
CD4 T Cell Immunity Is Critical for the Control of Simian Varicella Virus Infection in a Nonhuman Primate Model of VZV Infection
Primary infection with varicella zoster virus (VZV) results in varicella (more commonly known as chickenpox) after which VZV establishes latency in sensory ganglia. VZV can reactivate to cause herpes zoster (shingles), a debilitating disease that affects one million individuals in the US alone annually. Current vaccines against varicella (Varivax) and herpes zoster (Zostavax) are not 100% efficacious. Specifically, studies have shown that 1 dose of varivax can lead to breakthrough varicella, albeit rarely, in children and a 2-dose regimen is now recommended. Similarly, although Zostavax results in a 50% reduction in HZ cases, a significant number of recipients remain at risk. To design more efficacious vaccines, we need a better understanding of the immune response to VZV. Clinical observations suggest that T cell immunity plays a more critical role in the protection against VZV primary infection and reactivation. However, no studies to date have directly tested this hypothesis due to the scarcity of animal models that recapitulate the immune response to VZV. We have recently shown that SVV infection of rhesus macaques models the hallmarks of primary VZV infection in children. In this study, we used this model to experimentally determine the role of CD4, CD8 and B cell responses in the resolution of primary SVV infection in unvaccinated animals. Data presented in this manuscript show that while CD20 depletion leads to a significant delay and decrease in the antibody response to SVV, loss of B cells does not alter the severity of varicella or the kinetics/magnitude of the T cell response. Loss of CD8 T cells resulted in slightly higher viral loads and prolonged viremia. In contrast, CD4 depletion led to higher viral loads, prolonged viremia and disseminated varicella. CD4 depleted animals also had delayed and reduced antibody and CD8 T cell responses. These results are similar to clinical observations that children with agammaglobulinemia have uncomplicated varicella whereas children with T cell deficiencies are at increased risk of progressive varicella with significant complications. Moreover, our studies indicate that CD4 T cell responses to SVV play a more critical role than antibody or CD8 T cell responses in the control of primary SVV infection and suggest that one potential mechanism for enhancing the efficacy of VZV vaccines is by eliciting robust CD4 T cell responses.
Published in the journal:
. PLoS Pathog 7(11): e32767. doi:10.1371/journal.ppat.1002367
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1002367
Summary
Primary infection with varicella zoster virus (VZV) results in varicella (more commonly known as chickenpox) after which VZV establishes latency in sensory ganglia. VZV can reactivate to cause herpes zoster (shingles), a debilitating disease that affects one million individuals in the US alone annually. Current vaccines against varicella (Varivax) and herpes zoster (Zostavax) are not 100% efficacious. Specifically, studies have shown that 1 dose of varivax can lead to breakthrough varicella, albeit rarely, in children and a 2-dose regimen is now recommended. Similarly, although Zostavax results in a 50% reduction in HZ cases, a significant number of recipients remain at risk. To design more efficacious vaccines, we need a better understanding of the immune response to VZV. Clinical observations suggest that T cell immunity plays a more critical role in the protection against VZV primary infection and reactivation. However, no studies to date have directly tested this hypothesis due to the scarcity of animal models that recapitulate the immune response to VZV. We have recently shown that SVV infection of rhesus macaques models the hallmarks of primary VZV infection in children. In this study, we used this model to experimentally determine the role of CD4, CD8 and B cell responses in the resolution of primary SVV infection in unvaccinated animals. Data presented in this manuscript show that while CD20 depletion leads to a significant delay and decrease in the antibody response to SVV, loss of B cells does not alter the severity of varicella or the kinetics/magnitude of the T cell response. Loss of CD8 T cells resulted in slightly higher viral loads and prolonged viremia. In contrast, CD4 depletion led to higher viral loads, prolonged viremia and disseminated varicella. CD4 depleted animals also had delayed and reduced antibody and CD8 T cell responses. These results are similar to clinical observations that children with agammaglobulinemia have uncomplicated varicella whereas children with T cell deficiencies are at increased risk of progressive varicella with significant complications. Moreover, our studies indicate that CD4 T cell responses to SVV play a more critical role than antibody or CD8 T cell responses in the control of primary SVV infection and suggest that one potential mechanism for enhancing the efficacy of VZV vaccines is by eliciting robust CD4 T cell responses.
Introduction
Varicella zoster virus (VZV), a neurotropic alphaherpesvirus, is the causative agent of varicella (chickenpox). Following resolution of the acute infection, VZV establishes latency in sensory ganglia, and can reactivate years later, manifesting as dermatomal vesicular lesions known as herpes zoster (HZ, shingles) [1]. HZ is a painful and debilitating disease that causes significant morbidity such as post-herpetic neuralgia and HZ opthalmicus [2], [3] and occasionally mortality in the elderly and immune compromised [4]. HZ affects 1 million people each year in the United States [5], [6] and persons older than 60 year of age account for 40–50% of HZ cases reported each year [5], [6]. Given that by 2020 17% of the US population is estimated to be 65 years of age or older (US Census), the incidence of HZ and its associated morbidities is likely to increase.
There are currently two FDA approved VZV vaccines available that contain the live attenuated VZV Oka strain: Varivax, directed against chickenpox, and Zostavax, directed against shingles. The introduction of Varivax, and more specifically of the 2-dose regimen, has dramatically reduced the incidence of chickenpox and annual varicella-related hospitalizations and deaths in the US [7], [8]. Similarly, vaccination with Zostavax reduced the incidence of shingles by 51% in a 3-year study period and resulted in a 61% decrease in the burden of disease [9], [10]. However, it is important to note that the efficacy of Zostavax was reduced in individuals older than 70 years [9]. Moreover, recent studies showed that the cellular and humoral responses engendered by Zostavax significantly declined 2 years following Zostavax vaccination [11]. Given that the proportion of persons 85 years and older is expected to increase from 14% in 2010 to more than 21% in 2050 (US Census Bureau, 2010), new vaccination strategies against VZV should be explored.
Clinical observations suggest that the successful resolution of acute VZV infection is associated with the development of the host's VZV-specific cellular immunity rather than humoral immunity [1], [12], [13], [14], [15], [16]. Specifically, children suffering from congenital immune deficiencies affecting cellular immunity or from hematological malignancies, or undergoing immunosuppressive treatment are at risk for progressive varicella; whereas, children with agammaglobulinemia have uncomplicated varicella episodes [17], [18], [19], [20], [21]. Transfusion of lymphocytes from immune donors to children with immunodeficiencies can limit VZV infection and replication after the appearance of cutaneous lesions [22]. In contrast, the administration of immunoglobulins with high titers of IgG antibodies to VZV is only protective when administered within 72 hours of exposure [23], [24], [25]. While early production of VZV antibodies by HIV-infected children does not prevent progressive varicella [26], absence of cellular immunity in these children correlates with a risk of prolonged viremic phase, continued formation of skin lesions and dissemination of the virus to the lungs and other organs [16], [27]. However, these data are confounded by the fact that T cell-deficient patients lack both T cell-mediated and T cell-dependent B cell-mediated responses. Thus, more careful analyses of the contribution of each of the major lymphocyte subsets are required.
Both VZV-specific CD4 and CD8 T cells are detected during acute VZV infection [12], [28]. CD4 Th1 cells, which produce the anti-viral cytokine IFNγ and have cytotoxic potential, predominate the VZV-specific T cell response [12], [28], with little or no production of Th2 cytokines [12]. Several studies have shown that VZV-specific CD4 T cells recognize ORF4 [29], glycoprotein I [30], and ORF63 [31]. In contrast to CD4 T cells, the specificity and magnitude of VZV-specific CD8 T cell response are only partially understood. Several studies have suggested that VZV-specific CD8 T cells circulate at very low frequencies, possibly due to immune evasion strategies such as MHC class I downregulation [30], [32], [33]. However, it is also likely that the CD8 T cell response is under-appreciated because VZV lysate used in the majority of the studies is poorly presented via the MHC-I pathway [34]. Consequently, the role of CD8 versus CD4 T cell immunity in the control of VZV infection remains unclear.
Our understanding of the importance of the T and B cell responses to the resolution of VZV infection remains incomplete because of the lack of adequate animal models. VZV infection of small rodent models (guinea pigs, cotton rats, and mice) does not result in varicella [35], [36], [37], [38], [39]. Dr. Arvin and colleagues developed a SCID-humanized (SCID-hu) mouse model which has significantly advanced our understanding of VZV pathogenesis [40]. However, the immuno-deficient nature of the animals has precluded the characterization of immune response to VZV infection. Simian varicella virus (SVV) is a neurotropic alphaherpesvirus that naturally infects non-human primates (NHP) and shares 75% DNA homology and genome colinearity with VZV [41], [42], [43]. The exact mode of SVV transmission has not yet been experimentally determined but it is believed to occur through contact with skin lesions of an infected animal, or via exposure to virus-laden aerosolized droplets from infected animal(s), as described for VZV transmission in humans [44], [45]. We have recently shown that intrabronchial infection of rhesus macaques (RM) with SVV recapitulates the hallmarks of acute VZV infection in humans: (1) generalized varicella rash; (2) development of cellular and humoral responses; (3) resolution of acute infection; and (4) establishment of latency in sensory ganglia [46]. In our model, the introduction of SVV directly into the lungs potentially bypasses the initial stage of VZV replication that occurs in the oropharynx, head and neck regions [47], which could affect viral amplification within tonsilar memory T cell [40], [48], [49]. Despite this potential difference, T and B cell responses develop with similar kinetics relative to the onset of the exanthem in SVV inoculated animals and children infected with VZV, thereby providing a robust animal model with which to study anti-VZV immunity.
The goal of this study was to identify the host immune responses essential for protection against primary SVV infection in unvaccinated RM as a model of VZV infection in humans. To this end, we compared disease severity and immune response in four groups of young RM infected with SVV: (1) control animals; (2) CD20+ B cell depleted; (3) CD8+ T cell depleted; and (4) CD4+ T cell depleted. We show that loss of B cells during primary SVV infection does not alter viral loads or disease severity. In contrast, loss of CD8 T cells led to a slightly higher peak viral load and the loss of CD4 T cells led to significantly higher viral loads and disseminated varicella. These studies suggest that cellular immunity and more specifically CD4 T cell immunity plays a critical role in the control of SVV infection. These observations have important ramifications for the development of novel vaccination strategies to alleviate VZV associated diseases.
Results
Efficacy of depleting antibodies
The contributions of cellular versus humoral immune responses in the resolution of acute VZV infection have not yet been experimentally addressed. Therefore, using the infection of young RM with SVV as a model of acute VZV infection, we aimed to determine the role of T cell versus B cell responses during acute SVV infection. Prior to intrabronchial SVV inoculation, 16 young RM were divided into four groups of four animals each: (1) control; (2) CD8+ T cell depleted; (3) CD4+ T cell depleted; and (4) CD20+ B cell depleted. T and B cell depletion regimens were initiated 7 days prior to infection to ensure that the targeted lymphocyte population was not present on the day of infection (0 dpi). The frequency of CD4+ T cells, CD8+ T cells, and CD20+ B cells in peripheral blood mononuclear cells (PBMC) and bronchial alveolar lavage (BAL) samples were monitored throughout the study by flow cytometry (FCM). In contrast to peripheral blood, frequency of CD20 B cells is very low in BAL (Fig. 1). Treatment with CD20 depleting antibody resulted in the loss of B cells beginning at 0 dpi and lasting until 21 dpi in BAL (Fig. 1C) and 17 dpi in peripheral blood (Fig. 1F). Administration of CD8-depleting antibody resulted in complete loss of CD8+ T cells between 0 and 14 dpi in BAL (Fig. 1B) and between 0 and 17 dpi in peripheral blood (Fig. 1E) Administration of anti-CD4 depleting antibody decreased the frequency of CD4+ T cells on day 0 but complete loss was not achieved until 10 dpi (Fig 1A and D). This loss was very transient in BAL samples where the recovery started 14 dpi (Fig. 1A). In peripheral blood, CD4 depletion lasted until 17 dpi (Fig. 1D). In summary, CD20 and CD8 T cell depletions were more profound (achieving ∼100% loss by day 0) and lasted longer than CD4 T cell depletion. Following depletion, recovery was very slow in all three lymphocyte subsets and the frequencies did not return to baseline by the end of the study.

Impact of T and B cell loss on SVV viral loads and disease severity
To determine the impact of T and B cell loss on SVV replication, we measured viral loads in whole blood (WB), BAL and buccal swabs by qPCR. As previously reported [46], SVV viral loads in WB were detected in non-depleted control animals 3 dpi, peaked at 7 dpi, and resolved at 21 dpi in WB (Fig. 2A) and BAL (Fig. 2B). CD20 depleted animals showed comparable viral loads and viral replication kinetics as those observed in non-depleted animals (Fig. 2A and 2B). In contrast, CD4+ depleted animals showed the highest peak SVV viral loads in WB at 7 dpi (p = 0.05) and 10 dpi (p = 0.002) (Fig. 2A) and BAL at 7 dpi (p<0.001) (Fig. 2B) compared to non-depleted control animals. They also showed recurrent viremia as evidenced by the detection of SVV viral DNA in WB on days 56 and 70 (Fig. 2A). CD8 depleted animals also experienced higher viral loads 7 dpi in WB (Fig. 2A) and 3 dpi in the BAL (Fig. 2B) than non-depleted animals but these differences did not reach statistical significance. We also measured SVV shedding in saliva since it is well documented that VZV reactivation results in the shedding of infectious VZV in the saliva [50], [51]. SVV viral DNA was only detected in the buccal epithelial cells and not the saliva (Fig. 2C and data not shown). CD8 depleted animals experienced higher SVV viral loads in buccal epithelial cells at 7 dpi (p = 0.0418), while CD4 depleted animals showed higher SVV viral loads in buccal epithelial cells at 10 (p<0.001) and 14 dpi (p = 0.0182) in comparison to non-depleted control animals (Fig. 2C).

All 16 animals developed varicella (Fig. 3), but the severity and duration of the rash varied between groups (Table 1). As previously reported [46], control animals developed lesions beginning at 7 dpi that began to heal 14 dpi and were completely resolved 21 dpi (Table 1; Fig. 3A). CD20 depleted animals had comparable disease severity to control animals (Fig. 3B). However, CD8 depleted animals continued to display moderate to severe lesions at 14 dpi that did not resolve until 28 dpi (Table 1; Fig. 3C). CD4 depleted animals experienced severe and disseminated varicella rash (Fig. 3D), which also did not begin to heal until 42 dpi (Table 1). Taken together, these data show that in contrast to CD20 B cells, the absence of CD8 or CD4 T cells results in higher viral loads and increased disease severity during primary SVV infection.


Modulation of B cell response by T and B cell depletion
We next investigated the impact of lymphocyte depletion on the proliferative response of B cells and the generation of SVV-specific IgG, IgM, and neutralizing antibody responses. Following antigen encounter, naïve B cells undergo a proliferative burst and acquire the memory marker CD27. Therefore, we assessed the proliferative response of B cells by measuring changes in the expression of Ki67, a nuclear protein associated with G2-S cell cycle transition, within two major antigen-experienced B cell subsets: marginal zone-like (MZ-like) and memory B cell subsets, using FCM. In BAL from control animals, proliferation of MZ-like B cells began 10 dpi, peaked 14 dpi, and returned to baseline levels 17 dpi (Fig. 4A). Frequency of Ki67+ memory B cells in BAL of control animals increased at 7dpi, peaked 10dpi, and returned to baseline levels 17 dpi (Fig. 4B). In CD20-depleted animals, Ki67+ MZ-like and memory B cell in BAL were detected 14 dpi as the B cell compartment began to regenerate (Fig. 4A and 4B). Interestingly, we detected an earlier onset of proliferation of MZ-like and memory B cells in BAL in CD8 depleted animals compared to control animals (Fig. 4A and 4B). Proliferative burst of the MZ-like B cells peaked 7 days before control animals (7 dpi versus 14 dpi) and reached a higher level (p<0.0001) before returning to baseline levels 17 dpi (Fig. 4A). Similarly, the proliferative burst of the memory B cells was detected 4 days earlier (3dpi versus 7dpi) and reached a higher peak 7 dpi compared to control animals (p = 0.0003) (Fig. 4B). In contrast, we detected minimal B cell proliferation with no distinct kinetics (less than 10%) in both the MZ-like and memory subsets in CD4 depleted animals (Fig. 4A and 4B, and Table 2).


In PBMCs of control animals, MZ-like and memory B cell proliferation was detected 7 dpi, peaked 10–14 dpi, before returning to baseline levels 17 dpi (Fig. 4C and 4D). As described for BAL, Ki67+ MZ-like and memory B cells were detected 14 dpi in CD20 depleted animals (Fig. 4C and 4D), coinciding with the re-appearance of B cells in peripheral blood (Fig. 1). In CD8 depleted animals the proliferative burst of MZ-like and memory B cells was delayed by 3 days (10 dpi compared to 7 dpi, p = 0.008) (Fig. 4C and 4D) but reached higher peak value 14 dpi in the memory subset compared to control animals (p<0.001) (Fig. 4D). As observed in BAL, B cells from CD4 depleted animals showed minimal proliferation in response to SVV infection (p<0.05 at all time points examined between 7 and 14 dpi) (Fig. 4C and 4D, and Table 2).
To assess differences in the magnitude of the proliferative burst, we measured the area under the curve (AUC) for MZ-like and memory B cell proliferation between 3 and 17 dpi. This analysis revealed that the magnitude of the proliferative burst of MZ-like and memory B cells was significantly reduced in PBMC in CD4 depleted animals compared to control animals (p<0.001). In CD8 depleted animals, memory and MZ-like B cell proliferative burst was higher in BAL (p = 0.025) but MZ-like proliferation was lower in PBMC compared to controls (p = 0.013) (Table 2).
We also measured the impact of T and B cell depletion on antibody production. To that end, we measured SVV-specific IgM and IgG titers using standard ELISA and neutralizing antibody titer using plaque reduction assay. In control and CD8 depleted animals, SVV-specific IgM titers peaked 10 dpi (average titer 1∶2000 and 1∶2465, respectively) (Fig. 4E). In contrast, CD20 and CD4 depleted animals had significantly lower average titers of 1∶360 and 1∶190, respectively (Fig. 4E, p<0.05). Similarly, control and CD8 depleted animals generated a robust SVV-specific IgG response that peaked 21 dpi (average titer 1∶15800 and 1∶17300 respectively) (Fig. 4F), whereas CD20 and CD4 depleted animals had significantly reduced IgG antibody production 14, 17, 21, and 28 dpi compared to control animals (p<0.02 for all time points). Specifically, SVV-specific IgG titers 21 dpi were on average 1∶6200 and1∶2500 in CD20 and CD4 depleted animals respectively (Fig. 4F). Analysis of the AUC for SVV-specific IgG titers between 0 and 63 dpi showed that both CD20 depleted and CD4 depleted animals have significantly reduced IgG production compared to non-depleted control animals (p = 0.0245 and p = 0.0193, respectively) (Fig. 4F).
We also assessed neutralizing antibody titers following acute SVV infection 14 and 70 dpi in all animal groups by measuring the dilution at which 50% reduction in the number of SVV plaques was achieved (NT50, Table 3). NT50 titers were comparable between control and CD8 depleted animals 14 dpi, and by 70 dpi the titers increased 34 - to 90-fold and 8 - to 17-fold, respectively (Table 3). In contrast, CD4 depleted animals had considerably lower NT50 titers 14 dpi compared to control animals, which increased 6 - to 16-fold by 70 dpi (Table 3). Plasma obtained from CD20 depleted animals showed no neutralization ability 14 dpi and very negligible NT50 titers 70 dpi (Table 3). These data suggest that the resolution of varicella during primary SVV infection in unvaccinated animals does not require the presence of neutralizing antibodies.

Modulation of T cell response by B and T cell depletion
We assessed how loss of T and B cells modulated the kinetics and magnitude of T cell proliferation in BAL and PBMCs in response to SVV infection. Like B cell responses, after antigen encounter, naïve T cells become activated, differentiate into central and effector memory (CM and EM) T cells and undergo a robust proliferative burst. We assessed this proliferative burst by measuring frequency of Ki67+ cells within CM and EM T cell subsets in BAL and PBMC [46]. In BAL from control animals, T cell proliferation was detected in all subsets 7 dpi after which the frequency of Ki67+ T cells gradually declined reaching baseline levels 17 dpi (Fig. 5A–D, and Table 2). In CD20 depleted animals, the kinetics and magnitude of proliferation in all four T cell subsets was similar to that observed in control animals (Fig. 5A–D). The kinetics of the CD4 T cell proliferation in CD8 depleted animals was comparable to that observed in control animals. However, the peak frequency of Ki67+ CD4 CM at 7 dpi for CD8 depleted animals was significantly larger than that observed for control animals (p = 0.0028) (Fig. 5A). Similarly, the peak frequency of Ki67+ CD4 EM T cells 10 dpi and 14 dpi was also higher in CD8 depleted animals (p = 0.0054, p = 0.0058, respectively) (Fig. 5B). Interestingly the re-appearance of CD4 T cells in the BAL 21 dpi was not accompanied by an increased frequency of CD4 Ki67+ T cells (Fig. 5A and 5B). We detected proliferating CD8 T cells in CD8 depleted animals 17 dpi (Fig. 5C and 5D), which coincides with the re-appearance of CD8 T cells in these animals (Fig. 1). CD8 CM proliferation was significantly delayed (10 dpi versus 7 dpi in control animals) and reduced in CD4 depleted animals (p<0.001 at 7 dpi; p = 0.0059 at 14 dpi) (Fig. 5C). Similarly, proliferation of CD8 EM in CD4 depleted animals was also delayed (10 dpi versus 7 dpi in control animals, Fig. 5D).

We detected very little proliferation by the CD4 T cells in PBMC from control animals (Fig. 5E and 5F, and Table 2). On the other hand, CD8 T cell proliferation was easily detected in PBMC in control animals (Fig. 5G and 5H, and Table 2). The frequency of Ki67+ CD8 CM T cells in control animals peaked 7 dpi and returned to baseline 14 dpi, and the frequency of Ki67+ CD8 EM T cells peaked 10 dpi before returning to baseline levels 21 dpi (Fig 5G and 5H). The kinetics and magnitude of T cell proliferative burst in CD20 depleted animals were comparable to those described for control animals (Fig. 5E–H). As described for the BAL, CD4 T cell proliferation was more robust in CD8 depleted animals compared to controls. Specifically, the frequency of Ki67+ CD4 CM T cells was significantly higher 10 dpi (p = 0.0138) (Fig. 5E), and that of CD4 EM T cells was significantly higher 3, 7, 10, 14, and 17 dpi (p<0.01 for all days) (Fig. 5F) when compared to control animals. Ki67+ CD8 CM and EM T cells were detected 14 dpi (Fig. 5G and H), which coincided with the re-appearance of CD8 T cells (Fig. 1). As described for the BAL, CD4 depleted animals exhibited a significant reduction of proliferating CD8 CM (p = 0.001) and EM (p< 0.01 for 10,14 and 17 dpi) (Fig. 5G and 5H). As previously noted for the BAL, regeneration of the CD4 T cell compartment was not accompanied by an increase in CD4 T cell proliferation in these animals (Fig. 5G and 5H).
As described above for B cells, we measured the AUC from 3 to 17 dpi for T cell proliferation to assess the magnitude of the proliferative burst. This analysis showed that in BAL the proliferative burst of: 1) CD4 EM T cells was significantly higher in CD8 depleted animals (p = 0.026) and 2) that of CD8 CM was significantly lower in CD4 depleted animals (p<0.001) compared to control animals. In peripheral blood: 1) CD8 depleted animals experienced a larger proliferative burst in the CD4 CM (p = 0.08) and EM (p <0.001) compared to controls and 2) the CD4 depleted animals showed a smaller CD8 CM (p = 0.02) and EM (p <0.001) T cell proliferative burst compared to control animals (Table 3).
Effect of T and B cell depletions on responding T cell frequency
In addition to comparing the kinetics and magnitude of the T cell proliferative burst in response to SVV in the different groups, we determined the frequency of SVV-specific T cells within CM and EM subsets by measuring frequency of IFNγ+ or IFNγ+/TNFα+ following stimulation of PBMC and BAL cells with overlapping peptide pools that covered SVV ORFs 4, 31, 61, and 63 using intracellular cytokine staining (ICS) (Fig. 6, and Table 2).

Following stimulation with overlapping peptide pools, SVV-specific CD4 CM and EM T cells in BAL were detected at 7 dpi in control animals, and their frequencies remained stable until 56 dpi (Fig. 6A and 6B). SVV-specific CD8 CM and EM T cells were also detected 7 dpi. Whereas the frequency of SVV-specific CD8 CM T cells increased from 7 to 21 dpi after which it declined, that of CD8 EM T cells remained stable until 56 dpi (Fig. 6C and D). Frequency of SVV-specific CD4 CM T cells were lower in BAL from CD20 depleted animals 7–21 dpi compared to control animals (p = 0.01, 0.008 and 0.03 respectively, Fig. 6A). The frequencies of SVV-specific CD8 CM in BAL were initially comparable between CD20 depleted animals and controls but decayed faster in CD20 depleted animals 21 dpi (p<0.001, Fig. 6C). The frequency of SVV-specific CD8 EM was also initially lower in BAL in CD20 depleted animals 7 and 14 dpi (0.03 and 0.04 respectively, Fig. 7D). After day 21, frequency of SVV-specific T cells was comparable in CD20 depleted and control animals. In CD8 depleted animals the frequency of responding T cells within CD4 CM subset was lower than that detected in control animals 7–21 dpi (p = 0.04, p = 0.006 and p = 0.04 respectively, Fig. 6A). Similarly the frequency of responding T cells within CD4 EM was lower 14 dpi compared to controls (p = 0.0413) (Fig. 6B). SVV-specific CD8 T cells were first detected in CD8 depleted animals 21 dpi, which corresponds to the re-appearance of the CD8 T cells in these animals, albeit with lower frequency within the CD8 CM subset compared to control animals (p<0.001, Fig. 6C). SVV-specific CD4 T cells appeared in CD4 depleted animals 28 dpi and their frequencies within the CM and EM subsets were comparable to those observed in control animals. It should be noted that 21–28 dpi, the number of CD8 and CD4 T cells is still much lower in CD8 and CD4 T cell depleted animals respectively compared to controls. Therefore, although the relative frequencies of responding T cells within CM and EM subsets may be comparable, the absolute number of SVV specific CD8 and CD4 T cells is much lower in CD8 and CD4 depleted animals respectively compared to controls. In contrast, frequency of SVV-specific CD8 T cells within CM and EM subsets in CD4 depleted animals were lower than those observed in control animals at several time points (14 and 21 dpi in CD8 CM p = 0.007 and <0.001, respectively; and 7 and 14 dpi in CD8 EM p = 0.02 and 0.01, respectively, Fig. 6C and 6D).
In PBMCs, SVV-specific T cells were detected 7 dpi in control animals and their frequency remained stable until 56 dpi (Fig. 6E–H). In CD20 depleted animals, SVV-specific T cells were also detected 7 dpi, but the frequencies of CD4 EM and CD8 EM were lower compared to control animals (p = 0.03 and p = 0.007, respectively) (Fig. 6E–H). SVV-specific CD4 T cells in CD8 depleted animals were also detected with similar kinetics and frequencies as seen in control animals (Fig. 6E and 6F). Although SVV-specific CD8 T cells were also detected 7 dpi in CD4 depleted animals, their frequency was initially lower (7 dpi p<0.001 for both CD8 CM and EM, Fig. 6G and 6H). SVV-specific CD8 T cells in CD8 depleted animals were detected 21 dpi (Fig. 6G and 6H), concomitant with the re-appearance of CD8 T cells in these animals (Fig. 1). SVV-specific CD4 T cells were detected 28 dpi in CD4 depleted animals (Fig. 6E and 6F), corresponding with the re-appearance of CD4 T cells in these animals (Fig. 1). As stated above, although the relative percentage of responding T cells within CM and EM subsets in the T cell depleted animals may be comparable to those of control animals, the number of and CD8 and CD4 T cells is still much lower in CD8 and CD4 T cell depleted animals respectively compared to controls.
To determine the magnitude of the frequency of T cells in response to SVV peptide stimulation, we measured the AUC for SVV-specific T cells in all four animals groups between 7 and 35 dpi. This analysis showed that in BAL the magnitude of: (1) SVV-specific CD4 CM T cell response is significantly reduced in CD20 and CD8 depleted animals (p = 0.0155 and p = 0.0036, respectively); (2) SVV-specific CD8 CM response are significantly reduced in CD20 and CD4 depleted animals (p = 0.0146 and p = 0.0023, respectively); and (3) SVV-specific CD8 EM response is significantly reduced in CD4 depleted animals (p = 0.0445) compared to control animals.
The impact of T and B cell depletion on the development of cytolytic T cells
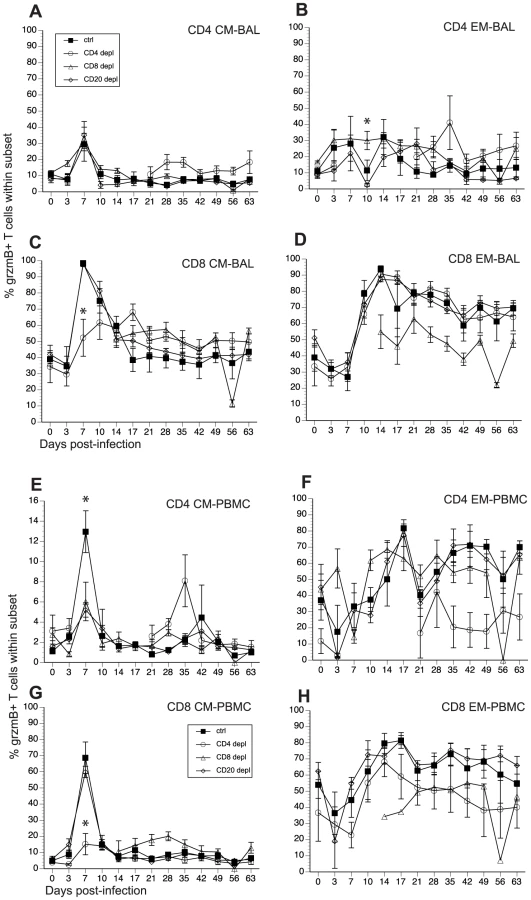
SVV and VZV infections are associated with the development of cytolytic CD4 and CD8 T cells [46], [52]. In order to determine how loss of lymphocyte subsets alters frequency of T cells with cytolytic potential, we measured the changes in granzyme B (grzmB) expressing CD4 and CD8 T cells (Fig 7, and Table 2). In BAL, we detected an increase in the frequency in grzmB+ CD4 CM 7 dpi in all animals (excluding CD4 depleted animals), which was followed by a quick return to baseline 14 dpi (Fig. 7A). Frequency of grzmB+ CD4 EM increased 3 dpi in control and CD8 depleted animals and 7 dpi in CD20 depleted animals (Fig. 7B). This increase in frequency was sustained until 28 dpi after which frequencies return to baseline levels (Fig. 7B). In CD4 depleted animals, grzmB+ CD4 CM and EM were detected 21 dpi (Fig. 7A and 7B) coinciding with the reappearance of CD4 T cells in these animals (Fig. 1) and peaked in frequency 35 dpi. As described for CD4 T cells, the frequency of grzmB+ CD8 CM T cells increased 7 dpi in all animals (excluding CD8 depleted animals) before returning to baseline 21 dpi (Fig. 7C). However, this increase was significantly reduced in CD4 depleted animals compared to control animals (p <0.001) (Fig. 7C). The frequency of grzmB+ CD8 EM increased 10 dpi in all groups (with the exception of CD8 depleted animals), followed by a small decrease and the establishment of a new set point (Fig. 7D). We detected grzmB+ CD8 CM and EM T cells in CD8 depleted animals 14 dpi, after which the frequency remained stable for the duration of the study (Figs. 7C and D).
A similar pattern of changes in grzmB+ T cell frequencies was observed in PBMCs. We detected a small increase in frequency of grzmB+ CD4 CM 7 dpi in control animals, followed by a return to baseline levels 10 dpi (Fig. 7E). This increase was significantly lower in CD8 and CD20 depleted animals compared to control animals (p <0.001 in both cases) (Fig. 7E). The frequency of grzmB+ CD4 EM T cells increased 10 dpi in CD8 depleted animals and 14 dpi in CD20 depleted and control animals (Fig. 7F), before reaching the same peak magnitude 17 dpi followed by the establishment of a new set point 21 dpi (Fig. 7F). In the CD8 CM subset, the frequency of grzmB+ peaked sharply 7 dpi in all groups, albeit at a significantly lower level in CD4 depleted animals (p<0.001), before returning to baseline levels 10 dpi (Fig. 7G). The frequency of grzmB+ CD8 EM increased 10 dpi in all groups, peaked 17 dpi before establishing a new set point (Fig. 7H). We detected grzmB+ CD8 CM and EM in CD8 depleted animals 14 dpi, which coincides with the re-appearance of CD8 T cells in these animals (Fig. 7H).
Finally, we measured the AUC between 3 and 21 dpi to assess the differences in grzmB expression between the different groups following SVV infection. In BAL, CD20 depleted animals have a significantly reduced frequency of grzmB+ CD4 EM T cells compared to control animals (p = 0.025). In addition, CD4 depleted animals have a significantly reduced frequency of grzmB+ CD8 CM T cells compared to control animals (p = 0.0147). In peripheral blood, the grzmB+ CD4 CM response was lower in both CD8 (p = 0.0265) and CD20 (p = 0.0356) depleted animals. Moreover, the magnitude of grzmB expression in the CD8 CM subset is lower in CD4 depleted animals (p = 0.0002).
Discussion
Several observations suggest that VZV infection is controlled via cellular immunity rather than humoral immunity. Specifically, children afflicted with T cell deficiencies are at increased risk from progressive varicella. In contrast, children diagnosed with B cell deficiencies such as agammaglobulinemia exhibit uncomplicated varicella [17], [18], [19], [20], [21]. Moreover, whereas VZV antibody administration after contagion but before the appearance of exanthem has been shown to block progression of systemic varicella [53], administration after the onset of varicella has no effect on disease progression [12]. Finally, despite the presence of high VZV-specific IgG titers, aged persons are at increased risk from VZV reactivation than young adults. However, the exact contributions of T cell and B cell immunity to the resolution of acute VZV infection in unvaccinated individuals have not yet been experimentally elucidated. This knowledge would play a critical role in the design of second-generation vaccines against VZV.
SVV is also a neurotropic α herpesvirus and a homologue of VZV that causes varicella in nonhuman primates. We have recently shown that SVV infection of juvenile rhesus macaques recapitulates the hallmarks of primary VZV infection in children including the development of a vesicular rash, the generation of T and B cell responses and the establishment of latency in sensory ganglia [46]. In this model, the animals are infected intrabronchially, which bypasses the early phase of VZV incubation/replication in the head and neck region, resulting in an incubation period of approximately 10 days. Direct inoculation of SVV into the lungs might also result in altered viral amplification since VZV replicates efficiently in tonsilar memory CD4 T cells [48], [49]. Although, this incubation period is slightly shorter than that of VZV (10–21 days), the detection of T and antibody responses in SVV-infected macaques in relation to the onset of the rash occurs with comparable kinetics as those described for VZV [12]. Moreover, whereas natural and experimental SVV infection results in significant morbidity and mortality in other nonhuman primate species, infection of rhesus macaques with SVV results in a milder disease that more closely resembles varicella in humans [54], [55]. These observations suggest that SVV infection of rhesus macaques is a robust model with which to investigate the contribution of CD4 T cell, CD8 T cells and B cell immunity to the control of acute VZV infection.
Our results show that depletion of CD20 B cells and loss of early antibody production does not alter viral loads or disease severity. As expected, CD20-depleted animals were significantly compromised in their ability to generate SVV-specific IgM and IgG antibodies. Our data also suggest that the production of neutralizing antibodies is not required for resolution of varicella as the CD20 animals failed to generate a robust neutralizing antibody response yet had uncomplicated varicella. These data are in agreement with clinical observations that the VZV specific antibody response generated during acute VZV infection contributes little to the immune response to VZV [47]. Indeed, children with congenital agammaglobulinemia experience uncomplicated varicella [56]. Moreover, passive antibody therapy is only successful when administered within the first 72 hours following VZV exposure [24], [25] and has no effect on disease progression if administered after the appearance of the rash [12]. We also observed similar kinetics and magnitude in the frequency of proliferating T cells in BAL and PBMCs of CD20 depleted animals compared to non-depleted controls. Although magnitude and kinetics of T cell proliferation were unchanged in CD20 depleted animals, we detected a lower frequency of SVV-specific T cells 7 and 14 dpi as detected by ICS following peptide stimulation (ORFs 4, 31, 61 and 63) in these animals. One possible explanation for these results is that B cells are an important source of antigen presenting cells in this in vitro assay, and their absence results in the detection of a lower frequency of responding T cells. Alternatively the absence of B cells in vivo could lead to a change in immunodominance profile, resulting in T cells in CD20 depleted animals preferentially responding to additional ORFs not included in our peptide pools. The SVV-induced increase in frequency of grzmB+ T cells in CD20 depleted animals was similar in kinetics and magnitude to that observed in control animals with the exception that frequency of grzmB+ CD4 CM T cells in PBMC 7 dpi was significantly lower than control animals. This decrease could be due to the fact that B cells contribute to the activation of CD4 T cells in the peripheral blood. Overall, these data are reminiscent of the documented clinical observations and strongly suggest that the development of antibody response during primary SVV/VZV infection is not critical to the resolution of the acute infection. Our data also suggest a potential role for B cells in modulating the specificity of the T cell response, but more conclusive analyses are required to specifically address that question.
Depletion of CD8 T cells resulted in slightly increased viral loads in whole blood and BAL samples but these differences did not reach statistical significance. Interestingly, the time points at which higher viral loads are observed in CD8 depleted animal differ between the BAL and WB, indicative of potentially different mechanisms of control. In the BAL slightly higher viral loads are detected 3 dpi. This is too early to suggest involvement of the CD8 T cell response. Indeed, the antibody that we used for CD8 T cell depletion targets all CD8 + lymphocytes, which in the macaque include NK cells. Thus, it is more likely that the higher viral load detected 3 dpi in CD8 depleted animals is a reflection of NK cell depletion rather than CD8 T cell depletion. In contrast, we observed a higher viral load 10 dpi in WB in CD8 depleted animals, which is more indicative of a defect in the CD8 T cell response. These observations are similar to clinical observations that severe varicella is associated with an absence of NK cells and primed CD8+ T cells responses in children [57]. CD8 depleted animals also experienced a somewhat more prolonged varicella episode and higher number of vesicles. Interestingly, we detected that depletion of CD8 T cells results in an increase in CD4 and B cell proliferation compared to control animals. This compensatory mechanism could have alleviated the consequences associated with loss of CD8 T cell function.
Finally, depletion of CD4 T cells resulted in disseminated varicella, higher viral loads and sustained viremia although complete CD4 T cell depletion was not achieved until 10 dpi. The difference in disease severity between CD4 and CD8 T cell depleted animals cannot be explained by differences in the duration of T cell depletion. In fact, complete CD4 T cell depletion was not achieved until 10 dpi and was very transient in the BAL compared to CD8 T cell depletion and lasted 7 days in PBMC (10–17 dpi), whereas CD8 T cells were depleted for 10 days in BAL and 17 days in PBMC (0–21 dpi). The severe varicella observed in CD4 depleted animals is in line with clinical observations that HIV+ children fail to generate a T cell response and suffer progressive-disseminated varicella as well as additional complications such as varicella pneumonia, hepatitis, coagulopathy and meningoencephalitis [16]. Additionally, HIV patients are more susceptible to herpes zoster during the phase in which absolute numbers of CD4 T cells decline [12], highlighting the importance of CD4 T cell responses in controlling VZV infection. It is possible that the use of anti-CD4 depleting antibody resulted in the depletion of CD4+ macrophages. However, we were not able to detect reduced frequency of monocytes/macrophages in CD4 depleted animals using CD14 and HLA-DR as markers (data not shown). It is also possible that the depleting antibody resulted in the loss of CD4+NK T cells. However, a difference in viral loads between CD4 depleted and control animals is not detected until 7 dpi, indicative of a defect in adaptive rather than innate immunity. Indeed, CD4-depleted animals experienced delayed and reduced CD8 T cell proliferative bursts in both peripheral blood and BAL. This delay correlated with a significant decrease in the frequency of SVV-specific CD8 T cells that were detected by ICS. Moreover, the appearance of SVV-induced grzmB+ CD8 CM T cells was also reduced in these animals. Similarly, B cell proliferation and IgG, IgM and neutralizing antibody production was compromised in CD4 depleted animals. Since data from our CD20 and CD8 depleted animals suggests a minimal role for these lymphocyte subsets in the resolution of acute SVV infection, it is very likely that the severe disease experiences by CD4-depleted animals is a consequence of a compromised CD4 T cell response.
Taken together, data presented in this manuscript suggest that the CD4 T cell response executes unique effector functions that are critical to the resolution of SVV/VZV infection. Although we cannot discount the possibility of synergism between the loss of B and CD8 T cell responses, our own depletion studies and data from additional studies support the hypothesis that CD4 T cells play a unique role during SVV/VZV infection. For instance, in addition to their role in recruiting CD8 CTL to sites of infection via release of Th1 cytokines [12], [58], CD4 T cells can kill VZV-infected cells [31], [59], [60], [61]. Moreover, studies that characterized the VZV-specific T cell response in patients with VZV-induced uveitis revealed that CD4 T cells were responsible for the bulk of the T cell response [62], [63]. VZV-specific CD4 T cells are also preferentially detected following re-exposure to VZV [64]. Taken together, the data presented in this manuscript and from previously published studies strongly suggest that CD4 T cells play a critical role in coordinating the anti-VZV/SVV response during primary infection that goes beyond providing help for CD8 and B cells. Additionally, a recent study investigating CMV-specific immune responses in CMV-seropositive renal transplant patients reported a positive correlation between low CD4 T cell counts and increased CMV DNA in the plasma of patients, suggesting a role for CD4 T cell immunity in controlling CMV reactivation [65]. This report, along with the data presented in this manuscript, suggest that the role of CD4 T cell responses in controlling herpesvirus infections may have previously been under-estimated. More investigations into this topic are needed to uncover the mechanisms by which CD4 T cells participate in the control of herpes viral infections.
In summary, using SVV infection of rhesus macaques as a model of VZV infection [46], we show for the first time in vivo the contribution of cellular immunity versus humoral immunity during acute SVV infection. Our results indicate that similar to clinical findings regarding VZV control in children, the ability of young rhesus macaques to control acute SVV infection is mediated by T cell immunity rather than humoral immunity. More importantly, our data strongly suggest that CD4 T cells mediate effector functions that are more important that providing help for antibody or CD8 T cell response in the resolution of acute SVV infection. Future studies will dissect the effector functions of CD4 T cells that are important for protection and elucidate the role of CD4 and CD8 T cell immunity in the protection against SVV reactivation.
Materials and Methods
Ethics statement
The study was carried out in strict accordance with the recommendations described in the Guide for the Care and Use of Laboratory Animals of the National Institute of Health, the Office of Animal Welfare and the United States Department of Agriculture. All animal work was approved by the Oregon National Primate Research Center Institutional Animal Care and Use Committee (IACUC protocol # 0779). The ONPRC has been continuously accredited by the American Association for Accreditation of Laboratory Animal Care since 1974 (PHS/OLAW Animal Welfare Assurance # A3304-01). All procedures were carried out under Ketamine anesthesia in the presence of veterinary staff and all efforts were made to minimize animal suffering.
Cells and virus
SVV was propagated in primary rhesus fibroblasts (1° RF) at 37°C in 175 cm2 flasks with DMEM supplemented with 10% FBS. SVV-infected 1° RF were frozen in FetalPlex with 10% DMSO, stored in LN2, and assayed by plaque assay. SVV cell lysate was obtained by scraping SVV-infected 1° RF at the height of cytopathic effect (CPE) followed by concentration, and then sonication using 7 pulses of 70–80W (Sonicator 3000, Misonix). The sonicated cell-resuspension was pelleted by centrifugation at 2000rpm for 5 min and frozen at −80°C.
Animals and sample collection
16 colony-bred Rhesus macaques (Macaca mulatta, RM) 3–4 years of age and of Indian origin were used in these studies. They were housed and handled in accordance with the Oregon National Primate Research Center Institutional Animal Care and Use Committee. To deplete CD4 T cells, 4 RM received the humanized monoclonal antibody OKT4-HulgG on −3, 0, and 7 days post-infection (dpi) at a dose of 50mg/Kg. To deplete CD8 T cells, 4 RM received the mouse-human chimeric monoclonal antibody cM-T807 on −3 dpi at a dose of 10mg/Kg, and on 0, 3, and 7 dpi at a dose of 5mg/Kg. To deplete B cells, 4 RM were administered the mouse-human chimeric antibody Rituxan on −7, 0, and 7 dpi at 20mg/Kg. All animals were inoculated intrabronchially with 4×105 PFU SVV as previously described [46]. Blood, bronchial alveolar lavage (BAL), and ∼1 ml saliva samples were collected on days −14, 0, 3, 7, 10, 14, 17, 21, 28, 35, 42, 49, 56, 63, and 70. The absolute numbers of lymphocytes/µL of blood were obtained using a Hemavet (Drew Scientific, Inc., Dallas, TX). BAL samples were pelleted and resuspended in RPMI supplemented with 10%FBS, streptomycin/penicillin, and L-glutamine. Peripheral blood mononuclear cells (PBMCs) and plasma were isolated by centrifugation over a histopaque gradient (Sigma) as per the manufacturer’s recommendation. Saliva samples were centrifuged in order to separate the buccal epithelial cells from the saliva.
DNA extraction and quantitative PCR
DNA was extracted from heparinized whole blood, BAL cells, saliva and buccal epithelial cells using the Qiagen genomic DNA extraction kit (Qiagen) and SVV DNA loads were determined by real-time PCR using primers and probes specific for ORF21 using the ABI 7700 and ABI StepOne instruments (Applied Biosystems, Foster City, CA) exactly as previously described [46].
Measurement of T and B cell proliferation
PBMC and BAL cells were surface stained with antibodies against CD8β (Beckman Coulter), CD4 (eBioscience, San Diego, CA), CD28, and CD95 (BioLegend, San Diego, CA) to delineate the naive (CD28+CD95−), central memory (CD28+CD95+), and effector memory (CD28−CD95+) T cell subsets. PBMC and BAL cells were also surface stained with antibodies against CD20 (Beckman Coulter, Brea, CA), CD3 (BD Pharmingen, San Diego, CA), IgD (Southern Biotech), and CD27 (BioLegend) to delineate the naïve (IgD+CD27−), MZ-like (IgD+CD27+), memory (IgD−CD27+) B cell subsets. Cells were fixed and permeabilized according to manufacturer recommendations (Biolegend) before the addition of Ki67-specific antibody (BD Pharmingen). The samples were analyzed using the LSRII instrument (Beckton, Dickinson and Company, San Jose, CA) and FlowJo software (TreeStar, Ashland, OR).
Granzyme B staining
PBMC and BAL cells were surface stained using antibodies against CD4, CD8β, CD28, and CD95 as described for Ki67 staining. Cells were fixed and permeabilized using fixation buffer (BioLegend), and then stained intracellularly using an antibody against granzyme B (BD Pharmingen). Samples were analyzed using the LSRII instrument and FlowJo software.
Intracellular cytokine staining
PBMC and BAL cells were either stimulated with SVV lysate (1 ug) for 12 h followed by incubation with Brefeldin A for 6 h, or were incubated with one of the following SVV peptides and Brefeldin A (Sigma, St Louis, MO) for 6 h: open reading frame (ORF) 4; ORF31; ORF61; or ORF63. After stimulation cells were surface stained with antibodies against CD4, CD8β, CD28, and CD95 as described for Ki67 staining. Samples were fixed and permeabilized using fixation buffer (BioLegend), and then stained using antibodies against IFNγ and TNFα (eBioscience). Samples were analyzed using the LSRII instrument and FlowJo software.
Enzyme-linked Immunosorbent Assay (ELISA)
ELISA plates were coated with SVV lysate overnight at 4°C, washed three times with 0.05% Tween-PBS, and incubated with heat-inactivated (56°C, 30 min) plasma samples in 3-fold dilutions in triplicate for 1 h. After washing three times with 0.05% Tween-PBS, horseradish peroxidase (HRP)-conjugated anti-rhesus IgG (Nordic Immunology, The Netherlands) or anti-rhesus IgM (Brookwood Biomedical, Birmingham, AL) was added for 1 h, followed by addition of o-phenylenediamine•2HCl (OPD) substrate (Sigma, St Louis, MO). The reaction was stopped with the addition of 1 M HCl. IgG and IgM endpoint titers were calculated using log-log transformation of the linear portion of the curve, and 0.1 optical density (OD) units as cut-off. IgG and IgM titers were standardized using a positive control sample that was included in every ELISA plate.
Plaque reduction assay
Neutralizing antibody titers were evaluated as previously described [66] by measuring the plasma dilution at which 50% reduction in SVV plaques was achieved (NT50). Serial two-fold dilutions of heat-inactivated monkey plasma from 0, 14, and 70 dpi were incubated with approximately 150pfu of SVV for 30 minutes at 37°C. Virus/plasma samples were then added to duplicate primary rhesus fibroblast cell monolayers seeded on 12-well plates and incubated for 4 days at 37°C. Monolayers were fixed with methanol and then stained with crystal violet to visualize the SVV plaques.
Statistical analysis
Repeated measures of ANOVA was used to explore differences between groups. Pair-wise comparisons at each time point were performed using contrast t-test. Statistical significance was determined at the level of 0.05. First order autoregressive covariance structure was used to account for within subject correlation. Due to the small sample size, other complicated covariance analyses was not considered.
Zdroje
1. ArvinAM 2000 Varicella-Zoster virus: pathogenesis, immunity, and clinical management in hematopoietic cell transplant recipients. Biol Blood Marrow Transplant 6 219 230
2. NagelMACohrsRJMahalingamRWellishMCForghaniB 2008 The varicella zoster virus vasculopathies: Clinical, CSF, imaging, and virologic features. Neurology 70 853 860
3. MuellerNHGildenDHCohrsRJMahalingamRNagelMA 2008 Varicella zoster virus infection: clinical features, molecular pathogenesis of disease, and latency. Neurol Clin 26 675 697, viii
4. RagozzinoMWMelton3rdLJKurlandLTChuCPPerryHO 1982 Population-based study of herpes zoster and its sequelae. Medicine (Baltimore) 61 310 316
5. InsingaRPItzlerRFPellissierJMSaddierPNikasAA 2005 The incidence of herpes zoster in a United States administrative database. J Gen Intern Med 20 748 753
6. WeaverBA 2009 Herpes Zoster Overview: Natural History and Incidence. J Am Osteopath Assoc 109 S2 6
7. GoulleretNMauvisseauEEssevaz-RouletMQuinlivanMBreuerJ 2010 Safety profile of live varicella virus vaccine (Oka/Merck): five-year results of the European Varicella Zoster Virus Identification Program (EU VZVIP). Vaccine 28 5878 5882
8. MarinMZhangJXSewardJF 2011 Near Elimination of Varicella Deaths in the US After Implementation of the Vaccination Program. Pediatrics 128 214 220
9. OxmanMNLevinMJJohnsonGRSchmaderKEStrausSE 2005 A vaccine to prevent herpes zoster and postherpetic neuralgia in older adults. N Engl J Med 352 2271 2284
10. OxmanMNLevinMJ 2008 Vaccination against Herpes Zoster and Postherpetic Neuralgia. J Infect Dis 197 Suppl 2 S228 236
11. LevinMJOxmanMNZhangJHJohnsonGRStanleyH 2008 Varicella-zoster virus-specific immune responses in elderly recipients of a herpes zoster vaccine. J Infect Dis 197 825 835
12. ArvinAM 1996 Immune responses to varicella-zoster virus. Infect Dis Clin North Am 10 529 570
13. SchmaderK 2001 Herpes zoster in older adults. Clin Infect Dis 32 1481 1486
14. BurkeBLSteeleRWBeardOWWoodJSCainTD 1982 Immune responses to varicella-zoster in the aged. Arch Intern Med 142 291 293
15. BergerRFlorentGJustM 1981 Decrease of the lymphoproliferative response to varicella-zoster virus antigen in the aged. Infect Immun 32 24 27
16. ArvinAMKoropchakCMWilliamsBRGrumetFCFoungSK 1986 Early immune response in healthy and immunocompromised subjects with primary varicella-zoster virus infection. J Infect Dis 154 422 429
17. NaderSBergenRSharpMArvinAM 1995 Age-related differences in cell-mediated immunity to varicella-zoster virus among children and adults immunized with live attenuated varicella vaccine. J Infect Dis 171 13 17
18. RedmanRLNaderSZerboniLLiuCWongRM 1997 Early reconstitution of immunity and decreased severity of herpes zoster in bone marrow transplant recipients immunized with inactivated varicella vaccine. J Infect Dis 176 578 585
19. WilsonASharpMKoropchakCMTingSFArvinAM 1992 Subclinical varicella-zoster virus viremia, herpes zoster, and T lymphocyte immunity to varicella-zoster viral antigens after bone marrow transplantation. J Infect Dis 165 119 126
20. ZerboniLNaderSAokiKArvinAM 1998 Analysis of the persistence of humoral and cellular immunity in children and adults immunized with varicella vaccine. J Infect Dis 177 1701 1704
21. ArvinAMPollardRBRasmussenLEMeriganTC 1978 Selective impairment of lymphocyte reactivity to varicella-zoster virus antigen among untreated patients with lymphoma. J Infect Dis 137 531 540
22. CamittaBChusidMJStarshakRJGottschallJL 1994 Use of irradiated lymphocytes from immune donors for treatment of disseminated varicella. J Pediatr 124 593 596
23. ParyaniSGArvinAMKoropchakCMDobkinMBWittekAE 1984 Comparison of varicella zoster antibody titers in patients given intravenous immune serum globulin or varicella zoster immune globulin. J Pediatr 105 200 205
24. BrunellPARossAMillerLHKuoB 1969 Prevention of varicella by zoster immune globulin. N Engl J Med 280 1191 1194
25. OrensteinWAHeymannDLEllisRJRosenbergRLNakanoJ 1981 Prophylaxis of varicella in high-risk children: dose-response effect of zoster immune globulin. J Pediatr 98 368 373
26. WeigleKAGroseC 1984 Molecular dissection of the humoral immune response to individual varicella-zoster viral proteins during chickenpox, quiescence, reinfection, and reactivation. J Infect Dis 149 741 749
27. JuraEChadwickEGJosephsSHSteinbergSPYogevR 1989 Varicella-zoster virus infections in children infected with human immunodeficiency virus. Pediatr Infect Dis J 8 586 590
28. AbendrothASlobedmanBLeeEMellinsEWallaceM 2000 Modulation of major histocompatibility class II protein expression by varicella-zoster virus. J Virol 74 1900 1907
29. JonesLBlackAPMalavigeGNOggGS 2006 Persistent High Frequencies of Varicella-Zoster Virus ORF4 Protein-Specific CD4+ T Cells after Primary Infection. J Virol 80 9772 9778
30. MalavigeGNJonesLBlackAPOggGS 2007 Rapid Effector Function of Varicella-Zoster Virus Glycoprotein I-Specific CD4 + T Cells Many Decades after Primary Infection. J Infect Dis 195 660 664
31. ArvinAMSharpMSmithSKoropchakCMDiazPS 1991 Equivalent recognition of a varicella-zoster virus immediate early protein (IE62) and glycoprotein I by cytotoxic T lymphocytes of either CD4+ or CD8+ phenotype. J Immunol 146 257 264
32. FreyCRSharpMAMinASSchmidDSLoparevV 2003 Identification of CD8+ T cell epitopes in the immediate early 62 protein (IE62) of varicella-zoster virus, and evaluation of frequency of CD8+ T cell response to IE62, by use of IE62 peptides after varicella vaccination. J Infect Dis 188 40 52
33. AsanumaHSharpMMaeckerHTMainoVCArvinAM 2000 Frequencies of memory T cells specific for varicella-zoster virus, herpes simplex virus, and cytomegalovirus by intracellular detection of cytokine expression. J Infect Dis 181 859 866
34. AbendrothAArvinAM 2001 Immune evasion as a pathogenic mechanism of varicella zoster virus. Semin Immunol 13 27 39
35. KinchingtonPR 1999 Latency of varicella zoster virus; a persistently perplexing state. Front Biosci 4 D200 211
36. MyersMGDuerHLHauslerCK 1980 Experimental infection of guinea pigs with varicella-zoster virus. J Infect Dis 142 414 420
37. WroblewskaZValyi-NagyTOtteJDillnerAJacksonA 1993 A mouse model for varicella-zoster virus latency. Microb Pathog 15 141 151
38. MyersMGConnellyBLStanberryLR 1991 Varicella in hairless guinea pigs. J Infect Dis 163 746 751
39. MyersMGConnellyBL 1992 Animal models of varicella. J Infect Dis 166 Suppl 1 S48 50
40. KuC-CBesserJAbendrothAGroseCArvinAM 2005 Varicella-Zoster Virus Pathogenesis and Immunobiology: New Concepts Emerging from Investigations with the SCIDhu Mouse Model. J Virol 79 2651 2658
41. GrayWLOakesJE 1984 Simian varicella virus DNA shares homology with human varicella-zoster virus DNA. Virology 136 241 246
42. GrayWLPumphreyCYRuyechanWTFletcherTM 1992 The simian varicella virus and varicella zoster virus genomes are similar in size and structure. Virology 186 562 572
43. GrayWLStarnesBWhiteMWMahalingamR 2001 The DNA sequence of the simian varicella virus genome. Virology 284 123 130
44. MahalingamRTraina-DorgeVWellishMSmithJGildenDH 2002 Naturally acquired simian varicella virus infection in African green monkeys. J Virol 76 8548 8550
45. GrayWL 2003 Pathogenesis of simian varicella virus. J Med Virol 70 Suppl 1 S4 8
46. MessaoudiIBarronAWellishMEngelmannFLegasseA 2009 Simian varicella virus infection of rhesus macaques recapitulates essential features of varicella zoster virus infection in humans. PLoS Pathog 5 e1000657
47. ArvinAM 1996 Varicella-zoster virus. Clin Microbiol Rev 9 361 381
48. KuCCPadillaJAGroseCButcherECArvinAM 2002 Tropism of varicella-zoster virus for human tonsillar CD4(+) T lymphocytes that express activation, memory, and skin homing markers. J Virol 76 11425 11433
49. KuC-CZerboniLItoHGrahamBSWallaceM 2004 Varicella-Zoster Virus Transfer to Skin by T Cells and Modulation of Viral Replication by Epidermal Cell Interferon-{alpha}. J Exp Med 200 917 925
50. MehtaSKCohrsRJForghaniBZerbeGGildenDH 2004 Stress-induced subclinical reactivation of varicella zoster virus in astronauts. J Med Virol 72 174 179
51. MehtaSKTyringSKGildenDHCohrsRJLealMJ 2008 Varicella-Zoster Virus in the Saliva of Patients with Herpes Zoster. J Infect Dis 197 654 657
52. HaywardAGillerRLevinM 1989 Phenotype, cytotoxic, and helper functions of T cells from varicella zoster virus stimulated cultures of human lymphocytes. Viral Immunol 2 175 184
53. GershonAASteinbergSBrunellPA 1974 Zoster immune globulin. A further assessment. N Engl J Med 290 243 245
54. SoikeKF 1992 Simian varicella virus infection in African and Asian monkeys. The potential for development of antivirals for animal diseases. Ann N Y Acad Sci 653 323 333
55. SoikeKFRanganSRGeronePJ 1984 Viral disease models in primates. Adv Vet Sci Comp Med 28 151 199
56. WellerTH 1983 Varicella and Herpes Zoster. N Engl J Med 309 1362 1368
57. VossenMTBiezeveldMHde JongMDGentMRBaarsPA 2005 Absence of circulating natural killer and primed CD8+ cells in life-threatening varicella. J Infect Dis 191 198 206
58. NakanishiYLuBGerardCIwasakiA 2009 CD8(+) T lymphocyte mobilization to virus-infected tissue requires CD4(+) T-cell help. Nature 462 510 513
59. DiazPSSmithSHunterEArvinAM 1989 T lymphocyte cytotoxicity with natural varicella-zoster virus infection and after immunization with live attenuated varicella vaccine. J Immunol 142 636 641
60. HicklingJKBorysiewiczLKSissonsJG 1987 Varicella-zoster virus-specific cytotoxic T lymphocytes (Tc): detection and frequency analysis of HLA class I-restricted Tc in human peripheral blood. J Virol 61 3463 3469
61. HaywardARPontesilliOHerbergerMLaszloMLevinM 1986 Specific lysis of varicella zoster virus-infected B lymphoblasts by human T cells. J Virol 58 179 184
62. MilikanJCMBaarsmaGSKuijpersRWAMOsterhausADMEVerjansGMGM 2009 Human Ocular-Derived Virus-Specific CD4+ T Cells Control Varicella Zoster Virus Replication in Human Retinal Pigment Epithelial Cells. Invest Ophthalmol Vis Sci 50 743 751
63. MilikanJCMKuijpersRWAMBaarsmaGSOsterhausADMEVerjansGMGM 2006 Characterization of the varicella zoster virus (VZV)-specific intra-ocular T-cell response in patients with VZV-induced uveitis. Exp Eye Res 83 69 75
64. VossenMTGentMRWeelJFde JongMDvan LierRA 2004 Development of virus-specific CD4+ T cells on reexposure to Varicella-Zoster virus. J Infect Dis 190 72 82
65. SundFLidehallAKClaessonKFossATottermanTH 2010 CMV-specific T-cell immunity, viral load, and clinical outcome in seropositive renal transplant recipients: a pilot study. Clin Transplant 24 401 409
66. SoikeKFFelsenfeldADGeronePJ 1981 Acyclovir treatment of experimental simian varicella infection of monkeys. Antimicrob Agents Chemother 20 291 297
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2011 Číslo 11
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- Multiple Candidate Effectors from the Oomycete Pathogen Suppress Host Plant Immunity
- The Splicing Factor Proline-Glutamine Rich (SFPQ/PSF) Is Involved in Influenza Virus Transcription
- A TNF-Regulated Recombinatorial Macrophage Immune Receptor Implicated in Granuloma Formation in Tuberculosis
- SH3 Domain-Mediated Recruitment of Host Cell Amphiphysins by Alphavirus nsP3 Promotes Viral RNA Replication
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy