncation of PrPSc [56]. It will be of interest to determine whether the extent of Sho depletion represents a consistent biochemical signature for a given protease-resistant prion strain that can be used to identify and classify different prion strains.
Whether Sho levels are also decreased in the brains of patients with sporadic or genetic prion disease remains to be determined. It will be interesting to compare Sho levels in the brains of patients with CJD and GSS with the variably protease-sensitive prionopathy (VPSPr) described recently [57]. These studies will require the generation of antibodies directed against the human Sho sequence that exhibit higher affinity than those currently available. A recent study has shown that secreted forms of Sho are generated in cells [28], suggesting that Sho may exit the brain and measurement of these secreted Sho forms might indicate total Sho levels in the brain. Furthermore, examination of Sho levels in biologically accessible fluids, such as CSF, from patients with prion disease might provide a more specific indicator of prion disease compared to tests currently used that determine 14-3-3 and total tau levels [58]. Sho may therefore represent the first non-PrP marker specific for prion disease in humans.
Potential mechanisms of Sho depletion in prion-infected brains
It is currently unknown whether decreased Sho levels in prion-infected brains result from decreased translation or increased turnover, although the latter seems more likely because PrPSc is primarily located at the cell surface and within endocytic vesicles [59]. Multiple lines of evidence argue that Sho is degraded via an endocytic pathway during prion disease by a PrPSc-mediated process. First, Sho levels were reduced to a much smaller extent in prion-infected Tg9949 mice expressing N-terminally truncated PrP(Δ23–88) compared to wt mice (Figure 9A and B). Deletion of residues 23–28 or residues 51–90 from PrP has been shown to reduce or eliminate the endocytosis of PrPC [60], [61], suggesting that endocytosis may be necessary for efficient Sho depletion. Second, Tg mice expressing anchorless PrP, in which endocytosis of PrP is likely to be impaired due to an absence of the GPI anchor and associated lipid raft targeting, also exhibited a less pronounced reduction in Sho levels following prion infection (Figure 9C and D). Third, Sho levels were inversely correlated with relative levels of the PrPSc C2 proteolytic fragment (Figure 10C), which is generated via the action of lysosomal proteases, including cathepsins [19]. Finally, coimmunoprecipation of Sho and PrPSc was observed ScN2a-Sho cells (Figure 6), suggesting that Sho may “piggyback” on aggregated PrPSc species in cells and be targeted towards intracellular degradation pathways. Aggregated PrPSc species are known to exhibit promiscuous binding to various surfaces and monoclonal antibodies due to nonspecific hydrophobic interactions [62], [63]. The observation that Sho levels were only marginally decreased in mice propagating protease-sensitive prions may be explained by decreased amounts of highly aggregated, protease-resistant PrP conformers or an increase in smaller, misfolded PrP species that are neurotoxic but are less prone to nonspecific hydrophobic interactions and more susceptible to protease digestion [64]. In agreement with this notion, the amount of PTA-precipitable PrPSc-like conformers in Tg(MoPrP,P101L) mice constitutes only ∼15% of total PrP [65], whereas a much greater proportion of PrP is PTA-precipitable in mice infected with laboratory prion strains, such as RML. Both full-length and N-terminally trimmed Sho species were decreased in the brains of prion-infected Tg(MoSho) mice, suggesting that the hydrophobic tract and C-terminal domain of Sho are sufficient for PrPSc-mediated interactions that result in Sho depletion during prion disease. Perhaps a direct interaction between the hydrophobic tract of Sho and the homologous region in PrPSc, which is conformationally altered in prion disease [13], is responsible for the significant decrease in Sho protein levels observed in prion-infected brains.
The role of Sho in prion disease
The quantitative link between Sho and PrPSc levels in the brain suggested that Sho might be capable of modulating prion replication. However, we found no alteration to the incubation period, neuropathology, or PrPSc levels in prion-infected Tg(MoSho) or Tg(HuSho) mice, arguing that increased levels of Sho do not modulate prion disease. Our findings also confirm results obtained using 22L prions and a different line of Tg(MoSho) mice [48]. Whether or not prion replication is altered in mice lacking Sho (Sprn0/0) remains to be determined. Because Sho demonstrated neuroprotective activity against toxicity caused by Dpl and PrP(Δ32–121) in cultured cerebellar granular neurons [1], it was speculated that a loss of Sho levels during prion disease (and any associated neuroprotective activity) in response to PrPSc accumulation may contribute to some of the clinical and/or neuropathological aspects of prion disease. However, in the absence of phenotypic data from studies on Sprn0/0 mice, a causative role for Sho depletion in prion disease seems unlikely since Sho levels in Tg(MoSho) mice following prion infection were higher than those present in uninfected, wt mice (Figure 8A).
Although Sho levels do not influence prion disease kinetics, further studies of Sho depletion during prion disease may reveal important clues about the mechanism of PrPSc-mediated neurotoxicity. For instance, many proteins are known to reside in close spatial proximity to PrPC (and presumably PrPSc) in the cell membrane [6], [8]. Nonspecific interactions between a subset of these proteins and aggregated PrPSc species may result in increased transport to endocytic compartments, increased turnover rates and, consequently, lower steady-state levels of neuronal membrane proteins. Similar mechanisms are known to be responsible for the sequestration of metastable proteins with important functions in the cytoplasm by amyloid-like protein species [66]. Interestingly, inhibition of the Na+/K+-ATPase, a protein that binds to PrPC [67], induces rapid spongiform change in the brains of rats similar to that observed in prion disease [68]. Thus, even small alterations to protein levels or activity by PrPSc may, over time, have deleterious consequences in the brain. Using Sho as a tool to dissect the behavior of PrPSc in the cell may therefore provide novel insight into the biology of prion disease.
Materials and Methods
Ethics statement
All mouse studies were carried out in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, National Academies Press, Washington, DC); protocols were reviewed and approved by the UCSF Institutional Animal Care and Use Committee: "Production of transgenic mice" (AN084871-01B) and “Incubation periods of prion diseases” (AN084950-01A).
Analysis of Sprn mRNA levels in prion-infected mice
Sprn mRNA data was extracted from the Prion Disease Database (PDDB) [26], which can be accessed at http://prion.systemsbiology.net.
Western blotting
Ten percent (wt/vol) brain homogenates were prepared in calcium - and magnesium-free PBS using an Omni Tip (Omni International, Marietta, GA) with a Fisher Scientific PowerGen homogenizer (Fisher Scientific, Pittsburg, PA). Homogenates were then subjected to detergent extraction using 0.5% sodium deoxycholate/0.5% NP-40 (in PBS) at 4°C for 30 min. Following low-speed centrifugation (2000 × g, 5 min, 4°C), protein concentration in the supernatant was determined by the bicinchoninic acid (BCA) assay (Pierce, Rockford, IL). For Sho blots, samples were prepared in Laemmli SDS loading buffer containing β-mercaptoethanol, boiled, and then separated using self-poured 14% polyacrylamide gels. For PrP blots, samples were prepared in 1× NuPAGE loading buffer (Invitrogen, Carlsbad, CA) containing β-mercaptoethanol and boiled for 5 min prior to loading on NuPAGE 10% Bis-Tris gels. Following SDS-PAGE, gels were transferred to PVDF membranes and then blocked for 2 h at room temperature with blocking buffer [5% nonfat milk in TBS containing 0.05% Tween-20 (TBST)]. Membranes were incubated with primary antibody at 4°C overnight in blocking buffer. Blots were rinsed three times with TBST, incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (BioRad, Hercules, CA) for 2 h, rinsed three times with TBST, and then developed using the enhanced chemiluminescent detection system (Amersham, Piscataway, NJ). The following primary antibodies were used: anti-mouse Sho antibodies 06rSH-1 [recognizes MoSho(30–61)] and 06SH-3a [recognizes MoSho(86–100), also used to detect sheep Sho] [1]; anti-human Sho antibody S-12 (Santa Cruz Biotechnology, Santa Cruz, CA); anti-PrP antibodies HuM-P [69], HuM-D18 [70], and 3F4 [71]; anti-Aβ antibody 6E10 (Covance, Princeton, NJ); and anti-APP antibody APPCT recognizing the C-terminus of both mouse and human APP (a generous gift from Paul Fraser). To confirm equal protein loading on the Sho blots, membranes were reprobed with the anti-actin antibody 20–33 (Sigma, St. Louis, MO).
Cell culture
Mouse N2a cells were cultured in Dulbecco's modified Eagle medium (DMEM) containing 10% (wt/vol) fetal bovine serum, 1× GlutaMax, and 0.2× penicillin/streptomycin (Invitrogen) and maintained in a 95% air/5% CO2-humidified environment. Cells were transfected with a mouse Sho cDNA cassette (pcDNA3.MoSho) [1] using Lipofectamine-2000 (Invitrogen) and single cell-derived stable clones selected using medium containing 1 mg/ml G418. High expressing clones, as determined by Western blotting, were selected for further analysis and were maintained in medium containing 0.2 mg/ml G418.
For prion infections, N2a-Sho cells were exposed to medium containing 1% (wt/vol) brain homogenate prepared from RML-infected CD1 mice for 3 days and then passaged 1∶10 five times; the resulting RML-infected cells were denoted ScN2a-Sho cells. In order to obtain more uniform populations of infected ScN2a-Sho cells, these cells were then subcloned further using limiting dilution. For experiments, cells were seeded at a density of 1.25 × 105 cells/well in 6-well tissue culture plates and incubated for 7 days. Culture medium was replenished as necessary. Cells were lysed using a buffer of 50 mM Tris-HCl, pH 7.4; 150 mM NaCl; 0.5% (wt/vol) sodium deoxycholate; 0.5% (vol/vol) NP-40, containing Complete protease inhibitor tablets (Roche, Palo Alto, CA). Post-nuclear supernatants were obtained following low-speed centrifugation (2000 × g, 5 min, 4°C) and then stored at −20°C.
Enzymatic digestion of proteins
For proteinase K (PK) digestions of brain homogenates, 200 µg of detergent-extracted protein was prepared in 60 µl PBS containing 50 µg/ml PK (PK:protein ratio of 1∶67). Digestions were performed at 37°C for 1 h and then stopped by the addition of NuPAGE sample buffer containing β-mercaptoethanol and subsequent boiling. For PK digestions of cell culture lysates, 20 µg/ml PK (PK:protein ratio of 1∶50) for 30 min was used. Sarkosyl and phosphotungstic acid (PTA; pH 7.4) were then added to final concentrations of 1% (vol/vol) and 0.7% (vol/vol), respectively. Samples were incubated at 37°C for 1 h and then centrifuged at 18,000 × g for 40 min. Pellets were resuspended in 1× NuPAGE sample buffer containing β-mercaptoethanol and then boiled. For thermolysin (TL) digestions of brain homogenates, 100 µg of detergent-extracted protein was prepared in 60 µl PBS containing 5 µg/ml TL. Digestions were performed at 37°C for 30 min, then stopped by the addition of PNGaseF denaturing buffer containing EDTA (5 mM final concentration) and subsequent boiling. Samples were then digested with PNGaseF according to the manufacturer's instructions (New England Biolabs).
Immunoprecipitations
Cell lysates were normalized using the BCA assay and then precleared with Protein G-coupled Dynabeads (Invitrogen) for 2 h to reduce nonspecific binding. Lysates (2 mg total protein) were then incubated with 2 µg of anti-Sho antibody 06SH-3a for 4 h at 4°C with end-over-end rotation. Antibody-antigen complexes were captured overnight at 4°C using Protein G–coupled Dynabeads and then washed 3 times with PBS containing 0.05% (vol/vol) Tween-20. Captured proteins were eluted in Laemmli SDS loading buffer by boiling and then analyzed by Western blotting as described above.
PTA precipitations
Post-nuclear supernatants were obtained from 10% brain homogenates by centrifugation at 700 × g for 5 min and then normalized for protein concentration. Sarkosyl and sodium phosphotungstic acid (pH 7.2) were then added sequentially to final concentrations of 2% each. Samples were incubated at 37°C with shaking for 1 h and then centifuged at 18,000 × g for 40 min. Pellets were resuspended in 1× NuPAGE loading buffer, boiled, and then analyzed by Western blotting.
Quantification of Sho levels
Samples were subjected to Western blotting as described above and then quantified by densitometry (ImageJ) using serial dilutions of Sho-containing samples as standards. All statistical analysis was performed using GraphPad Prism software (GraphPad Software, La Jolla, CA). Statistical differences between groups were assessed using the Student's t-test or one-way ANOVA (with Tukey's Multiple Comparison test) with a significance threshold of P<0.05.
Quantification of PrPC by ELISA
Relative PrPC levels in the brains of wt FVB, Tg(MoSho), and Tg(HuSho) mice were determined on detergent-extracted, BCA assay–normalized samples by sandwich ELISA. Immulon 4HBX plates (Nunc, Rochester, NY) were coated overnight at 4°C with the capture antibody HuM-D18 at a concentration of 5 µg/ml. Following blocking for ∼2 h with 1% BSA diluted in phosphate-buffered saline containing 0.05% Tween-20 (PBST), samples (diluted in PBS containing 0.5% Triton X-100) were added and then incubated overnight at 4°C. After 4 washes with PBST, the detection antibody (HRP-labeled HuM-P diluted in blocking buffer) was added and the plate incubated for 2 h at room temperature. Following 5 washes with PBST, the plate was developed using TMB-Blue (Dako, Carpinteria, CA), stopped by the addition of 1 N HCl, and then read at 450 nm using a Spectramax 384 Plus plate reader.
Real-time quaking-induced conversion (RT-QuIC)
RT-QuIC experiments were carried out essentially as described [46]. Briefly, 10% brain homogenates were extracted on ice with 1% (v/v) Triton X-100 for 30 min and then centrifuged at 1,000 × g for 5 min. The supernatant was then diluted 1∶100 into PBS containing 0.1% SDS and 1× N2 Supplement (Invitrogen). In each well of a 96-well plate (BD Biosciences, Bedford, MA), 2 µl of the diluted, detergent-extracted brain homogenate was added to 98 µl of a reaction mixture consisting of 10 mM phosphate buffer (pH 7.4) containing 50 µg/ml recombinant mouse PrP(89–230) [47], 130 mM NaCl, 10 µM EDTA, and 10 µM Thioflavin T. Lyophylized samples of recombinant PrP were resuspended initially in 10 mM phosphate buffer (pH 5.8), aliquoted, and stored at −80°C. Plates were sealed with a clear film (Nunc) and then incubated in a Spectramax M2 plate reader set at 42°C. Samples were subjected to repeated rounds of 1-min rest and 1-min shaking, and top-read fluorescence measurements (444-nm excitation and 485-nm emission filters) were taken every 2 min. Fluorescence values for different samples were compared after 5 h of incubation. Each brain sample was assayed in 8 replicates.
Mouse lines
The following lines of mice were used in this study: wt FVB or CD-1 mice expressing the PrP-A allotype; B6.I mice expressing the PrP-B allotype [72]; Prnp0/0 or Prnd0/0 mice lacking PrP or Dpl expression, respectively [73], [74]; Rcm0 Prnp0/0 mice with ectopic Dpl expression [9]; Tg(MoPrP)B4053 mice overexpressing mouse PrP [75]; Tg(ElkPrP)L12584 mice expressing elk PrP [76]; Tg(OvPrP,V136)N14882 mice expressing sheep PrP [77]; Tg(HuPrP,M129)S2667 mice expressing human PrP with the M129 polymorphism (Watts et al., manuscript in preparation); Tg(HuPrP,V129)152 mice expressing human PrP with the V129 polymorphism [78]; Tg(MoPrP,P101L)A2866 and Tg(MoPrP,P101L)464 mice expressing MoPrP with the analogous P102L mutation causing GSS in humans [42], [44]; Tg(MoPrPΔ23–88)H9949 mice that express N-terminally truncated PrP [49]; Tg(PrP-ΔGPI) mice expressing MoPrP lacking its GPI anchor [50]; Tg(MoDpl)10329 mice [38]; Tg(APP23) and Tg(CRND8) mice, which express mutant human amyloid precursor protein [39], [40]; Tg(SNCA,A53T) mice that express mutant human alpha-synuclein [79]; and Tg(MAPT,P301S) mice that express mutant human tau [80].
Generation of transgenic mice
The murine Sho open reading frame (ORF) was first modified by site-directed mutagenesis to remove a NotI site, amplified from pcDNA3.MoSho with flanking SalI restriction sites by PCR using the primers 5′-CTATATGTCGACACCATGAACTGGACTGCTGCC-3′ (forward) and 5′-CTATATGTCGACCTAAGGCCGAAGCAGTTCTA-3′ (reverse), digested with SalI, purified by agarose gel electrophoresis, and then inserted into SalI-digested and dephosphorylated cos.Tet cosmid vector [81] using T4 DNA ligase. In cos.Tet, neuronal expression of the protein of interest is driven by the hamster Prnp promoter. Ligation mixtures were electroporated into bacteria and clones carrying the correct DNA molecule identified by colony PCR and DNA sequencing.
The human Sho ORF was amplified from an IMAGE cDNA clone (ID #4816858) with flanking HindIII and XbaI restriction sites and then inserted into the pcDNA3 vector. Following removal of three NotI sites using the QuikChange Multi Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA), the HuSho ORF was amplified by PCR with flanking SalI restriction sites using the primers 5′-CTATATGTCGACACCATGAACTGGGCACCCGCA-3′ (forward) and 5′-CTATATGTCGACCTAGGGCCGCAGCAGCCCCA-3′ (reverse), and then inserted into cos.Tet as described above.
Vectors containing Sho constructs were linearized by digestion with NotI, purified by agarose gel electrophoresis, and then microinjected into the pronuclei of fertilized eggs obtained from FVB mice. Southern blotting of genomic DNA samples was used to identify potential founder animals using a probe located in the 3′ untranslated region of the hamster Prnp gene, and the sequences of the integrated transgenes were verified by DNA sequencing. Tg(MoSho) and Tg(HuSho) lines were maintained by backcrossing to wt FVB mice. Relative transgene expression levels in the brain were determined by Western blotting and densitometry using serial dilutions of extracts prepared from Tg(MoSho) mice in comparison to FVB mice. Tg(NSE-MoPrP) mice, which express MoPrPC selectively in neurons under the control of the NSE promoter, were generated similarly, except that microinjection was performed in FVB/Prnp0/0 eggs.
Prion inoculations
The following prion inocula were used in this study: mouse-adapted scrapie strains RML, 22L, and Me7 (maintained in mice expressing the PrP-A allotype) as well as 87V (maintained in mice expressing the PrP-B allotype); hamster-adapted scrapie strains Sc237 and 139H; hamster-adapted TME strains HY and DY; mouse-adapted BSE strain 301V (passaged in mice expressing either PrP-A or PrP-B); SSBP/1 sheep scrapie prions derived from a pool of scrapie-infected sheep brains; CWD prions derived from the brain of a naturally-infected elk; and human sCJD prions obtained from the brains of patients exhibiting either the MM1 or VV2 disease subtypes.
Brain homogenates were diluted to 1% (wt/vol) in 5% BSA and then 30 µl was inoculated into the right parietal lobe of weanling mice or meadow voles (obtained from a breeding colony at the University of California Berkeley) using a 27-gauge syringe. For hamsters, the inoculation volume was 50 µl. Inoculated animals were monitored daily for routine health and assessed three times per week for neurological dysfunction. Mice, hamsters, or meadow voles were euthanized following the onset of neurological symptoms based on standard diagnostic criteria. Brains were removed and snap-frozen prior to storage at -80°C. All animal studies were performed in accordance with protocols approved by the UCSF Institutional Animal Care and Use Committee.
Neuropathology
Samples were immersion-fixed in 10% buffered formalin and then embedded in paraffin using standard procedures. Sections (8 µm) were cut, deparaffinized, and then either stained with haematoxylin and eosin, or processed for immunohistochemistry. Endogenous peroxidase activity was blocked by incubation in 3% hydrogen peroxide (in methanol) and then sections to be stained with anti-PrP antibodies were subjected to hydrolytic autoclaving (121°C for 10 min in citrate buffer). Following blocking with 10% normal goat serum, sections were incubated with primary antibody overnight at 4°C. The following antibodies were used: anti-PrP HuM-P and anti-GFAP (Dako). Antibody binding was detected using a Vectastain ABC peroxidase kit (Vector Laboratories, Burlingame, CA) and visualized using 3-3′-diaminobenzidine (DAB).
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. WattsJCDrisaldiBNgVYangJStromeB 2007 The CNS glycoprotein Shadoo has PrPC-like protective properties and displays reduced levels in prion infections. EMBO J 26 4038 4050
2. PrusinerSB 1998 Prions. Proc Natl Acad Sci USA 95 13363 13383
3. PrusinerSB 1982 Novel proteinaceous infectious particles cause scrapie. Science 216 136 144
4. BüelerHAguzziASailerAGreinerR-AAutenriedP 1993 Mice devoid of PrP are resistant to scrapie. Cell 73 1339 1347
5. SailerABüelerHFischerMAguzziAWeissmannC 1994 No propagation of prions in mice devoid of PrP. Cell 77 967 968
6. Schmitt-UlmsGHansenKLiuJCowdreyCYangJ 2004 Time-controlled transcardiac perfusion cross-linking for the study of protein interactions in complex tissues. Nat Biotechnol 22 724 731
7. WattsJCWestawayD 2007 The prion protein family: diversity, rivalry, and dysfunction. Biochim Biophys Acta 1772 654 672
8. WattsJCHuoHBaiYEhsaniSJeonAH 2009 Interactome analyses identify ties of PrP and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathog 5 e1000608
9. MooreRCLeeIYSilvermanGLHarrisonPMStromeR 1999 Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol 292 797 817
10. BehrensAGenoudNNaumannHRülickeTJanettF 2002 Absence of the prion protein homologue Doppel causes male sterility. EMBO J 21 3652 3658
11. PremzlMSangiorgioLStrumboBMarshall GravesJASimonicT 2003 Shadoo, a new protein highly conserved from fish to mammals and with similarity to prion protein. Gene 314 89 102
12. MoHMooreRCCohenFEWestawayDPrusinerSB 2001 Two different neurodegenerative diseases caused by proteins with similar structures. Proc Natl Acad Sci USA 98 2352 2357
13. PeretzDWilliamsonRAMatsunagaYSerbanHPinillaC 1997 A conformational transition at the N-terminus of the prion protein features in formation of the scrapie isoform. J Mol Biol 273 614 622
14. BaumannFTolnayMBrabeckCPahnkeJKlozU 2007 Lethal recessive myelin toxicity of prion protein lacking its central domain. EMBO J 26 538 547
15. LiAChristensenHMStewartLRRothKAChiesaR 2007 Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J 26 548 558
16. SakthiveluVSeidelRPWinklhoferKFTatzeltJ 2011 Conserved stress-protective activity between prion protein and shadoo. J Biol Chem 286 8901 8908
17. ChenSGTeplowDBParchiPTellerJKGambettiP 1995 Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem 270 19173 19180
18. TaraboulosARaeberAJBorcheltDRSerbanDPrusinerSB 1992 Synthesis and trafficking of prion proteins in cultured cells. Mol Biol Cell 3 851 863
19. DronMMoudjouMChapuisJSalamatMKBernardJ 2010 Endogenous proteolytic cleavage of disease-associated prion protein to produce C2 fragments is strongly cell - and tissue-dependent. J Biol Chem 285 10252 10264
20. YoungRPassetBVilotteMCribiuEPBeringueV 2009 The prion or the related Shadoo protein is required for early mouse embryogenesis. FEBS Lett 583 3296 3300
21. BeckJACampbellTAAdamsonGPoulterMUphillJB 2008 Association of a null allele of SPRN with variant Creutzfeldt-Jakob disease. J Med Genet 45 813 817
22. LloydSEGrizenkovaJPotaHCollingeJ 2009 Shadoo (Sprn) and prion disease incubation time in mice. Mamm Genome 20 367 374
23. DaudeNWohlgemuthSRogaevaEFaridAHHeatonM 2009 Frequent missense and insertion/deletion polymorphisms in the ovine Shadoo gene parallel species-specific variation in PrP. PLoS ONE 4 e6538
24. StewartPShenCZhaoDGoldmannW 2009 Genetic analysis of the SPRN gene in ruminants reveals polymorphisms in the alanine-rich segment of shadoo protein. J Gen Virol 90 2575 2580
25. HopeJWoodSCBirkettCRChongABruceME 1999 Molecular analysis of ovine prion protein identifies similarities between BSE and an experimental isolate of natural scrapie, CH1641. J Gen Virol 80 1 4
26. GehlenborgNHwangDLeeIYYooHBaxterD 2009 The Prion Disease Database: a comprehensive transcriptome resource for systems biology research in prion diseases. Database (Oxford) bap011
27. GossnerAGBennetNHunterNHopkinsJ 2009 Differential expression of Prnp and Sprn in scrapie infected sheep also reveals Prnp genotype specific differences. Biochem Biophys Res Commun 378 862 866
28. DaudeNNgVWattsJCGenovesiSGlavesJP 2010 Wild-type Shadoo proteins convert to amyloid-like forms under native conditions. J Neurochem 113 92 104
29. DickinsonAGFraserH 1979 An assessment of the genetics of scrapie in sheep and mice. PrusinerSBHadlowWJ Slow Transmissible Diseases of the Nervous System, Vol 1 New York Academic Press 367 386
30. DickinsonAGMeikleVMH 1969 A comparison of some biological characteristics of the mouse-passaged scrapie agents, 22A and ME7. Genet Res 13 213 225
31. BruceMChreeAMcConnellIFosterJPearsonG 1994 Transmission of bovine spongiform encephalopathy and scrapie to mice: strain variation and the species barrier. Philos Trans R Soc Lond B Biol Sci 343 405 411
32. BruceMEDickinsonAG 1985 Genetic control of amyloid plaque production and incubation period in scrapie-infected mice. J Neuropathol Exp Neurol 44 285 294
33. KimberlinRHColeSWalkerCA 1987 Temporary and permanent modifications to a single strain of mouse scrapie on transmission to rats and hamsters. J Gen Virol 68 1875 1881
34. BessenRAMarshRF 1992 Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J Gen Virol 73 329 334
35. BosquePJPrusinerSB 2000 Cultured cell sublines highly susceptible to prion infection. J Virol 74 4377 4386
36. MahalSPBakerCADemczykCASmithEWJuliusC 2007 Prion strain discrimination in cell culture: the cell panel assay. Proc Natl Acad Sci USA 104 20908 20913
37. SaborioGPPermanneBSotoC 2001 Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411 810 813
38. MooreRCMastrangeloPBouzamondoEHeinrichCLegnameG 2001 Doppel-induced cerebellar degeneration in transgenic mice. Proc Natl Acad Sci USA 98 15288 15293
39. Sturchler-PierratCAbramowskiDDukeMWiederholdKHMistlC 1997 Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA 94 13287 13292
40. ChishtiMAYangDSJanusCPhinneyALHorneP 2001 Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 276 21562 21570
41. HsiaoKKScottMFosterDGrothDFDeArmondSJ 1990 Spontaneous neurodegeneration in transgenic mice with mutant prion protein. Science 250 1587 1590
42. NazorKEKuhnFSewardTGreenMZwaldD 2005 Immunodetection of disease-associated mutant PrP, which accelerates disease in GSS transgenic mice. EMBO J 24 2472 2480
43. HsiaoKKGrothDScottMYangS-LSerbanH 1994 Serial transmission in rodents of neurodegeneration from transgenic mice expressing mutant prion protein. Proc Natl Acad Sci USA 91 9126 9130
44. TellingGCHagaTTorchiaMTremblayPDeArmondSJ 1996 Interactions between wild-type and mutant prion proteins modulate neurodegeneration in transgenic mice. Genes Dev 10 1736 1750
45. ColbyDWWainRBaskakovIVLegnameGPalmerCG 2010 Protease-sensitive synthetic prions. PLoS Pathog 6 e1000736
46. WilhamJMOrruCDBessenRAAtarashiRSanoK 2010 Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog 6 e1001217
47. ColbyDWZhangQWangSGrothDLegnameG 2007 Prion detection by an amyloid seeding assay. Proc Natl Acad Sci USA 104 20914 20919
48. WangHWanJWangWWangDLiS 2011 Overexpression of Shadoo protein in transgenic mice does not impact the pathogenesis of scrapie. Neurosci Lett 496 1 4
49. SupattaponeSMuramotoTLegnameGMehlhornICohenFE 2001 Identification of two prion protein regions that modify scrapie incubation time. J Virol 75 1408 1413
50. StöhrJWattsJCLegnameGOehlerALemusA Spontaneous generation of anchorless prions in transgenic mice. Proc Natl Acad Sci USA (In press)
51. NonnoRDi BariMACardoneFVaccariGFazziP 2006 Efficient transmission and characterization of Creutzfeldt-Jakob disease strains in bank voles. PLoS Pathog 2 e12
53. CronierSGrosNTattumMHJacksonGSClarkeAR 2008 Detection and characterization of proteinase K-sensitive disease-related prion protein with thermolysin. Biochem J 416 297 305
54. YadavalliRGuttmannRPSewardT Centers AP, Williamson RA, et al. 2004 Calpain-dependent endoproteolytic cleavage of PrPSc modulates scrapie prion propagation. J Biol Chem 279 21948 21956
54. WestawayDGenovesiSDaudeNBrownRLauA 2011 Down-regulation of shadoo in prion infections traces a pre-clinical event inversely related to PrPSc accumulation. PLoS Pathog 7 e1002391
55. LampoEVan den BroeckWWillemarckNVan PouckeMCasteleynCR 2011 Distribution of the Shadoo protein in the ovine brain assessed by immunohistochemistry. Res Vet Sci 90 372 378
56. MiyazawaKManuelidisL 2010 Agent-specific Shadoo responses in transmissible encephalopathies. J Neuroimmune Pharmacol 5 155 163
57. ZouWQPuotiGXiaoXYuanJQingL 2010 Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol 68 162 172
58. OttoMWiltfangJCepekLNeumannMMollenhauerB 2002 Tau protein and 14-3-3 protein in the differential diagnosis of Creutzfeldt-Jakob disease. Neurology 58 192 197
59. GodsaveSFWilleHKujalaPLatawiecDDeArmondSJ 2008 Cryo-immunogold electron microscopy for prions: toward identification of a conversion site. J Neurosci 28 12489 12499
60. PereraWSHooperNM 2001 Ablation of the metal ion-induced endocytosis of the prion protein by disease-associated mutation of the octarepeat region. Curr Biol 11 519 523
61. SunyachCJenADengJFitzgeraldKTFrobertY 2003 The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J 22 3591 3601
62. WeissmannCEnariMKlohnPCRossiDFlechsigE 2002 Transmission of prions. Proc Natl Acad Sci USA 99 16378 16383
63. MorelNSimonSFrobertYVollandHMourton-GillesC 2004 Selective and efficient immunoprecipitation of the disease-associated form of the prion protein can be mediated by nonspecific interactions between monoclonal antibodies and scrapie-associated fibrils. J Biol Chem 279 30143 30149
64. PastranaMASajnaniGOniskoBCastillaJMoralesR 2006 Isolation and characterization of a proteinase K-sensitive PrP(Sc) fraction. Biochemistry 45 15710 15717
65. TremblayPBallHLKanekoKGrothDHegdeRS 2004 Mutant PrPSc conformers induced by a synthetic peptide and several prion strains. J Virol 78 2088 2099
66. OlzschaHSchermannSMWoernerACPinkertSHechtMH 2011 Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144 67 78
67. KleeneRLoersGLangerJFrobertYBuckF 2007 Prion protein regulates glutamate-dependent lactate transport of astrocytes. J Neurosci 27 12331 12340
68. BignamiAPalladiniG 1966 Experimentally produced cerebral status spongiosus and continuous pseudorhythmic electroencephalographic discharges with a membrane-ATPase inhibitor in the rat. Nature 209 413 414
69. SafarJGScottMMonaghanJDeeringCDidorenkoS 2002 Measuring prions causing bovine spongiform encephalopathy or chronic wasting disease by immunoassays and transgenic mice. Nat Biotechnol 20 1147 1150
70. WilliamsonRAPeretzDPinillaCBallHBastidasRB 1998 Mapping the prion protein using recombinant antibodies. J Virol 72 9413 9418
71. KascsakRJRubensteinRMerzPATonna-DeMasiMFerskoR 1987 Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol 61 3688 3693
72. CarlsonGAGoodmanPALovettMTaylorBAMarshallST 1988 Genetics and polymorphism of the mouse prion gene complex: control of scrapie incubation time. Mol Cell Biol 8 5528 5540
73. BüelerHFisherMLangYBluethmannHLippH-P 1992 Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356 577 582
74. TamgüneyGGilesKGliddenDVLessardPWilleH 2008 Genes contributing to prion pathogenesis. J Gen Virol 89 1777 1788
75. CarlsonGAEbelingCYangS-LTellingGTorchiaM 1994 Prion isolate specified allotypic interactions between the cellular and scrapie prion proteins in congenic and transgenic mice. Proc Natl Acad Sci USA 91 5690 5694
76. TamgüneyGGilesKBouzamondo-BernsteinEBosquePJMillerMW 2006 Transmission of elk and deer prions to transgenic mice. J Virol 80 9104 9114
77. TamgüneyGMillerMWGilesKLemusAGliddenDV 2009 Transmission of scrapie and sheep-passaged bovine spongiform encephalopathy prions to transgenic mice expressing elk prion protein. J Gen Virol 90 1035 1047
78. TellingGCScottMHsiaoKKFosterDYangS-L 1994 Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc Natl Acad Sci USA 91 9936 9940
79. GiassonBIDudaJEQuinnSMZhangBTrojanowskiJQ 2002 Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron 34 521 533
80. AllenBIngramETakaoMSmithMJJakesR 2002 Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci 22 9340 9351
81. ScottMRKöhlerRFosterDPrusinerSB 1992 Chimeric prion protein expression in cultured cells and transgenic mice. Protein Sci 1 986 997
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 11
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Multiple Candidate Effectors from the Oomycete Pathogen Suppress Host Plant Immunity
- The Splicing Factor Proline-Glutamine Rich (SFPQ/PSF) Is Involved in Influenza Virus Transcription
- A TNF-Regulated Recombinatorial Macrophage Immune Receptor Implicated in Granuloma Formation in Tuberculosis
- SH3 Domain-Mediated Recruitment of Host Cell Amphiphysins by Alphavirus nsP3 Promotes Viral RNA Replication
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy