
Proteolysis of Human Thrombin Generates Novel Host Defense Peptides
The coagulation system is characterized by the sequential and highly localized activation of a series of serine proteases, culminating in the conversion of fibrinogen into fibrin, and formation of a fibrin clot. Here we show that C-terminal peptides of thrombin, a key enzyme in the coagulation cascade, constitute a novel class of host defense peptides, released upon proteolysis of thrombin in vitro, and detected in human wounds in vivo. Under physiological conditions, these peptides exert antimicrobial effects against Gram-positive and Gram-negative bacteria, mediated by membrane lysis, as well as immunomodulatory functions, by inhibiting macrophage responses to bacterial lipopolysaccharide. In mice, they are protective against P. aeruginosa sepsis, as well as lipopolysaccharide-induced shock. Moreover, the thrombin-derived peptides exhibit helical structures upon binding to lipopolysaccharide and can also permeabilize liposomes, features typical of “classical” helical antimicrobial peptides. These findings provide a novel link between the coagulation system and host-defense peptides, two fundamental biological systems activated in response to injury and microbial invasion.
Published in the journal:
. PLoS Pathog 6(4): e32767. doi:10.1371/journal.ppat.1000857
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000857
Summary
The coagulation system is characterized by the sequential and highly localized activation of a series of serine proteases, culminating in the conversion of fibrinogen into fibrin, and formation of a fibrin clot. Here we show that C-terminal peptides of thrombin, a key enzyme in the coagulation cascade, constitute a novel class of host defense peptides, released upon proteolysis of thrombin in vitro, and detected in human wounds in vivo. Under physiological conditions, these peptides exert antimicrobial effects against Gram-positive and Gram-negative bacteria, mediated by membrane lysis, as well as immunomodulatory functions, by inhibiting macrophage responses to bacterial lipopolysaccharide. In mice, they are protective against P. aeruginosa sepsis, as well as lipopolysaccharide-induced shock. Moreover, the thrombin-derived peptides exhibit helical structures upon binding to lipopolysaccharide and can also permeabilize liposomes, features typical of “classical” helical antimicrobial peptides. These findings provide a novel link between the coagulation system and host-defense peptides, two fundamental biological systems activated in response to injury and microbial invasion.
Introduction
The innate immune system, largely based on antimicrobial peptides, provides a first line of defense against invading microbes [1], [2], [3], [4], [5]. During recent years it has become increasingly evident that many cationic and amphipathic antimicrobial peptides, such as defensins and cathelicidins, are multifunctional, also mediating immunomodulatory roles and angiogenesis [6], [7], [8], thus motivating the recent and broader definition host defense peptides (HDP) for these members of the innate immune system. The family of HDPs has recently been shown to encompass various bioactive peptides with antimicrobial activities, including proinflammatory and chemotactic chemokines [9], neuropeptides [10], peptide hormones [11], [12], growth factors [13], the anaphylatoxin peptide C3a [14], [15], and kininogen-derived peptides [16], [17], [18].
The coagulation cascade represents a fundamental host defense system activated in response to injury and infection [19], [20]. Through a series of cascade-like proteinase activation steps, thrombin is formed, leading to fibrinogen degradation and clot formation [20]. In addition, thrombin has other physiologic functions in hemostasis; i.e., mediating clot stabilization by activation of TAFI and activation of transglutaminase (FXIII), providing anticoagulant and antifibrinolytic activities in complex with thrombomodulin, and causing platelet aggregation due to PAR cleavage [19], [20]. Moreover, thrombin elicits numerous cellular responses, including increased CAM expression and growth factor and cytokine release by endothelial cells, as well as growth stimulation of both smooth muscle and fibroblast cells [20]. These pivotal functions of thrombin in host defense, its ubiquitous occurrence in blood and in fibrin networks, the high evolutionary conservation of the enzyme, as well as presence of an amphipathic, cationic and helical C-terminus in the protein [19], made us raise the question whether thrombin could constitute a source of HDPs released at sites of wounding and infection. Our results indeed show that C-terminal peptides of thrombin constitute a previously undisclosed and significant class of HDPs, generated in humans during wounding and with therapeutic potential against infection and septic shock.
Results
Proteolysis of prothrombin and thrombin generates antimicrobial activity
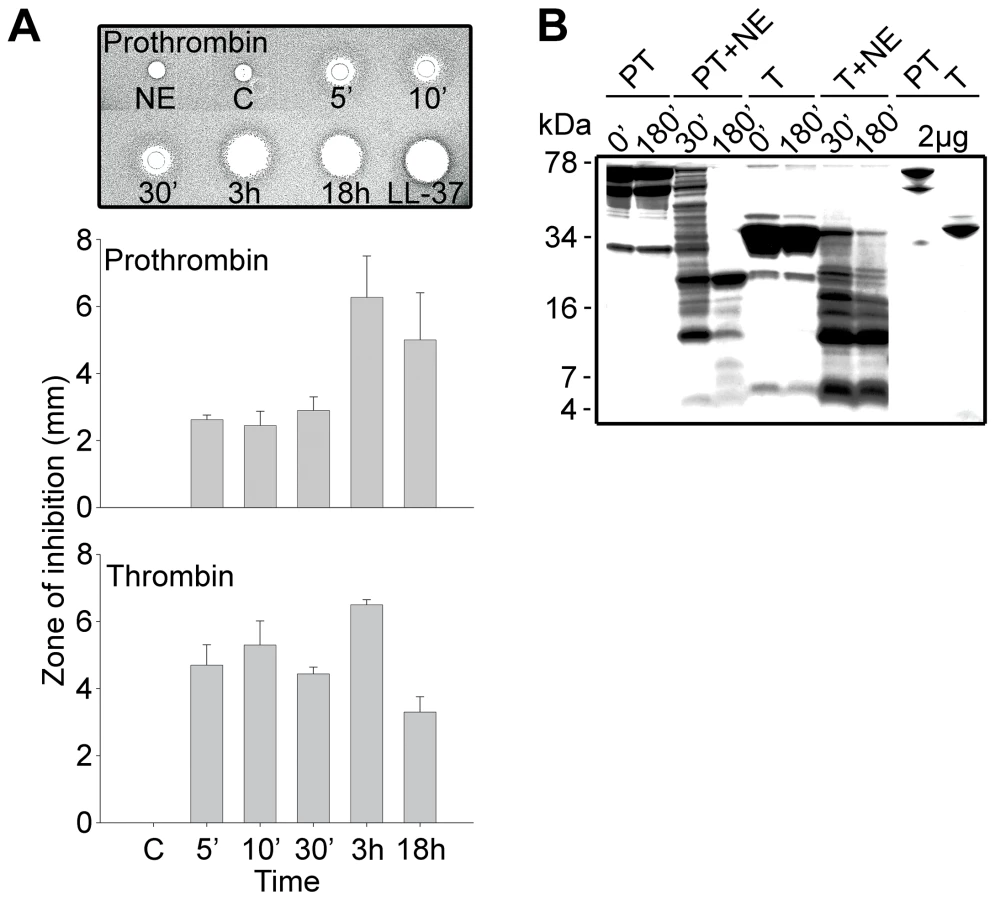
To test the hypothesis that prothrombin or its activated forms may generate antimicrobial peptides upon fragmentation, we incubated human prothrombin and thrombin with neutrophil elastase, a major neutral protease released by leukocytes during blood coagulation and inflammation or in response to bacterial products such as endotoxins. Earlier studies have shown that neutrophil elastase acts on proteinase-sensitive regions in human thrombin, generating smaller fragments [21]. As judged by the RDA assays (Figure 1A), digestion of the proteins yielded antimicrobial activity already after 5 min of incubation with the enzyme. In contrast, the intact mother proteins were inactive. The activity following proteolysis was still observed after several hours of incubation, suggesting the presence of relatively stable intermediates. Noteworthy, the maximum observed inhibition zones were similar in size to those generated by the classical antimicrobial peptide LL-37. Analysis by SDS-PAGE (Figure 1B) showed that the degradation generated several low molecular weight fragments in the range of 5–15 kDa. In spite of the known amidolytic properties of thrombin, no detectable antimicrobial activity was detected after prolonged incubation of the enzyme form alone (not shown). The observation that the zymogen as well as the activated forms generated similar activities, suggests that the antimicrobial epitopes localize to regions in the active enzyme after R271 (prothrombin numbering).
Structure-based screening for identification of antimicrobial epitopes
In order to identify possible antibacterial peptide regions of prothrombin/thrombin, overlapping peptide sequences comprising 20mers (Figure 2A) were synthesized and screened for antibacterial activities against the two test bacteria E. coli and P. aeruginosa. Properties common for most antimicrobial peptides include minimum levels of cationicity, amphipathicity, and hydrophobicity [5]. Taking these structural prerequisites into account, additional peptides comprising regions of high net charge and/or presence of amphipathic helical regions, such as those encompassing the C-terminus, were selected and synthesized (Figure 2A). The experiments showed that particularly peptides derived from the C-terminal region (peptides 45–48) were antimicrobial, although other active peptides were also identified (eg. 9 and 31) (Figure 2B). However, at high ionic strength (0.1 M NaCl), only the C-terminal peptides retained their antimicrobial activity against E. coli as well as P. aeruginosa (Figure S1) demonstrating that only this region, characterized by a high relative hydrophobicity (μHrel), positive net charge (znet = +2 for the most active C-terminal peptides) (Figure 2B) and amphipathicity (Figure 2C), features typical of classical antimicrobial peptides [1], [2], [3], [4], may generate peptides active against bacteria at physiologic conditions. Corresponding to the antimicrobial activities observed above, only peptides derived from the C-terminal part significantly bound to E. coli LPS (Figure 2D).

Since the absence of activity of the holoproteins in RDA could possibly be attributed to their high molecular weight (compromising diffusion during the assay), the antibacterial results above were further substantiated by matrix-free viable count assays. The results demonstrated that in contrast to the holoproteins, the selected model peptides VFR17 (Figure 2A; peptide 48) and the longer 25mer peptide GKY25 (indicated in Figure 2A) from the C-terminal part of the enzyme (both peptides indicated by colors in the 3D model of thrombin; Figure 2E), demonstrated significant antibacterial activity (Figure 2F), thus corroborating the RDA assays above (Figure 1A and Figure 2B). In conclusion, LPS-binding and antimicrobial data, combined with structural and biophysical considerations clearly indicate a pivotal role of Thrombin-derived C-terminal Peptides, in the following text denoted “TCP”, for mediating the antimicrobial activity.
Definition of low molecular weight fragments generated by degradation of thrombin
In parallel to the above structure-based screening approach, studies were undertaken to identify active fragments generated after subjection of thrombin to neutrophil elastase. RP-HPLC separation of elastase-digested thrombin, followed by antibacterial assays using E. coli identified several antibacterial peptide fractions (Figure 3A). Combined analyses using MALDI-TOF, ESI-MS/MS, and N - and C-terminal sequencing of fraction no. 38, which contained the majority of the activity comprising peptides active in high salt, unambiguously identified a major 11041 Da fragment comprising the C-terminal 96 amino acids of thrombin (amino acid 527–622, predicted pI 8.4, indicated in Figure 2A). Correspondingly, SDS-PAGE identified one single peptide of ∼11 kDa, that contained the C-terminal epitope, as shown by immunoblot analysis using polyclonal antibodies against the C-terminal peptide VFR17 (Figure 3A, rightmost upper panel). Gel overlay assays demonstrated that the major antimicrobial peptide of fraction 38 corresponded to one major active peptide, also identified in neutrophil elastase-digested thrombin (Figure 3A, rightmost lower panel), showing significantly lower mobility when compared to the C-terminal peptide GKY25, thus reflecting its higher molecular mass. Interestingly, MALDI-TOF and ESI-MS/MS of fraction no. 30 identified the peptide HVFRLKKWIQKVIDQFGE, described in the previous in vitro screening experiments (Figure 2A and B, peptide 47), as well as a shorter 16 aa long variant (Figure 3A, FRLKKWIQKVIDQFGE), both peptides from the C-terminus of thrombin. The analyses of the less hydrophobic material (fractions 20 and 21) yielded several low molecular weight fragments corresponding to internal, and cationic, sequences of low hydrophobicity and amphipathicity, matching antibacterial regions identified by the previous 20-mer screening (Table S1 available online and Figure 2A). Taken together, these results showed that neutrophil elastase generates antimicrobial TCPs, of which the major forms comprise a ∼11 kDa fragment of 96 amino acids, but also smaller fragments from the distal helical and amphipathic terminus.

Thrombin-derived C-terminal fragments are generated by human and bacterial proteinases
During inflammation, neutrophils release a multitude of enzymes, which could have activity on either thrombin or its proform. Therefore, thrombin was incubated with supernatants from activated neutrophils and the material analysed for antimicrobial activity and generation of TCPs. Indeed, antimicrobial activity was found upon proteolysis, while immunoblotting identified several TCPs of similar molecular weights as those generated by neutrophil elastase alone (Figure 3B). Similar fragments (Figure 3C) and corresponding antimicrobial activity (Figure 3D) were also detected when prothrombin was subjected to neutrophil elastase, cathepsin G as well as the bacterial thermolysin-like proteinase of Pseudomonas aeruginosa, lasB (also denoted P. aeruginosa elastase) [22]. Interestingly, low molecular weight peptides (∼3–4 kDa), generated by the latter P. aeruginosa enzyme, co-migrated with the model peptide GKY25 (Figure 3C). These results demonstrated that both human and bacterial enzymes may generate TCPs, irrespective of the activation state of prothrombin.
C-terminal thrombin peptides are generated ex vivo and in vivo and are protective against infection
Prothrombin, as many other proteins in plasma, is under meticulous control by antiproteinases in the normal state, preventing its activation and/or degradation. Therefore, we hypothesized that favorable environments promoting TCP formation should comprise i) localisation as well as concentration of thrombin, and ii) local release of enzymes, such as neutrophil elastase or bacterial proteinases. These environments are typical of sites of injury and infection, such as skin wounds, comprising thrombin activation, fibrin formation, bacterial colonization or infection, and subsequent neutrophil influx. Earlier studies have shown that thrombin binds to fibrin clots, and that fibrin acts as a reservoir for active thrombin [23]. Furthermore, human neutrophils release elastase during clotting, and neutrophils also penetrate fibrin [21]. Bacteria, such as S. aureus and P. aeruginosa frequently colonize and infect skin wounds, accompanied by excessive proteolysis and activation of neutrophils [24], [25]. Given this, the production of TCPs in fibrin, as well as in sterile or infected biological fluids and wounds was investigated. The results showed that TCPs were formed when fibrin clots were subjected to neutrophil elastase in vitro (Figure 4A). Furthermore, a similar ∼11 kDa fragment was detected in fibrin “slough” from a patient with a non-infected chronic venous leg ulcer, indicating that TCPs can be found in fibrin in vivo (Figure 4A). Analogous results, showing rapid formation of TCPs, were obtained using human plasma subjected to proteolysis by neutrophil elastase, thus simulating the high elastase activity observed during wounding (Figure 4B). Importantly, TCPs were also identified in wound fluid from patients post-surgery, as well as in wound fluid from patients with chronic (non-infected) venous leg ulcers (Figure 4B). The latter wounds are always colonized by bacteria such as S. aureus and P. aeruginosa [24].

Next, a series of experiments were performed in order to study the physiological role of TCPs in relation to bacterial infection. First, initial experiments showed that immunoreactive thrombin fragments, including the ∼11 kDa TCP peptide, are proteolytically generated and exclusively bind to bacteria during P. aeruginosa infection of plasma and in presence of fibrin (Figure S2A). FACS analysis utilizing antibodies against the C-terminal part of thrombin showed that TCPs, either occurring in wound fluid from acute wounds, or generated in human plasma during P. aeruginosa infection, bind to the bacteria similarly to the above described antibacterial C-terminal peptide GKY25 (Figure 4C). Similar results were obtained using S. aureus (Figure S2B). Second, electron microscopy studies employing gold-labeled antibodies demonstrated that TCPs are predominantly associated with bacterial surfaces ex vivo and in vivo. Thus, P. aeruginosa grown in plasma or acute wound fluid exhibited disintegrated areas as shown by ejected cytoplasmic material and membrane blebs (Figure 4D, see P and AWF). Furthermore, C-terminal epitopes of thrombin were found particularly in association with these damaged zones. Similar findings were seen after incubating P. aeruginosa with wound fluid derived from patients with chronic ulcers (Figure 4D, CWF), as well after incubating the bacteria with the C-terminal thrombin peptide GKY25 (Figure S3). Bacteria grown in plasma or acute wound fluid supplemented with heparin (shown to block the antimicrobial effects of the two C-terminal, heparin-binding peptides of thrombin described above, GKY25 and VFR17) were similar to the control bacteria, and did not exhibit either binding of TCPs or membrane damage (Figure S3). Additionally, analysis of fibrin slough from a patient with a chronic ulcer infected with P. aeruginosa and S. aureus, identified multiple coccoid bacteria both extracellularly and inside phagocytes (Figure 4D, CWS; Figure S4), that all displayed significant binding of immunogold antibodies, demonstrating the existence of TCPs at bacterial surfaces in vivo in fibrin from human wounds. Figure 4E (upper panel) further shows that the growth of P. aeruginosa is significantly enhanced in plasma depleted of prothrombin, when compared with native control plasma. Furthermore, addition of physiological concentrations of prothrombin, or the peptide GKY25 (at 1.5 µM, equivalent to the physiological prothrombin concentration), restored the suppressive effect of plasma on bacterial growth. Acute wound fluid depleted of thrombin and C-terminal fragments also showed increased growth of P. aeruginosa when compared with the control (Figure 4E, lower panel). Similar results were obtained with S. aureus (Figure S2C). Taken together, these results, and given the above findings on the generation and binding of TCPs to bacteria ex vivo and in vivo, unequivocally demonstrate a direct link between occurrence of TCPs and suppression of bacterial growth in plasma.
Further experiments with the model C-terminal peptide GKY25, exhibiting antibacterial activities similar to the endogenously produced C-terminal peptide HVF18 (peptide 47, Figure 2A), were employed in order to further study a physiological, as well as therapeutic role of TCPs. The MIC-levels of GKY25, according to standard NCSLA-protocols, were comparable to, and in some cases lower than, those observed for LL-37 and omiganan (Table S2 available online). Since the latter is a highly active and broad-spectrum designed antimicrobial peptide now in Phase III clinical studies, the data on GKY25 also implied a possible therapeutic role for TCPs. Initial studies revealed no significant permeabilizing effects of GKY25 on human erythrocytes (60–120 µM peptide) as well as keratinocytes (up to 60 µM peptide) in plasma and serum conditions, respectively (Figure S5). In order to investigate a possible in vivo function of GKY25, we therefore injected this peptide subcutaneously into mice infected intraperitoneally with P. aeruginosa. Compared to the controls, treatment with GKY25 yielded a significant increase in survival (Figure 4F) and significantly lower bacterial numbers in the spleen and kidney of the animals (Figure 4G). Taken together, these results demonstrate that TCPs constitute a previously undisclosed neo-structure of thrombin, formed in vitro as well as ex vivo in plasma, but also in vivo in human wound fluid and fibrin, exerting antimicrobial activities at physiological concentrations in plasma, and finally, showing significant therapeutic potential.
Immunomodulatory roles of TCPs
As mentioned above, recent evidence shows that HDPs trigger a range of immunomodulatory responses. The observation of LPS-binding of TCPs (Figure 2D), prompted us to investigate possible endotoxin-neutralizing effects of the model peptide GKY25. Slot-binding experiments showed that the peptides bound heparin as well as LPS from E. coli and P. aeruginosa (Figure 5A). In a mouse macrophage model, GKY25 significantly inhibited NO-release of LPS-stimulated macrophages (Figure 5B), as well as release of TNF-α at concentrations below 2 µM (Figure S6). Similar effects on TNF-α were noted using human monocyte-derived macrophages (not shown). In a mouse model of LPS-induced shock, GKY25 displayed a dramatic improvement on survival (Figure 5C). Analyses of cytokines 6 hours after LPS injection, showed significant reductions of proinflammatory IL-6, IFN-γ, TNF-α, and IL-12p70, whereas IL-10 remained unchanged (Figure 5D). SEM analyses of lungs from LPS-treated animals demonstrated pulmonary leakage of protein and red blood cells (see inset in Figure 5E), an effect completely blocked by GKY25 (Figure 5E). The results thus demonstrate that GKY25, like many HDPs, is multifunctional; in addition to its antimicrobial activity it also exerts potent anti-endotoxic and immunomodulatory effects.

Functional and structural studies of thrombin-derived C-terminal peptides
To examine possible peptide-induced permeabilization of bacterial plasma membranes, P. aeruginosa and S. aureus was incubated with GKY25 at concentrations yielding complete bacterial killing (30 µM), and analyzed by electron microscopy (Figure 6A). Clear differences in morphology between peptide-treated bacteria and the control were demonstrated. The peptide caused local perturbations and breaks along P. aeruginosa and S. aureus plasma membranes, and intracellular material was found extracellularly. These findings were similar to those seen after treatment with LL-37 (Figure 6A). The data suggest that GKY25 acts on bacterial membranes, but do not demonstrate the exact mechanistic events following peptide addition to bacteria, as secondary metabolic effects on bacteria may also trigger bacterial death and membrane destabilization. However, analogous results were also obtained using the impermeant dye FITC and E. coli as test bacterium (Figure 6B) demonstrating membrane permeabilisation after exposure to GKY25. Kinetic studies showed that GKY25 killed >90% of bacteria after 10 minutes, compatible with a direct action on bacterial membranes (Figure S7). Furthermore, circular dichroism (CD) spectroscopy was used to study the structure and the organization of the peptides GKY25 and VFR17 in solution and on interaction with negatively charged (bacteria-like) liposomes as well as E. coli LPS. Neither GKY25 nor VFR17 adopted an ordered conformation in aqueous solution, however the CD spectra revealed significant structural change, largely induction of helicity, taking place in the presence of negatively charged liposomes (Figure 6C), and E. coli LPS (Figure 6D). Compatible with earlier results, LL-37 displayed some helicity also in buffer solution [26]. Similarly to LL-37, the two thrombin-derived peptides induced leakage of liposomes, also at high ionic strength (Figure 6E). Kinetic analysis showed that ∼80% of the maximum leakage was reached within ∼200 seconds for the two thrombin-derived peptides (at 1 µM) (not shown). Considering the above results with GKY25 and VFR17, both containing the crucial helical (and antimicrobial) epitope, the results therefore indicate that TCPs function like most helical AMPs such as LL-37 [5], [27], by interactions with both the lipid membrane and LPS (possibly also peptidoglycan) at bacterial surfaces, leading to induction of an α-helical conformation, which in turn facilitates membrane interactions, membrane destabilization, and bacterial killing.

The TCP structure complies with a γ-core motif and is evolutionary conserved
Recently, a multidimensional signature, the γ-core motif, was identified in multiple classes of cystein-containing AMPs [28]. Analysis showed that the 96aa TCP is closely related to this fundamental motif so common in various HDPs (Figure S8). Furthermore, this region of thrombin is highly conserved in various species (Figure S9). Next, we compared the antibacterial activities of C-terminal peptide GKY25 of thrombin with corresponding peptides from other closely related human coagulation factors (Figure 6F, for sequences see Table S3 available online). Whereas the peptide from thrombin (Factor II), as well as peptides from factors X and IX were antimicrobial against Gram-negative P. aeruginosa and E. coli, Gram-positive S. aureus and B. subtilis, as well as the fungus C. albicans (Figure 6F), corresponding peptides from factor XI and kallikrein were inactive against these microbes (not shown). As seen in the 3D model (Figure 6G), the coagulation factors (II, X, IX) share a similar overall structure with a helical C-terminal “tail”. Indeed, C-terminals of these factors, as well as factor XI and kallikrein, contain a pattern sequence {DS}-X-[PFY]-G-[FIV]-Y-T-X-V-{C}-[AEQRY]-X-{R}-X-W-[IL]-X-{H}-X(4,24), which describes an amphipathic structure. However, only factors II, X, IX have a positive net charge (+3 or more) in this region (Table S3 available online), thus in perfect agreement with the data obtained on the antimicrobial activity (Figure 6F). Taken together, these analyses show that the TCP molecule represents a novel structural entity, which is related to other cysteine-linked HDPs containing the γ-core motif, and also found in closely related coagulation factors.
Discussion
The key finding in this report is the discovery of a novel function of thrombin-derived C-terminal peptides in host defense. The findings expand the field of innate immunity to thrombin and the coagulation system. From an evolutionary perspective, this function of thrombin is logical, since injury and infection both represent situations necessitating an optimized innate immune system. Hence, from the perspective of wounding, thrombin, after fulfilling its primary function in generating a first line of defense, the fibrin clot, adds expanded functionality this natural physical shield by subsequent generation of antimicrobial and anti-endotoxic HDPs upon proteolysis. The significant and curative effect of a thrombin-derived peptide in a model of LPS-induced shock underscores the anti-inflammatory role this novel peptide, and contrasts to the pro-inflammatory actions of other HDPs, such as the anaphylatoxin C3a and chemotactic defensins [14], [29]. Thus, during injury and infection, different pathways are activated, employing HDPs with multiple and sometimes opposite roles, all balancing and fine-tuning inflammation while counteracting microbial invasion. Recent evidence showing a significant cross-talk between the coagulation and complement systems [30] further adds biological relevance to the observed generation of C3a and various TCPs during inflammation.
TCPs further add to the increasingly recognized redundancy of host defense mechanisms, enforcing optimized control of the microbial flora by minimizing the risk for resistance development against one particular HDP, as well as protecting against detrimental effects due to specific HDP deficiencies. Notably, the observation of proteolytic formation of multiple TCP fragments of different lengths parallel previous findings on LL-37 and C3a [14], [31], showing that these molecules are further processed while retaining their antimicrobial activity. Although not shown in this study, additional HDPs may be released from C-termini of factor X and XI, further increasing the arsenal of HDPs and adding to redundancy. From an investigatory standpoint, such concerted action is challenging when it comes to defining roles of a given peptide in vivo. Nevertheless, the suppressive effects of formed TCPs on bacterial growth ex vivo, their association with bacterial surfaces ex vivo and in vivo, as well as significant effects of the TCP GKY25 under standard NCSLA conditions, as well as in an animal model of P. aeruginosa sepsis, clearly indicate an in vivo role for released TCPs. These findings, in concert with the anti-endotoxic and immunomodulatory effects of the peptides in vitro and in vivo, infer interesting therapeutic possibilities for TCPs in treatment of local and systemic infections, as well as sepsis. Recent evidence, showing a higher susceptibility to S. aureus infection in mice rendered thrombin-deficient [32], is also compatible with the new role in host-defense of thrombin-derived C-terminal peptides revealed here. Of relevance is also the increased susceptibility to infection after inhibition of the contact system, linked to abrogated release of kininogen-derived HDPs [16], but possibly also due to a reduced capability to form TCPs and other antimicrobial molecules associated with fibrin networks. Of particular clinical relevance is also the finding that TCPs are detected in wound fluids from patients with acute surgical wounds, as well as in patients with non-healing wounds. The latter patient group is characterized by an excessive bacterial colonization e.g. by P. aeruginosa, extensive proteolysis and inflammation [24]. Although speculative, the noted absence of TCPs in some patients could therefore be indicative of a defective host-defense and diminished control of released endotoxins (local or systemic). Thus, apart from therapeutic possibilities, the present findings provide a potential diagnostic marker for inflammation, which is currently under evaluation in larger patient groups. Concerning the immunomodulatory role of TCPs, it should be noted that although a direct binding and thus inhibition of LPS activity was demonstrated, the observed anti-inflammatory effects could also depend on additional effects by TCPs on various intracellular signaling pathways including inhibition of NF-κB activation.
From a structural standpoint, the TCP structure relates to the previously reported γ-core signature that characterize many cysteine-containing AMPs [28], further supporting the concept of multidimensional signatures in antimicrobial peptides and extending these also to HDPs of coagulation factors. The high degree of conservation of the cysteine-constrained TCP-molecule during evolution also suggests that the TCP structure is, although novel to science, not necessarily new. Interestingly, the 96-amino acid TCP also contains a peptide region responsible for the well-known growth promoting activity of thrombin [33], further adding biological importance to the findings. It remains to be investigated whether thrombin fragments comprising this region promote cell-growth. If so, these TCPs, generated in response to injury mediate not only microbial evasion and immunomodulation, but also wound closure, three fundamental aspects of host defense.
Materials and Methods
Ethics statement
The use of human wound materials was approved by the Ethics Committee at Lund University (LU 708-01, LU 509-01). Written informed consent was obtained from the participants. The animal experiments were conducted according to national guidelines (Swedish Animal Welfare Act SFS 1988 : 534), and were approved by the Laboratory Animal Ethics Committee of Malmö/Lund.
Peptides and proteins
Prothrombin and thrombin were from Innovative Research, USA. The coagulation factor-derived peptides (Table S3) and omiganan (ILRWPWWPWRRK-amide) were synthesized by Biopeptide Co. The purity (>95%) and molecular weight of these peptides was confirmed by mass spectral analysis (MALDI.TOF Voyager). LL-37 (LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES) was from Innovagen AB. 20mer peptides corresponding to various overlapping regions of prothrombin (Figure 2) were from Sigma (Custom Peptide Libraries, SigmaGenosys).
Biological materials
Wound fluids (100–600 µl) from patients with chronic venous leg ulcers were collected under a Tegaderm dressing for 2 h as previously described [25]. Fibrin slough from two chronic venous leg ulcers (chronic wound slough/surface, denoted CWS) was collected by a sterile spatula, and was immediately put into the fix solution for electron microscopy. Sterile wound fluids were obtained from surgical drainages after mastectomy. Collection was for 24 h, 24 to 48 h after surgery. Wound fluids were centrifuged, aliquoted and stored at −20°C.
Microorganisms
Escherichia coli ATCC 25922, Pseudomonas aeruginosa ATCC 27853, Pseudomonas aeruginosa 15159, Staphylococcus aureus ATCC 29213, Bacillus subtilis ATCC 6633 bacterial isolates, and the fungal isolate Candida albicans ATCC 90028 were from the Department of Bacteriology, Lund University Hospital.
Radial diffusion assay
Essentially as described earlier [34], [35], bacteria were grown to mid-logarithmic phase in 10 ml of full-strength (3% w/v) trypticase soy broth (TSB) (Becton-Dickinson). The microorganisms were then washed once with 10 mM Tris, pH 7.4. Subsequently, 4×106 cfu were added to 15 ml of the underlay agarose gel, consisting of 0.03% (w/v) TSB, 1% (w/v) low electroendosmosis type (EEO) agarose (Sigma-Aldrich) and 0.02% (v/v) Tween 20 (Sigma-Aldrich). The underlay was poured into a Ø 144 mm petri dish. After agarose solidification, 4 mm-diameter wells were punched and 6 µl peptide solution of required concentration added to each well. Plates were incubated at 37°C for 3 h to allow peptide diffusion. The underlay gel was then covered with 15 ml of molten overlay (6% TSB and 1% Low-EEO agarose in distilled H2O). Antimicrobial activity of a peptide was visualized as a zone of clearing around each well after 18–24 h of incubation at 37°C.
Viable count assays and analysis of bacterial growth
E. coli ATCC 25922 bacteria were grown to mid-logarithmic phase in Todd-Hewitt (TH) medium. Bacteria were washed and re-suspended in 10 mM Tris, pH 7.4 containing 5 mM glucose. E. coli ATCC 25922 (50 µl; 2×106 cfu/ml) were incubated, at 37°C for 2 h with prothrombin, thrombin, GKY25, or VFR17 at 3 and 6 µM. For assessment of bacterial growth in plasma and effect of TCPs, overnight cultures (in TH) of P. aeruginosa 15159 and S. aureus ATCC 29213 bacteria were incubated (17 µl in 450 µl) with control plasma, acute wound fluid (AWF) or depleted AWF, plasma depleted of prothrombin (DP, Innovative Research), or DP supplemented with the peptide GKY25 (at 1.5 µM) for 0, 2, 4, and 6 h at 37°C. Serial dilutions of the incubation mixtures were plated on TH agar, followed by incubation at 37°C overnight and cfu determination. In order to deplete acute wound fluids (AWF) of thrombin and fragments containing C-terminal peptides, AWF was diluted with an equal volume of PBS, and passaged 5 times through an affinity column (0.3 ml, Thermo Scientific) having coupled IgG antibodies specific for VFR17 (Innovagen AB). For control, a column with rabbit IgG was used.
Flow cytometry analysis
50 µl of overnight bacteria was added to 450 µl of human plasma, AWF, or DP either alone or supplemented with the peptide GKY25 (at 1.5 µM). Samples were incubated for 4 h at 37°C, centrifuged, washed with PBS, resuspended in 100 µl PBS with polyclonal antibodies against VFR17 (1∶100), and subsequently incubated for one hour at room temperature. Bacteria were pelleted and washed twice with PBS, incubated in 100 µl PBS with goat anti rabbit IgG FITC-labeled antibodies (1∶500, Sigma) for 30 min at room temperature and washed twice with PBS. Flow cytometry analysis (Becton-Dickinson, Franklin Lakes, NJ) was performed using a FACS-Calibur flow cytometry system equipped with a 15 mW argon laser turned a 488 nm. The bacterial population was selected by gating with appropriate settings of forward scatter (FSC) and sideward scatter (SSC).
Slot-blot assay
LPS-binding ability of the peptides was examined by a slot-blot assay. Peptides (2 and 5 µg) were bound to nitrocellulose membranes (Hybond-C, GE Healthcare BioSciences), pre-soaked in PBS. Membranes were then blocked by 2 wt% BSA in PBS, pH 7.4, for 1 h at room temperature, and subsequently incubated with 125I-labeled LPS (40 µg/ml; 0.13×106 cpm/µg) for 1 h in PBS. After incubation, the membranes were washed 3 times, 10 min each time, in PBS and visualized for radioactivity on Bas 2000 radioimaging system (Fuji). Unlabeled heparin (6 mg/ml) was added for competition of binding.
Liposome preparation and leakage assay
Dry lipid films were prepared by dissolving either dioleoylphosphatidylethanolamine (Avanti Polar Lipids, Alabaster, AL) (70 mol%) and dioleoylphosphatidylglycerol (30 mol%) in chloroform, and then removing the solvent by evaporation under vacuum overnight. Subsequently, buffer solution containing 10 mM Tris, pH 7.4, either with or without additional 150 mM NaCl, was added together with 0.1 M carboxyfluorescein (CF) (Sigma). After hydration, the lipid mixture was subjected to eight freeze-thaw cycles consisting of freezing in liquid nitrogen and heating to 60°C. Unilamellar liposomes with a diameter of about 130 nm (as found with cryo-TEM; results not shown) were generated by multiple extrusions through polycarbonate filters (pore size 100 nm) mounted in a LipoFast miniextruder (Avestin). Untrapped carboxyfluorescein was then removed by filtration through two subsequent Sephadex G-50 columns with the relevant Tris buffer as eluent. Both extrusion and filtration was performed at 22°C. The CF release was monitored by fluorescence at 520 nm from a liposome dispersion (10 mM lipid in 10 mM Tris pH 7.4). An absolute leakage scale is obtained by disrupting the liposomes at the end of the experiment through addition of 0.8 mM Triton ×100 (Sigma), thereby causing 100% release and dequenching of CF. A SPEX-fluorolog 1650 0.22-m double spectrometer (SPEX Industries) was used for the liposome leakage assay.
CD-spectroscopy
Circular dichroism (CD) spectra were measured by a Jasco J-810 Spectropolarimeter (Jasco, Easton, USA). The measurements were performed in triplicate at 37°C in a 10 mm quartz cuvette under stirring with a peptide concentration of 10 µM. The effect on peptide secondary structure of liposomes at a lipid concentration of 100 µM, and of E. coli LPS at a concentration of 0.02 wt%, was monitored in the range 200–250 nm. To account for instrumental differences between measurements, the background value (detected at 250 nm, where no peptide signal is present) was subtracted. Signals from the bulk solution were also corrected for.
Fluorescence microscopy
For study of membrane permeabilization using the impermeant probe FITC, E. coli ATCC 25922 bacteria were grown to mid-logarithmic phase in TSB medium. The bacteria were washed and resuspended in either 10 mM Tris, pH 7.4, 10 mM glucose, to yield a suspension of 1×107 cfu/ml. 100 µl of the bacterial suspension was incubated with 30 µM of the respective peptides at 30°C for 30 min. Microorganisms were then immobilized on poly (L-lysine)–coated glass slides by incubation for 45 min at 30°C, followed by addition onto the slides of 200 µl of FITC (6 µg/ml) in the appropriate buffers and incubated for 30 min at 30°C. The slides were washed and bacteria fixed by incubation, first on ice for 15 min, then in room temperature for 45 min in 4% paraformaldehyde. The glass slides were subsequently mounted on slides using Prolong Gold antifade reagent mounting medium (Invitrogen). For fluorescence analysis, bacteria and fungi were visualized using a Nikon Eclipse TE300 (Nikon, Melville, NY) inverted fluorescence microscope equipped with a Hamamatsu C4742-95 cooled CCD camera (Hamamatsu) and a Plan Apochromat ×100 objective (Olympus, Orangeburg, NY). Differential interference contrast (Nomarski) imaging was used for visualization of the microbes themselves.
Electron microscopy
For transmission electron microscopy and visualization of peptide effects on bacteria, P. aeruginosa ATCC 27853 and S. aureus ATCC 29213 (1–2×106 cfu/sample) were incubated for 2 h at 37°C with the peptide GKY25 at 30 µM. LL-37 (30 µM) was included as a control. Samples of P. aeruginosa and S. aureus suspensions were adsorbed onto carbon-coated copper grids for 2 min, washed briefly on two drops of water, and negatively stained on two drops of 0.75% uranyl formate. The grids were rendered hydrophilic by glow discharge at low pressure in air. For analysis of effects on biological fluids on bacterial integrity as well as detection of bound TCPs, P. aeruginosa 15159 bacteria, grown overnight in TH, were washed and resuspened in PBS (1×109 cfu/ml). Equal volumes of bacterial suspension and chronic wound fluids were incubated together for 30 min at 37°C. For control, 2 µM of GKY25 was incubated with bacteria for 30 min at 37°C. In another experiment, P. aeruginosa 15159 bacteria were directly added to citrate plasma or AWF in the absence or presence of heparin (100 µg/ml), and further incubated for 4 h at 37°C. All the samples were centrifuged and washed with PBS and re-suspended in 4% paraformaldehyde and stored at 4°C, followed by gold labeling. Fibrin slough from patients with chronic venous ulcers (CWS) was fixed (1.5% PFA, 0.5% GA in 0.1 M phosphate buffer, pH 7.4) for 1 h at room temperature, followed by washing with 0.1 M phosphate buffer, pH 7.4. The fixed and washed samples were subsequently dehydrated in ethanol and further processed for Lowicryl embedding [36]. Sections were cut with a LKB ultratome and mounted on gold grids. For immunostaining, the grids were floated on top of drops of immune reagents displayed on a sheet of parafilm. Free aldehyde groups were blocked with 50 mM glycine, and the grids were then incubated with 5% (vol/vol) goat serum in incubation buffer (0.2% BSA-c in PBS, pH 7.6) for 15 minutes. This blocking procedure was followed by overnight incubation with 1 µg/ml of VFR17 polyclonal antibodies at 4°C. Controls without these primary antibodies were included. After washing the grids in a large volume (200 ml) of incubation buffer, floating on drops containing the gold conjugate reagents, 1 µg/ml EM goat antiRabbit IgG 10nm Au (BBI) in incubation buffer was performed for 2 h at 4°C. After further washes by an excess volume of incubation buffer, the sections were postfixed in 2% glutaraldehyde. Finally, sections were washed with distilled water and poststained with 2% uranyl acetate and lead citrate. All samples were examined with a Jeol JEM 1230 electron microscope operated at 80 kV accelerating voltage. Images were recorded with a Gatan Multiscan 791 charge-coupled device camera.
Degradation of prothrombin and thrombin
Prothrombin and thrombin (Innovative Research) (27 µg, 0.6 mg/ml) was incubated at 37°C with human neutrophil elastase (NE) (0.6 µg, 20 units/mg) (Sigma) and prothrombin also with cathepsin G (0.5 µg, 2 units/mg) (BioCol GmbH) or P. aeruginosa elastase (PAE) (30 mU, a generous gift from Dr. H. Maeda, Kumamoto University, Japan) in a total volume of 45 µl 10 mM Tris, pH 7.4 for different time periods as indicated in the figures. Neutrophils were prepared by routine procedures (Polymorphprep) from blood obtained from healthy human donors. The cells were disrupted by freeze-thawing and addition of 0.3% Tween 20. Neutrophil extracts (corresponding to 4.8×107 cells) were incubated at 37° with thrombin (27 µg, 0.6 mg/ml) for 180 min. The reaction was stopped by boiling at 95°C for 3 min. 6 µl (3.6 µg) of the material was analyzed by RDA and 20 µl (12 µg) fractions analyzed by SDS-PAGE using 16.5% precast Tris-tricine gels (Bio-rad), run under non-reducing or reducing conditions. The gels were stained with Coomassie brilliant blue and destained.
Definition of cleavage products of thrombin
Peptide fragments of thrombin, digested by neutrophil elastase for 30 min, were separated by hplc (PerkinElmer Series 200) on a reversed phase column (Vydac 218TPC18, 250×4.6 mm, 5 µm) (Dalco chromtech AB). After injection, samples were eluted with a gradient of acetonitrile in 0.1% aqueous trifluroacetic acid at 1 ml per minute. Fractions were collected and stored at −80°C. Samples were freeze-dried, redissolved in water, and analyzed by RDA, SDS-PAGE, immunoblotting and gel-overlay assay (Figure 3). Active fractions were analyzed by combinations of MALDI-TOF MS, ESI-MS/MS, N - and C-terminal sequencing at the Karolinska Institutet Protein Analysis Center (PAC Stockholm). See also legend to Figure 3 for additional information.
SDS-PAGE and immunoblotting
Prothrombin and thrombin, either intact or subjected to enzymes, were analyzed by SDS-PAGE on 16.5% Tris-tricine gels (Bio-Rad). Proteins and peptides were transferred to nitrocellulose membranes (Hybond-C). Membranes were blocked by 3% (w/v) skimmed milk, washed, and incubated for 1 h with rabbit polyclonal antibodies recognizing the peptide VFR17 (1∶800) (Innovagen AB) or rabbit antibodies of similar specificity (1∶1000) (Dako), washed three times for 10 min, and subsequently incubated (1 h) with HRP-conjugated secondary antibodies (1∶2000) (Dako), and then washed again three times, each time for 10 min. C-terminal thrombin peptides were visualized by an enhanced chemiluminesent substrate (LumiGLO®) developing system (Upstate cell signaling solutions). For identification of TCPs in human fibrin, normal citrate-plasma was supplemented with 10 mM Ca2+ in eppendorf tubes at 37°C overnight. The clots formed were washed three times with PBS and incubated with human neutrophil elastase (20 units/mg) for 0, 1, and 3 h at 37°C. Samples were centrifuged at 10000 RPM for 10 min, after which supernatants and pellets were separated. Samples were freeze-dried and then redissolved in 60% acetonitrile and 0.1% aqueous TFA. Pooled samples were freeze-dried, redissolved in water and analyzed by SDS-PAGE followed by immunoblotting as above. For identification of TCPs in human citrate plasma, 1.5 µl of citrate-plasma or patient would fluids were analysed by SDS-PAGE under reducing conditions, followed by immunoblotting as above. For identification of TCPs bound to bacteria, overnight cultures of P. aeruginosa 15159 bacteria were washed, resuspended, and incubated with human plasma or a preformed fibrin clot [37] for 4 h at 37°C. The bacterial cells were collected, washed wih PBS, and bound proteins were eluted with 0.1 M glycine-HCL, pH 2.0. The pH of the eluted material was raised to 7.5 with the addition of 1 M Tris. Eluted proteins were precipitated with 5% trichloroacetic acid (TCA) for 30 min on ice followed by centrifugation at 15 000g (4°C for 20 min). Precipitated material was dissolved in SDS sample buffer and subjected to Tris-Tricine SDS-PAGE under reducing conditions, followed by immunoblotting as above.
Gel-overlay assay
Gel overlay assay was performed essentially as described previously [35]. Briefly, duplicate samples were run on non-denaturing acid urea (AU-PAGE) gels in 5% acetic acid at 100 V for 1 h 15 min. Bacteria were grown overnight in TH broth, inoculated, and grown until the OD was 0.4. The bacteria were washed and resuspended in 10 mM Tris, pH 7.4. Bacteria (4×106) were added to 12 ml melted underlay agarose (10 mM Tris, pH 7.4, 0.03% TH broth, 1% agarose type 1 (Sigma-Aldrich)) and poured into a square petri dish. One AU gel was stained with Coomassie brilliant blue and one AU gel was washed three times for 4 min in 10 mM Tris, pH 7.4 and then placed on top of the agarose gel and incubated for 3 h at 37°C. The AU gel was then removed and an overlay agarose (6% TH broth, 1% agarose type 1) was poured on top of the underlay and incubated overnight at 37°C. To make the clearing zones more visible, the agarose was stained with Coomassie brilliant blue and then destained with water.
Animal infection model
Animals were housed under standard conditions of light and temperature and had free access to standard laboratory chow and water. P. aeruginosa 15159 bacteria were grown overnight, harvested, washed in PBS, diluted in the same buffer to 2×108 cfu/ml, and kept on ice until injection. Hundred microliter of the bacterial suspension were injected intraperitoneally (ip) into female BALB/c mice. Sixty minutes after the bacterial injection, 0.5 mg GKY25 or buffer alone was injected sc into the mice. This was repeated after 24 hours. In this Pseudomonas infection model, infected mice develop severe signs of sepsis within 1–2 days and usually do not recover from the infection. In order to study bacterial dissemination to target organs, the mice were infected as previously described and after a time period of 8 h, spleen and kidney were harvested, placed on ice, homogenized, and colony-forming units determined. The P-value was determined using the Mann-Whitney U-test. Data from three independent experiments were pooled.
LPS effects on macrophages in vitro
3.5×105 cells were seeded in 96-well tissue culture plates (Nunc, 167008) in phenol red-free DMEM (Gibco) supplemented with 10% FBS and antibiotics. Following 6 h of incubation to permit adherence, cells were stimulated with 100 or 10 ng/mL E. coli (0111∶B4) or P. aeruginosa LPS (Sigma), with and without peptide of various doses. The levels of NO in culture supernatants were determined after 24 hours from stimulation using the Griess reaction [38]. Briefly, nitrite, a stable product of NO degradation, was measured by mixing 50 µl of culture supernatants with the same volume of Griess reagent (Sigma, G4410) and reading absorbance at 550 nm after 15 min. Phenol-red free DMEM with FBS and antibiotics were used as a blank. A standard curve was prepared using 0–80 µM sodium nitrite solutions in ddH20.
LPS model in vivo
C57BL/6 mice (8–10 weeks, 22+/−5g), divided into weight and sex matched groups, were injected intraperitoneally with 18 mg E. coli 0111∶B4 LPS (Sigma) per kg of body weight. Thirty minutes after LPS injection, 0.2 mg GKY25 or buffer alone was injected intraperitoneally into the mice. Survival and status was followed during seven days. For the cytokine assay, mice were sacrificed 6 h post LPS challenge, and blood was collected by cardiac puncture. For SEM, mice were sacrificed 20 h after LPS challenge, and lungs were removed and fixed.
Cytokine assay
The cytokines IL-6, IL-10, MCP-1, INF-γ, TNF-α, and IL-12p70 were measured in plasma from LPS-infected mice (with or without GKY25 treatment) using the Cytometric bead array; mouse inflammation kit (Becton Dickinson AB) according to the manufacturer's instructions.
Statistical analysis
Bar diagrams (RDA, VCA) are presented as mean and standard deviation, from at least three independent experiments. Animal data are presented as dot plots, with mean for normally distributed data, or median for data, which do not meet the criteria for normal distribution. Outliers were not excluded from the statistical analysis. Differences with P<0.05 were considered statistically significant.
MIC, hemolysis, MTT, and LDH assay
MIC assay was carried out by a microtiter broth dilution method as previously described in the NCSLA guidelines [39]. Hemolysis, MTT, and LDH assays were performed as previously described [40] (See Text S1).
Alignment of TCPs
See Text S1.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. LehrerRI
GanzT
2002 Cathelicidins: a family of endogenous antimicrobial peptides. Curr Opin Hematol 9 18 22
2. HarderJ
GlaserR
SchröderJM
2007 Review: Human antimicrobial proteins effectors of innate immunity. J Endotoxin Res 13 317 338
3. ZasloffM
2002 Antimicrobial peptides of multicellular organisms. Nature 415 389 395
4. TossiA
SandriL
GiangasperoA
2000 Amphipathic, alpha-helical antimicrobial peptides. Biopolymers 55 4 30
5. YountNY
BayerAS
XiongYQ
YeamanMR
2006 Advances in antimicrobial peptide immunobiology. Biopolymers 84 435 458
6. ZanettiM
2004 Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol 75 39 48
7. ElsbachP
2003 What is the real role of antimicrobial polypeptides that can mediate several other inflammatory responses? J Clin Invest 111 1643 1645
8. GanzT
2003 Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol 3 710 720
9. ColeAM
GanzT
LieseAM
BurdickMD
LiuL
2001 Cutting edge: IFN-inducible ELR - CXC chemokines display defensin-like antimicrobial activity. J Immunol 167 623 627
10. BrogdenKA
2005 Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 3 238 250
11. KowalskaK
CarrDB
LipkowskiAW
2002 Direct antimicrobial properties of substance P. Life Sci 71 747 750
12. MorA
AmicheM
NicolasP
1994 Structure, synthesis, and activity of dermaseptin b, a novel vertebrate defensive peptide from frog skin: relationship with adenoregulin. Biochemistry 33 6642 6650
13. MalmstenM
DavoudiM
WalseB
RydengårdV
PasupuletiM
2007 Antimicrobial peptides derived from growth factors. Growth Factors 25 60 70
14. NordahlEA
RydengårdV
NybergP
NitscheDP
MörgelinM
2004 Activation of the complement system generates antibacterial peptides. Proc Natl Acad Sci U S A 101 16879 16884
15. PasupuletiM
WalseB
NordahlEA
MörgelinM
MalmstenM
2007 Preservation of antimicrobial properties of complement peptide C3a, from invertebrates to humans. J Biol Chem 282 2520 2528
16. FrickIM
ÅkessonP
HerwaldH
MörgelinM
MalmstenM
2006 The contact system–a novel branch of innate immunity generating antibacterial peptides. Embo J 25 5569 5578
17. NordahlEA
RydengårdV
MörgelinM
SchmidtchenA
2005 Domain 5 of high molecular weight kininogen is antibacterial. J Biol Chem 280 34832 34839
18. RydengårdV
Andersson NordahlE
SchmidtchenA
2006 Zinc potentiates the antibacterial effects of histidine-rich peptides against Enterococcus faecalis. Febs J 273 2399 2406
19. DavieEW
KulmanJD
2006 An overview of the structure and function of thrombin. Semin Thromb Hemost 32 Suppl 1 3 15
20. BodeW
2006 The structure of thrombin: a janus-headed proteinase. Semin Thromb Hemost 32 Suppl 1 16 31
21. BrowerMS
WalzDA
GarryKE
FentonJW2nd
1987 Human neutrophil elastase alters human alpha-thrombin function: limited proteolysis near the gamma-cleavage site results in decreased fibrinogen clotting and platelet-stimulatory activity. Blood 69 813 819
22. SchmidtchenA
HolstE
TapperH
BjörckL
2003 Elastase-producing Pseudomonas aeruginosa degrade plasma proteins and extracellular products of human skin and fibroblasts, and inhibit fibroblast growth. Microb Pathog 34 47 55
23. LiuCY
NosselHL
KaplanKL
1979 The binding of thrombin by fibrin. J Biol Chem 254 10421 10425
24. LundqvistK
HerwaldH
SonessonA
SchmidtchenA
2004 Heparin binding protein is increased in chronic leg ulcer fluid and released from granulocytes by secreted products of Pseudomonas aeruginosa. Thromb Haemost 92 281 287
25. SchmidtchenA
2000 Degradation of antiproteinases, complement and fibronectin in chronic leg ulcers. Acta Derm Venereol 80 179 184
26. OrenZ
LermanJC
GudmundssonGH
AgerberthB
ShaiY
1999 Structure and organization of the human antimicrobial peptide LL-37 in phospholipid membranes: relevance to the molecular basis for its non-cell-selective activity. Biochem J 341 501 513
27. ZelezetskyI
TossiA
2006 Alpha-helical antimicrobial peptides-Using a sequence template to guide structure-activity relationship studies. Biochim Biophys Acta 1758 1436 1449
28. YountNY
YeamanMR
2004 Multidimensional signatures in antimicrobial peptides. Proc Natl Acad Sci U S A 101 7363 7368
29. OppenheimJJ
YangD
2005 Alarmins: chemotactic activators of immune responses. Curr Opin Immunol 17 359 365
30. AmaraU
RittirschD
FlierlM
BrucknerU
KlosA
2008 Interaction between the coagulation and complement system. Adv Exp Med Biol 632 71 79
31. MurakamiM
Lopez-GarciaB
BraffM
DorschnerRA
GalloRL
2004 Postsecretory processing generates multiple cathelicidins for enhanced topical antimicrobial defense. J Immunol 172 3070 3077
32. MullinsES
KombrinckKW
TalmageKE
ShawMA
WitteDP
2009 Genetic elimination of prothrombin in adult mice is not compatible with survival and results in spontaneous hemorrhagic events in both heart and brain. Blood 113 696 704
33. GlennKC
FrostGH
BergmannJS
CarneyDH
1988 Synthetic peptides bind to high-affinity thrombin receptors and modulate thrombin mitogenesis. Pept Res 1 65 73
34. AnderssonE
RydengårdV
SonessonA
MörgelinM
BjörckL
2004 Antimicrobial activities of heparin-binding peptides. Eur J Biochem 271 1219 1226
35. LehrerRI
RosenmanM
HarwigSS
JacksonR
EisenhauerP
1991 Ultrasensitive assays for endogenous antimicrobial polypeptides. J Immunol Methods 137 167 173
36. CarlemalmE
VilligerW
HobotJA
AcetarinJD
KellenbergerE
1985 Low temperature embedding with Lowicryl resins: two new formulations and some applications. J Microsc 140 55 63
37. RydengårdV
ShannonO
LundqvistK
KacprzykL
ChalupkaA
2008 Histidine-rich glycoprotein protects from systemic Candida infection. PLoS Pathog 4 e1000116 doi:10.1371/journal.ppat.1000116
38. PollockJS
ForstermannU
MitchellJA
WarnerTD
SchmidtHH
1991 Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc Natl Acad Sci U S A 88 10480 10484
39. WiegandI
HilpertK
HancockRE
2008 Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat Protoc 3 163 175
40. PasupuletiM
WalseB
SvenssonB
MalmstenM
SchmidtchenA
2008 Rational design of antimicrobial C3a analogues with enhanced effects against Staphylococci using an integrated structure and function-based approach. Biochemistry 47 9057 9070
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 4
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- The Effect of Vaccination on the Evolution and Population Dynamics of Avian Paramyxovirus-1
- Reconstitution of SARS-Coronavirus mRNA Cap Methylation
- Increased Monocyte Turnover from Bone Marrow Correlates with Severity of SIV Encephalitis and CD163 Levels in Plasma
- Deficiencies in Jasmonate-Mediated Plant Defense Reveal Quantitative Variation in Pathogenesis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy