
A Genomic Survey of Positive Selection in Provides Insights into the Evolution of Accidental Virulence
Certain environmental microorganisms can cause severe human infections, even in the absence of an obvious requirement for transition through an animal host for replication (“accidental virulence”). To understand this process, we compared eleven isolate genomes of Burkholderia pseudomallei (Bp), a tropical soil microbe and causative agent of the human and animal disease melioidosis. We found evidence for the existence of several new genes in the Bp reference genome, identifying 282 novel genes supported by at least two independent lines of supporting evidence (mRNA transcripts, database homologs, and presence of ribosomal binding sites) and 81 novel genes supported by all three lines. Within the Bp core genome, 211 genes exhibited significant levels of positive selection (4.5%), distributed across many cellular pathways including carbohydrate and secondary metabolism. Functional experiments revealed that certain positively selected genes might enhance mammalian virulence by interacting with host cellular pathways or utilizing host nutrients. Evolutionary modifications improving Bp environmental fitness may thus have indirectly facilitated the ability of Bp to colonize and survive in mammalian hosts. These findings improve our understanding of the pathogenesis of melioidosis, and establish Bp as a model system for studying the genetics of accidental virulence.
Published in the journal:
. PLoS Pathog 6(4): e32767. doi:10.1371/journal.ppat.1000845
Category:
Research Article
doi:
https://doi.org/10.1371/journal.ppat.1000845
Summary
Certain environmental microorganisms can cause severe human infections, even in the absence of an obvious requirement for transition through an animal host for replication (“accidental virulence”). To understand this process, we compared eleven isolate genomes of Burkholderia pseudomallei (Bp), a tropical soil microbe and causative agent of the human and animal disease melioidosis. We found evidence for the existence of several new genes in the Bp reference genome, identifying 282 novel genes supported by at least two independent lines of supporting evidence (mRNA transcripts, database homologs, and presence of ribosomal binding sites) and 81 novel genes supported by all three lines. Within the Bp core genome, 211 genes exhibited significant levels of positive selection (4.5%), distributed across many cellular pathways including carbohydrate and secondary metabolism. Functional experiments revealed that certain positively selected genes might enhance mammalian virulence by interacting with host cellular pathways or utilizing host nutrients. Evolutionary modifications improving Bp environmental fitness may thus have indirectly facilitated the ability of Bp to colonize and survive in mammalian hosts. These findings improve our understanding of the pathogenesis of melioidosis, and establish Bp as a model system for studying the genetics of accidental virulence.
Introduction
Burkholderia pseudomallei (Bp), the causative agent of the often-fatal disease melioidosis, represents one of the most complex bacterial genomes sequenced to date [1]. Comprising two circular chromosomes with a combined length of 7.2 Mb, the Bp genome contains an estimated ∼5800 genes involved in a myriad of functions, allowing microbial survival in extreme environments and virulence in diverse host species including humans, gorillas, pigs, and fish [2]–[3]. Epidemiological and genetic evidence suggests that Bp is likely an ‘accidental pathogen’, in that adaptations incurred by Bp in its natural environmental reservoir (soil) may have indirectly contributed to its ability to colonize a mammalian host [4]–[7]. Understanding the genetic basis of these environmental adaptations may thus provide important insights into the pathogenesis of melioidosis, and shed light on how environmental microorganisms are able to acquire novel traits enhancing their ability to cause opportunistic disease.
The evolutionary success of Bp as a thriving soil microbe suggests that most Bp strains are likely to possess a common repertoire of genes (the Bp core genome, or BpCG) regulating survival and fitness in this highly competitive environmental niche. Specific selective pressures encountered in soil, such as evading phagocytosis by amoebae [8] or ingestion by nematodes [9] might further enhance Bp environmental fitness by inducing modifications in BpCG genes, and some of these modifications might also contribute indirectly to mammalian virulence. Indeed, many classical virulence genes such as adhesins, fimbrae, exopolysaccharides and Type III secretion (TTS) systems are part of the BpCG [7], suggesting a plausible link between the BpCG and mammalian pathogenicity. Currently, little is known regarding the extent of genetic variation in the Bp core genome (BpCG) and whether BpCG variations might underlie potential virulence phenotypes. In this study, we undertook a comprehensive qualitative and quantitative survey of the BpCG across a panel of eleven Bp genomes, comprising nine independently derived strains, and two related strain pairs isolated from human patients at primary infection and disease relapse. We found evidence for the presence of several new genes in the Bp genome, and discovered a sizeable degree of genetic variation in BpCG genes. We identified over two hundred BpCG genes with signatures of positive selection, likely reflecting the activity of multiple distinct environmental pressures. Finally, we provide experimental evidence that some of these positively selected genes may have indirectly contributed to Bp pathogenesis in mammals, by facilitating interactions with host cellular pathways or the use of host nutrients.
Results
Genome Sequencing and Annotation
We analyzed whole-genome sequences from eleven Bp strains, comprising ten clinical isolates from four countries (Australia, Thailand, Singapore, and Vietnam) and one soil isolate (S13) from Singapore. To achieve maximal genetic diversity, we elected to analyze all Bp strains regardless of their source of isolation (clinical or environmental). Notably, environmental Bp isolates have also been shown to exhibit high levels of virulence in animal models [10]. Among the clinical isolates, strain pairs 1106a–1106b and 1710a–1710b were isolated from the same patients during either primary infection or disease relapse (Table S1). Reflecting the genetic diversity in this panel, the Bp isolates belong to different multi-locus subtypes (MLST) with an overall MLST allele/subtype ratio of 2.67, markedly higher than the allele/subtype ratio of the general Bp population (0.43, as of Jan 2009). Ten genomes were sequenced by conventional Sanger based shotgun methods (coverage range 7.75x – 11.4x), while strain Bp 22 was sequenced using next-generation instrumentation (GS20-454, average read length 100 bp, 20× coverage) followed by de novo assembly using a custom 454 large-insert paired-end sequencing protocol (CN and YR, manuscript in prep). The genome sequences were uniformly annotated by a FGENESB gene prediction pipeline [11], and predicted protein-coding regions, tRNAs, rRNAs, and potential promoters, terminators and operons were identified. Predicted genes were comprehensively annotated against known proteins in the NR, COG, KEGG and STRING databases (details in Methods). All genomes revealed similar benchmark data such as genome size, GC content, and numbers of predicted genes (Table 1).

Chromosomal Organization
Both chromosomes (1 and 2) were highly syntenic across the Bp genomes (Figure 1 [12]–[13] and Figure S1). No evidence for inter-chromosomal exchange of genetic material across the two chromosomes was observed. We identified three large-scale inversions of 1.6 Mb, 1.2 Mb and 880 Kb on Chromosome 1, largely flanked either by rRNAs, tRNAs, or inverted protein units (Text S1). The 1.2 Mb inversion was observed in two strains, 1655 and Pasteur 52237, hailing from distinct geographic origins (Australia and Vietnam) and belonging to unrelated MLSTs, suggesting that this rearrangement may have independently occurred at least twice during Bp genome evolution. The other two inversions were only observed in single strains (406e and K96243), however it is worth noting that K96243 represents the original Bp reference genome described in 2004 [1].

An Updated Bp Annotation Reveals Additional Genomic Complexity
Our comparative analysis allowed us to revisit the original 2004 genome analysis with updated annotation protocols. Our annotation pipeline identified 6332 protein coding genes in Bp K96243 (Datasets S1 and S2), a considerably higher number (∼10%) than the 5855 genes originally described [1]. The vast majority (90%) of genes, however, were commonly identified in both annotation pipelines (Figure 2A), indicating that differences in the two annotation sets are likely due to subtle differences in the prediction algorithms used [14]–[15] (FGENESB vs GeneMark/Glimmer). Deciding to investigate these previously unreported genes, we sought to distinguish between likely bona-fide new genes and those arising due to computational over-prediction (false positives). We manually curated a set of 519 novel predicted genes exhibiting non-overlapping start-stop boundaries to the previously reported genes (see Figure 2B for an example), and subjected the 519 putative novel genes to three independent lines of analysis (mRNA transcript information, homology to previously reported genes, and presence of ribosomal binding sites, RBSs).

First, using whole genome tiling microarrays covering the entire non-repetitive Bp K96243 genome, we identified transcription units from Bp cultures isolated from six distinct growth conditions (see Methods, [16]). Confirming the accuracy of the microarray, many mRNA transcripts were tightly associated with the boundaries of previously-identified genes (Figure S2). Of the 519 novel genes, we found that 280 (53%) were associated with discrete mRNA transcripts. 178 novel genes exhibited mRNA transcripts in at least 1 out of 6 different growth conditions, indicating that they are differentially-regulated (Figure 2C), while the remaining 102 were constitutively expressed across the six conditions. The presence of several novel gene transcripts was also directly confirmed by targeted RT-PCR assays (Figure S3). To investigate if any of the novel genes might correspond to non-coding RNAs (ncRNAs), we used Rfam, a public database of non-coding RNA families [17], to identify ncRNAs in the BpK96243 reference genome. Of 82 small ncRNAs identified by Rfam analysis, 8 ncRNAs corresponded to the novel genes.
Second, using matching criteria similar to other studies [18]–[19] (see Methods, [20]), approximately 46% of the novel genes (239) were associated with at least one other matching protein in the COG, KEGG, STRING and NR databases (Figure 2D, [21]). 138 novel genes had matching proteins previously observed in other Bp strains, and 97 novel genes had matches to other Burkholderia species. A small fraction (∼1%) exhibited homology to other non-Burkholderia species (eg Xanthomonas oryzae pv. oryzae MAFF, Sodalis glossinidius str morsitans).
Third, using the RBSfinder program [22]–[24], we checked the novel genes for the presence of ribosome binding sites (RBS). The ability of RBSfinder to detect true RBSs in the Bp genome was confirmed by benchmarking the numbers of RBS predictions using previously-identified Bp genes against a set of background randomized sequences [25]–[26] (Text S2). Of the 519 novel genes, we identified high-confidence RBSs in 309 genes (59.5%), without requiring alteration of the predicted gene start/stop coordinates.
Combining these three lines of supporting evidence (mRNA transcripts, database matches, presence of RBS), we identified 282 novel genes supported by two lines of evidence (“dual evidence genes”), and 81 novel genes supported by all three lines (Table S2). A comparison of compositional features (length, G+C content, CAI, hydrophobicity [27]) between the 282 dual evidence genes and 5728 protein-coding genes from the original 2004 annotation revealed striking differences in gene length between the sets (average gene length 98±56 aa vs 348±307 aa between novel and 2004 genes, p = 1.23×10−304) (Figure 2E). Significant differences in G+C content, CAI, and hydrophobicity were also observed (eg G+C content 0.63±0.1 vs 0.68±0.05, p = 9.69×10−17) (Table S3). Interestingly, some of these latter compositional differences might be indirectly related due to the short lengths of the novel genes, as significant G+C content, CAI, and hydrophobicity differences were also observed when a set of “short length” genes from the original annotation (<200 aa) were compared against the entire 5728 set (Table S3). Because compositional differences can often influence gene prediction accuracy [28]–[29], it is possible that some of these differences might have contributed to the novel genes being missed in the original annotation. To facilitate integration with existing genome features, we assigned identities to the 282 novel genes based on their proximity to existing genes (eg BPSL2192.1) (Table S2).
We also investigated the 120 genes missed in the current gene prediction analysis but identified by the previous 2004 genome annotation (Table S4). Of these 120 genes, 87 genes (73%) were categorized either as “doubtful CDs”, “gene remnants”, or “pseudogenes” in the original 2004 annotation, indicating that these genes were likely regarded as ambiguous in the previous annotation as well. Of the remaining 33 genes, 21 genes encode hypothetical proteins while another 6 appear to have bacteriophage origins that may contain coding signals distinct from the rest of the Bp genome. The ambiguous nature for three-quarters of these genes, coupled with presence of atypical coding signals, provides the most likely explanation for their failure to be detected by the current automated prediction pipeline.
The availability of multiple Bp genomes also permitted the analysis of pseudogene dynamics within a species. Of 26 previously-described pseudo-genes in Bp K96243 [1], at least 6 were ‘resurrected’ in >6 other Bp genomes. For example, the BPSL2828 pseudo-gene exhibits a premature truncation due to a stop codon at position 107 (TGG → TGA). This mutation, however, was only observed in Bp K96243 and Bp Pasteur 52237; while the other 9 Bp genomes had an extended gene sequence to position 147 (Figure S4). The differential presence of multiple pseudogenes across the Bp strains suggests that pseudogene formation in Bp is likely to be an active and highly dynamic process, consistent with its role as a recently evolved pathogen.
Comparative Analysis of the Bp Core Genome
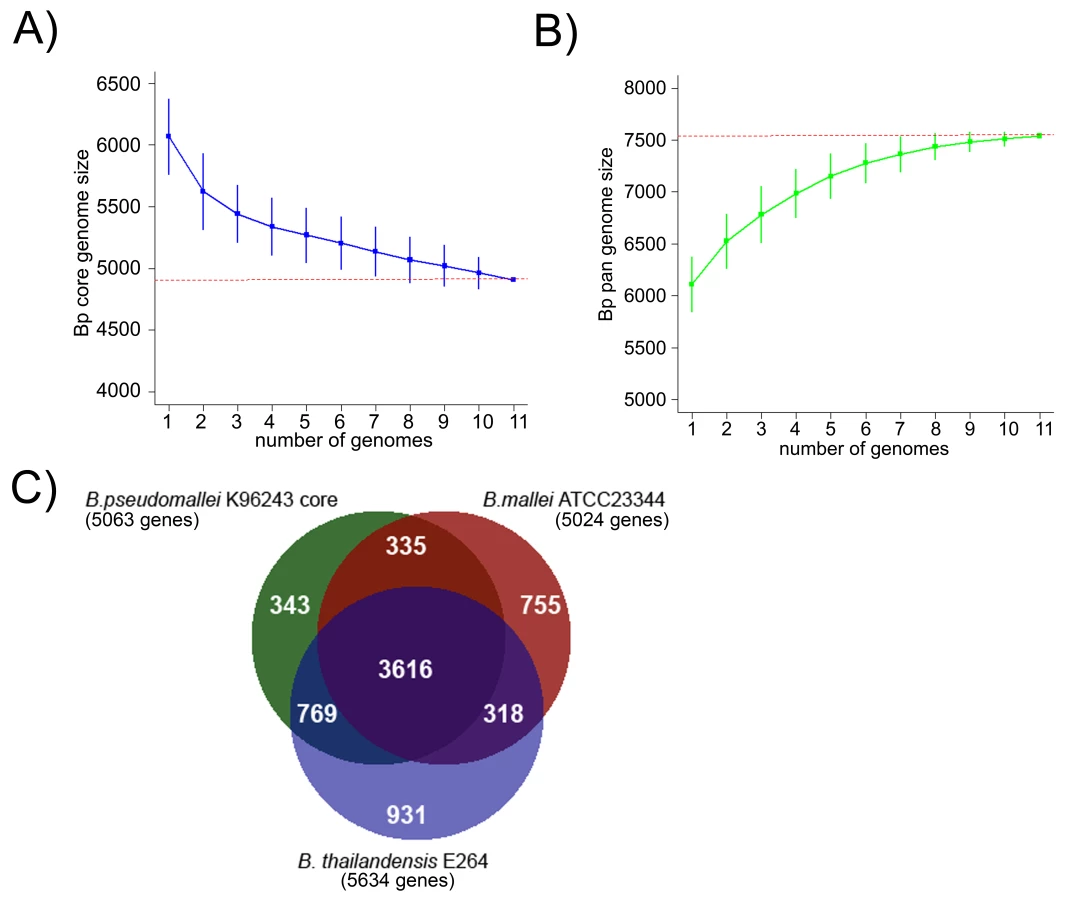
An analysis of gene orthologs across the Bp genomes identified a BpCG of 4908 genes present in all 11 strains (Figure 3A, [30]), with slight variations in individual genomes due to the presence of gene duplications and paralogs (range 5049–5139 genes). Similar core genome estimates were obtained when the analysis was confined to the nine independently derived isolates (Figure S5). We confirmed the robustness of this BpCG estimate using the method of Tettelin et al [31]. An evolutionary comparison of the BpCG against two closely related Burkholderia species with highly distinct niches - B. mallei ATCC23344 (Bm), a intracellular pathogen specific to horses [32], and B. thailandensis E264 (Bt), a non pathogenic, environmental bacterium [33]–[34], defined a common set of ∼3616 genes found in all three species (Figure 3C). 270 out of 335 genes are common to Bp and Bm with no orthologs in Bt, while 641 out of 769 genes are common to Bp and Bt with no ortholog in Bm. Besides the core genes, gene accumulation curves also project the global gene repertoire of Bp (the Bp pangenome) to be ∼7,500 genes (Figure 3B), a number close to 1.5x the size of the Bp core genome. A detailed analysis of the Bp pangenome will be described elsewhere.
Genetic Variation in the Bp Core Genome
To survey the landscape of genetic variation in Bp, we focused on a high quality ortholog set of 4673 BpCG genes (one orthologous gene per genome with >50% sequence similarity, each member exhibiting positional conservation to every other member, and excluding paralogs). We catalogued single-nucleotide polymorphisms (SNPs) and insertion/deletion sequences (indels) in the BpCG. Each Bp strain exhibited an average of ∼8594 SNPs compared to the K96243 reference genome, resulting in an overall SNP/Kb frequency of ∼2.0 for BpCG genes, while indels account for 0.1% and 0.3% of the total genetic variation in chromosomes 1 and 2 respectively. We confirmed the reliability of the genetic variation data by several methods. First, we confirmed by targeted resequencing >100 randomly-selected SNPs and 25 randomly-selected indels (data not shown). Second, 83% of identified SNPs are either (a) recurrently observed across multiple genomes (Table S5) [35], or (b) observed in Bp genomes of particularly high sequence quality (1106a, 1710b, 22, K96243 and 406e) (Table S5). Third, the SNP distributions are entirely consistent with geographic models in that strains with the highest levels of genetic variation compared to K96243 were observed in isolates from Australia, the most geographically distant locale (Figure 4A). This is consistent with previous proposals that strains from Australia are genetically distinct from their Asian counterparts [36] and form an ancestral population [35]. The existence of a deep genetic distinction between the South East Asian and Australian strains was further supported by phylogenetic analysis of 14,544 shared orthologous SNPs across 23 Bp genomes (including the genomes analyzed in this study), and also by an MLST population structure analysis involving >1800 Bp strains (647 sequence types) (Figure S6).

Among the clinical isolates, strain pairs 1106a–1106b and 1710a–1710b were isolated from the same patients during either primary infection or disease relapse, with intervening periods of approximately three years (Table S1). Surprisingly, a comparison of the primary and relapse strain genomes in both pairs failed to reveal a significant number of newly acquired mutations in relapsed strains (4 variants in 1106a vs 1106b, 6 variants in 1710a vs 1710b, none recurrent between both pairs) (Table S6). This lack of genetic variation between the primary and relapsed strains suggests that the former may have remained dormant in the human host during this intervening period, supporting the notion that that the Bp genome is likely to exhibit a high degree of stability during in vivo infection and persistence.
Positive Selection in the Bp Core Genome
To assess the functional implications of BpCG variation, we divided the BpCG SNPs into subsets predicted to cause either synonymous (Ks) or nonsynonymous (Ka) nucleotide substitutions. The Ks rate was similar between Bp Chr 1 and 2, indicating comparable levels of background genetic diversity between the two chromosomes. However, the Ka rate of Chr 2 was significantly higher than Chr 1 (P = 2.42×10−21, unpaired t-test, under a one-ratio model (M0) assuming a constant Ka/Ks ratio, Figure 4B), indicating that BpCG genes on Chr 2 are experiencing a higher degree of functional substitution than Chr 1. These chromosomal differences support the model of Holden et al [1] that Chr 1 of Bp represents the ancestral chromosome, with genes primarily related to housekeeping functions while Chr 2 contains genes involved in accessory functions and secondary adaptation.
We identified BpCG genes with signatures of positive selection using established methods [37]–[39] (Figure S7 and Methods, [40]). A maximum likelihood analyses was performed on each Bp core gene to detect coding sequence sites displaying features of differential selective pressure (positive selection) using two different likelihood ratio (LR) models (M1a-M2a, or M7-M8). Out of 4673 genes, Model M1a-M2a was significant for 212 genes, while model M7 -M8 test was significant for 239 genes (Ka/Ks>1; ∼2% FDR; P<0.001, LR Test). In total, 211 genes were commonly identified by both models as being positively selected (Table S7). Consistent with these 211 genes exhibiting above-background rates of functional variation (median Ka/Ks = 60.07 and P<0.001, LR Test), the average Ks value of the 211 positively selected genes was similar to the Ks value of non-PS genes (Ks = 0.2 for PS and non-PS genes, p = 0.56), while in contrast, Ka, the rate of non-synonymous substitution was 3 times greater in the positively-selected genes compared to genes under neutral selection (p = 0.5×10−5, t-test). The Ka/Ks value of the positively selected genes was also markedly higher compared to seven housekeeping genes typically used in MLST analysis (ace, gltB, gmhD, lepA, lipA, narK and ndh) (P<0.001, LR Test). A significantly greater fraction of positively-selected genes were identified on Chr 2 than Chr 1 (P = 0.006, χ2 test, 10000 simulations). These observations suggest that a significant proportion of the Bp core genome (∼4.5%) may be under positive selection.
We investigated whether the elevated Ka/Ks rate of the 211 positively selected genes might be due to mutation or recombination between the genomes in this strain panel. All 4673 core genome alignments were tested for the potential presence of recombination using two different methods (GENECONV [41], and the Pairwise Homoplasy Index (Phi)) [42]. Combining both methods, 56 out of 4673 core genes were identified as exhibiting a recombination signature. Of these 56, only 3 belong to the 211 positively selected genes, indicating that only a relatively minor component of the 211 genes are associated with a recombination signature. We also assessed rho/theta, the recombination/mutation ratio, of the Bp genomes analyzed in this study [43]. Using the Clonalframe algorithm [43], an inspection of 4294032 variation sites estimated rho/theta to be 0.012–0.015 (95% credibility region) for Chr 1 and 0.015–0.019 for Chr 2 respectively. This low value suggests that mutation rather than recombination appears to be the predominant evolutionary process explaining the patterns of genetic variation observed in the current panel of Bp strains.
Consistent with the BpCG responding to multiple selective pressures, the positively selected genes were widely dispersed across a wide variety of functions, including metabolic processes, membrane functions, signal transduction, and gene expression regulation (Table 2). A functional category analysis subsequently revealed that positively selected genes in the Bp core genome were significantly enriched in COG categories related to secondary metabolism (P = 0.036) and carbohydrate metabolism (P = 0.01, binomial test after correction for multiple hypotheses) (Figure 4C), highlighting these two metabolic pathways as major processes experiencing selective pressure.

Positively Selected Genes May Contribute to Mammalian Virulence
We were intrigued by the possibility that the positively selected genes, while overtly responding to environmental pressures encountered by Bp in soil, might indirectly facilitate the colonization of mammalian hosts. Supporting this notion, the positively selected genes were significantly enriched in genes previously identified as putative virulence-related genes [1] (20 genes, P = 0.019, based on 10,000 empirical permutations). For example, one representative class of virulence-related genes are Type IV pili (TFP), which are bacterial surface proteins implicated in multiple cellular processes, including motility, cell adhesion, microcolony formation, and virulence [44]. Of eight previously identified TFP loci in Bp K96243 [45], positively selected genes were associated with three TFP loci (TFP2, TFP4 and TFP7), with the TFP4 Type IVA minor pilin locus containing two positively selected genes (BPSL2754 pilW and BPSL2755 pilV). To evaluate if TFP4 might be involved in mammalian virulence, we generated isogenic Bp mutant strains deleted in the TFP4 locus, and tested the virulence of TFP4 deletion strains in a BALB/c mouse intranasal infection assay [46]. TFP4 deleted strains exhibited significantly reduced virulence compared to parental Bp K96243 wild-type controls (p = 0.048, Mantel-Haenszel log-rank test, Figure 5A), supporting a role for Type IV minor pilin activity in murine virulence. These results suggest that a subset of positively selected genes in Bp may influence virulence in mammals.

To further explore if other positively selected genes might conceivably provide traits facilitating successful mammalian infection, we then investigated two other features typically associated with successful intracellular human pathogens - a) the ability to interact with host cellular processes, and b) the ability to utilize host metabolites as nutrients. Previous studies have shown that many microbial pathogens can alter host cytoskeletons and cell morphology during infection, using proteins such as TTS factors to induce actin stress fibers, lamellipodia, and filapodia [46]–[48]. To examine the role of positive selection in this process, we curated a list of ten positively selected genes, either related to TTS biology (BPSS1552) or present in Bp and Bm (both pathogenic species) but absent from Bt (non-pathogenic) (Table S8). We cloned and expressed these ten genes in Hela cells, and examined the transfected cells for cytoskeletal perturbations. As a positive control, we also included BopE (BPSS1525), a TTS effector protein capable of inducing actin rearrangements [49]. Nine of the positively selected genes were successfully expressed in Hela cells but did not induce any significant differences in actin morphology compared to vector controls (eg BPSS0415, Figure 5B). In contrast, cells transfected with BPSL1057F1, a hypothetical protein and one of the novel genes identified in this study, exhibited a marked increase in actin stress fiber formation in the majority (60%) of transfected cells, with phenotypes very similar to BopE transfection (Figure 5B and 5C). Protein analysis of BPSL1057F1 revealed the presence of a twin-arginine signal peptide sequence, often found in proteins exported into an extra-cellular environment [50]. These results suggest that some positively selected genes in Bp may provide Bp with the potential to interact with host cellular pathways.
We also analyzed the list of positively selected genes for potential genes involved in host metabolite catabolism. Of metabolites linked to the 10 positively selected secondary metabolism genes, we focused on taurine (2-aminoethanesulfonate), since taurine is an amino acid found at high levels in potential mammalian hosts in muscles, bile, and white blood cells, but absent or present at only trace levels in bacteria and plants [51]. Supporting the notion that Bp has developed an ability to metabolize taurine, the taurine dixoygenase gene BPSS0161 (tauD) exhibited a significant degree of positive selection across the eleven Bp genomes (P<0.001, Ka/Ks = 57.6, EC 1.14.11.17). Prompted by this finding, we further explored the role of taurine metabolism genes in Bp and discovered a previously-unreported species-specific expansion of additional tauD gene members in Bp. Specifically, compared to Bt or Bm which have three tauD genes on Chr 2, the Bp Chr 2 genomes harbor eight-nine tauD genes, a three-fold expansion (Figure 5D [52]–[53], also on Chr 2). The Bp tauD genes all share the same tauD pfam family domain (PF02668) but otherwise exhibit low sequence similarity between each other (average nucleotide homology of 36%), arguing against this expansion occurring by gene duplication. Instead, sequence analysis suggests that many of the Bp tauD genes were likely acquired by lateral gene transfer. For example, BPSS0665, another tauD gene, is localized to genomic island 14 (GI14), a region of codon bias deviation and atypical % GC content (Figure S8). Intriguingly, despite exhibiting many features of mobile elements, GI14 has been previously shown to be consistently present across a large panel of natural Bp isolates in contrast to other GIs [7] (Figure S8). It is possible that a selective requirement for maintaining levels of tauD activity might have contributed to GI14 behaving as a conserved feature of the Bp genome.
In other bacterial species, tauD is required to metabolize taurine as a sulphur source [54]–[55]. Experimental assays comparing the growth Bp and Bt strains confirmed that Bp also exhibits a significantly enhanced ability to efficiently utilize taurine as a sulphur source compared to Bt (p = 0.002, Figure 5E). The ability of Bp to metabolize taurine for sulphur utilization is specific, as Bp was unable to use taurine as an alternative carbon or nitrogen source, activities which are not mediated by tauD (Figure S8). Finally, to investigate the molecular response of Bp to taurine, we generated whole-genome transcriptome profiles of Bp exposed to high levels of taurine (250 uM). Here, the taurine concentrations used were based on previous reports studying taurine metabolism in E. coli [54]–[55]. Compared to Bp grown in standard laboratory media, taurine-exposed Bp exhibited transcriptional up-regulation of ∼280 genes, of which 40% (126 genes) have been previously associated with pathogenicity, host–cell interaction, or survival in diverse and challenging environments [1]. Specific examples of taurine-regulated genes implicated in virulence included several flagella gene clusters (BPSL0024-BPSL0032, BPSL0224-BPSL0236, BPSL0266-BPSL0282, BPSL3288 - BPSL3330) [56], siderophore biosynthesis and iron metabolism genes (BPSL1771 - BPSL1787, BPSS0239 - BPSS0244, BPSS0581 - BPSS0588) [57], and fimbrae/pili (BPSL2026 - BPSL2031, BPSS1593 - BPSS1605) [45] (Figure 5F, Table S9A and S9B). Taken collectively, these findings suggest that altered taurine metabolism likely mediated by tauD may represent a species-specific adaptation of Bp that may have also facilitated its ability to survive in infected mammalian hosts [58].
Discussion
In this, the first nucleotide-scale comparative analysis of multiple Bp genomes, we expanded the known gene repertoire of Bp, defined the BpCG, and described the extent of genetic variation in BpCG genes. We identified a set of genes exhibiting positive selection, and examined how such variations can impact genomic organization and structure. Our results suggest that a significant proportion of the BpCG may be experiencing functional selection, and that a large aspect of this selection involves the modification of preexisting metabolic circuits related to carbohydrate and secondary metabolism. Importantly, we also provide evidence that a subset of these genes may have also facilitated the ability of Bp to interact with mammalian hosts, either structurally or nutritionally.
In our analysis, we have proposed that many of the genetic alterations observed in the positively selected genes were primarily driven by environmental pressures outside the human or mammalian host. Nevertheless, if Bp undergoes cryptic cycling through normal humans or other potential mammalian hosts, such as livestock or wild cattle [59], it remains possible that certain survival and virulence traits were directly selected for in mammals. In melioidosis-endemic NE Thailand, the majority of healthy individuals have antibodies to Bp by the age of 4 years, indicating constant exposure to the bacterium that may occur by inoculation, inhalation or ingestion [4]. Within such hosts, Bp might spend periods of time being exposed to the mammalian immune response and various physiologic traits. Subsequent return to the environment in a viable state, through skin desquamation or in urine and stool, could also lead to the selection of factors that promote survival in vivo. However, because we a) consider the mammalian host to be a relatively minor component of Bp ecology, b) such cryptic cycling through mammalian hosts has yet to be documented, and c) the lack of genetic variation between the primary and relapsed strains suggests that the Bp genome is likely to exhibit a high degree of stability during mammalian infection, we argue that this scenario is, on balance, possible but less likely.
A large proportion of Bp genes are still unannotated or poorly characterized, raising the need for systematic approaches to link discrete sets of Bp genes to their specific biological and cellular functions. The genomic identification of these positively selected genes should facilitate the process of targeted experimentation to elucidate the pathogenesis of melioidosis. The prioritization of candidate genes for targeted experimentation is particularly relevant for Bp due to its classification as a potential biothreat agent. Under international biosafety regulations, Bp research is typically conducted in high containment (Category 3) facilities and limited to highly focused projects [60] (http://www.selectagents.gov/). Finally, it is worth noting that the ability of this approach to uncover candidate host interaction genes and pathways from a genome as complex as Bp suggests that similar approaches should prove equally fruitful in elucidating novel aspects of biology in other recently emergent pathogens as well.
Methods
Ethics Statement
This research was approved by the Genome Institute of Singapore Institutional Review Board. All animal experimentation was conducted at DSTL (Defence Science and Technology Laboratory) in the United Kingdom (UK) under Animal (Scientific Procedures) Act 1986.
Genome Annotations and Comparative Analysis
Bp genes were predicted using FGENESB [http://linux1.softberry.com/berry.phtml?topic=fgenesb&group=help&subgroup=gfindb (Softberry)]. tRNA genes were identified using tRNAScan-SE [20], and rRNA genes by sequence conservation (blastn, e-value threshold: 1e-08). Operons were identified based on a) distances between genes, b) likelihood of neighboring genes also appearing in other bacterial genomes as neighbors, and c) locations of predicted promoters and terminators. Genes were annotated against the NR, COG, KEGG and STRING [www.ncbi.nlm.nih.gov (NR); www.ncbi.nlm.nih.gov/COG (COG); www.genome.jp/kegg (KEGG); http://string.embl.de/ (STRING)] databases using the following criteria: i) BLASTP e-value threshold of <1e-10; ii) percent identify threshold of >60%, and iii) a percentage coverage threshold of 80%. These criteria were used based on previous studies [18]–[19]. Ribosome binding sites (RBSs) were identified using RBSfinder [22]–[24]. Notably, the consensus RBS sequences between E. coli and Bp are similar [25]–[26]. Non-coding RNAs were identified using the Rfam database [17]. CodonW (http://codonw.sourceforge.net/) was used to identify codon adaptation indexes (CAI), Kyte and Doolittle scales of hydrophobicity [27], GC percentages and gene lengths. Multiple whole-genome alignments were performed using Mauve 2.2.0 [61].
Transcriptome Profiling
Bp K96243 cultures were isolated from six conditions: Luria-Bertani broth (mid-logarithmic, early stationary and late stationary phases, conditions 1–3), minimal media (mid-log and early stationary, conditions 4–5), or exposure to 1x PBS solution (condition 6). Bacterial mRNAs were profiled on a high-density Bp tiling array representing both strands of the Bp K96243 genome (7.2 Mb) (Nimblegen) (50-mers, 15-base overlap). All transcriptome profiles are the average of 2 biological replicates. Three distinct criteria were employed to consider a novel gene as “expressed”. First, an “expressed” novel gene was required to exhibit a minimum of 3 consecutive array probes with fluorescence intensities above the array median intensity. Second, for genes covered by more than five array probes, the combined pseudo-median expression value of the novel gene was assessed using the SIGN Test, a statistical method previously used to measure the transcriptional activity of genes using tiling microarrays [16]. Only novel genes passing the SIGN test were considered as “expressed” (p<0.05). Third, short novel genes covered by less than five probes that did not qualify for the SIGN Test were manually curated to confirm the presence of contiguous expression signals for each gene. For analyses of differential gene expression, ratios of normalized probe signals were computed. Probe identities with more than 2-fold up-regulation or down-regulation were matched to Bp gene identities. Genes that have 50% or more probes showing at least 2-fold up-regulation or down-regulation were taken as differentially expressed between the conditions compared.
Bp Core Genome and Pan Genome
Gene orthologs across the Bp genomes were determined using OrthoMCL [62]. An all-against-all BLASTp [63] was performed, followed by a reciprocal BLAST to define putative ortholog pairs or recent paralogs (genes within the same genome that are reciprocally more similar to each other than any sequence from another genome). Reciprocal BLASTp values were converted to a normalized similarity matrix that was analyzed by the Markov Cluster algorithm MCL to define ortholog clusters. OrthoMCL was run with a BLAST e-value cut-off of 1e-5, and an inflation parameter of 1.5. The OrthoMCL output was used to construct tables of shared orthologs and strain-specific genes.
Bp Core Genome Variation and Positive Selection
Orthologs exhibiting positional conservation across the Bp genomes were aligned at the DNA level with ClustalW [21] and manually confirmed. SNAP.pl was used to calculate the number of synonymous vs. non-synonymous base substitutions (Nei and Gojobori method) for all pairwise comparisons of ortholog sequences [40]. Ambiguous codons or codons with insertions were excluded from the tally of compared codons. Base-substitutions were also manually inspected to remove from consideration substitutions indirectly caused by upstream frame-shifts. GENECONV [41] was used to identify recombination breakpoints, and genes exhibiting a recombination signature were fragmented at the predicted breakpoints. The recombination sub-fragments (total 152 sub-fragments) were individually applied to the PHYLIP pipeline to infer maximum parsimony trees. The core gene alignments were also tested for the presence of recombination using the Pairwise Homoplasy Index (Phi) as implemented in the HYPHY package (100000 permutations, cutoff at ∼1% FDR) [42]. ClonalFrame version 1.1 was used to compute rho/theta, the recombination/mutation ratio [43]. Protein sequences were aligned using ClustalW (‘ktuple’ ⇒ 2 and ‘matrix’ ⇒ ‘BLOSUM’). PAL2NAL [64] Perl scripts were used to convert the multiple sequence protein alignments and corresponding DNA sequences into codon alignments. Maximum parsimony (MP) trees were generated using PHYLIP (‘dnapars’ module) using default values (http://evolution.genetics.washington.edu/phylip.html). Codon alignments and MP trees were analyzed by PAML 4.0 [38] to calculate Ka/Ks (or ω) ratios and test different evolutionary models. The following nested models were used: M1a-M2a and M7-M8 [39]. A likelihood ratio test was used to compare model M2a with M1a, and model M8 with M7, at a significance cutoff of ∼2% FDR [38]. The nested model M0 (one-ratio)-M3 (discrete) was also used to confirm heterogeneity of Ka/Ks in the cohort of positively selected genes [65].
Construction of Isogenic Mutant Strains
Isogenic unmarked mutant Bp strains carrying a 3.7 kb deletion of the TFP4 gene cluster were generated as previously described in Boddey et al., 2006 [66]. Briefly, a TFP4 (BPSL2749-BPSL2755) targeting vector was constructed and conjugated into Bp K96243. Integrants were selected on chloramphenicol plates (100 ug/ml) and confirmed by PCR. Merodiploid integrants were then cultured without selection and plated onto medium lacking sodium chloride but containing 15% sucrose to enrich for colonies carrying a deleted chromosomal locus. Bp TFP4 mutants were confirmed both by PCR and Southern blotting.
Mouse Virulence Studies
Virulence of wild-type and mutant Bp strains were assessed using an intranasal BALB/c mouse model as previously described [45]. Briefly, groups of six age-matched BALB/c female mice were anesthetized and infected intranasally with 10-fold dilutions (101–106) of either wild-type Bp K96243 or TFP5 deletion strains grown overnight at 37degC with shaking. Mice were recovered and survival was recorded for up to 51 days. The survival data was analyzed using the Mantel-Haenszel log rank test in GraphPad Prism 4 or by Regression with Life Data in MIniTAB v13.0, using a significance threshold of α = 0.05.
Cell Culture, DNA Transfection and Immunoflouresence
Positively selected genes were PCR-amplified from Bp genomic DNA and subcloned into Vivid Colors®pcDNA® 6.2/N-EmGFP-GW/TOPO® mammalian expression vectors (Invitrogen). Hela cells were transfected using Gene Juice (Novagen), and cultured for 24 h after tranfection. Cells were fixed in 3.7% paraformaldehyde/PBS (pH 7.0). After washing and preincubation, cells were stained with Alexa Flour 555 phalloidin (Invitrogen) and DAPI (Sigma-Aldrich). Stained cells were visualized using a confocal Zeiss LSM 150 inverted laser scanning microscope and analyzed using Zeiss LSM Image Browser software (Carl Zeiss, Oberkochen, Germany).
Taurine Utilization
2 Bp and 2 Bt strains (Bp K96243, Bp 22, Bt ATCC700388 and Bt E305) were cultured in modified M63 media, or media supplemented with 250 µM taurine or 250 µM Na2SO4. Cultures were grown at 37°C, 150 rpm and OD600 readings were taken every 2 hrs for 72 hrs. To study differential gene expression, Bp K96243 was cultured in modified M63 medium with 250 µM taurine at 37°C, 150 rpm for 48 hrs to reach stationary phase. The expression profile obtained was compared with that obtained for Bp K96243 cultured in LB at stationary phase. All transcriptome profiles are the average of 2 biological replicates.
Supporting Information
Zdroje
1. HoldenMT
TitballRW
PeacockSJ
Cerdeno-TarragaAM
AtkinsT
2004 Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc Natl Acad Sci U S A 101 14240 14245
2. WiersingaWJ
van der PollT
WhiteNJ
DayNP
PeacockSJ
2006 Melioidosis: insights into the pathogenicity of Burkholderia pseudomallei. Nat Rev Microbiol 4 272 282
3. CurrieBJ
2008 Advances and remaining uncertainties in the epidemiology of Burkholderia pseudomallei and melioidosis. Trans R Soc Trop Med Hyg 102 225 227
4. CasadevallA
PirofskiLA
2007 Accidental virulence, cryptic pathogenesis, martians, lost hosts, and the pathogenicity of environmental microbes. Eukaryot Cell 6 2169 2174
5. U'Ren JM
HornstraH
PearsonT
SchuppJM
LeademB
2007 Fine-scale genetic diversity among Burkholderia pseudomallei soil isolates in northeast Thailand. Appl Environ Microbiol 73 6678 6681
6. TumapaS
Burkholderia pseudomallei genome plasticity associated with genomic island variation. BMC Genomics 9 190 (2008)
7. SimSH
YuY
LinCH
KaruturiRK
WuthiekanunV
2008 The core and accessory genomes of Burkholderia pseudomallei: implications for human melioidosis. PLoS Pathog 4 e1000178 doi:10.1371/journal.ppat.1000178
8. InglisTJ
RobertsonT
WoodsDE
DuttonN
ChangBJ
2003 Flagellum-mediated adhesion by Burkholderia pseudomallei precedes invasion of Acanthamoeba astronyxis. Infect Immun 71 2280 2282
9. GanYH
ChuaKL
ChuaHH
LiuB
HiiCS
2002 Characterization of Burkholderia pseudomallei infection and identification of novel virulence factors using a Caenorhabditis elegans host system. Mol Microbiol 44 1185 1197
10. UlettGC
CurrieBJ
ClairTW
MayoM
KetheesanN
2001 Burkholderia pseudomallei virulence: definition, stability and association with clonality. Microbes Infect 3 621 631
11. FrigaardNU
MartinezA
MincerTJ
DeLongEF
2006 Proteorhodopsin lateral gene transfer between marine planktonic Bacteria and Archaea. Nature 439 847 850
12. BensonG
1999 Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27 573 580
13. SiguierP
PerochonJ
LestradeL
MahillonJ
ChandlerM
2006 ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res 34 D32 36
14. MavromatisK
IvanovaN
BarryK
ShapiroH
GoltsmanE
2007 Use of simulated data sets to evaluate the fidelity of metagenomic processing methods. Nat Methods 4 495 500
15. ZhuH
HuGQ
YangYF
WangJ
SheZS
2007 MED: a new non-supervised gene prediction algorithm for bacterial and archaeal genomes. BMC Bioinformatics 8 97
16. BertoneP
StolcV
RoyceTE
RozowskyJS
UrbanAE
2004 Global identification of human transcribed sequences with genome tiling arrays. Science 306 2242 2246
17. GardnerPP
DaubJ
TateJG
NawrockiEP
KolbeDL
2009 Rfam: updates to the RNA families database. Nucleic Acids Res 37 D136 140
18. LoftusB
AndersonI
DaviesR
AlsmarkUC
SamuelsonJ
2005 The genome of the protist parasite Entamoeba histolytica. Nature 433 865 868
19. KalmanS
MitchellW
MaratheR
LammelC
FanJ
1999 Comparative genomes of Chlamydia pneumoniae and C. trachomatis. Nat Genet 21 385 389
20. LoweTM
EddySR
1997 tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25 955 964
21. LarkinMA
BlackshieldsG
BrownNP
ChennaR
McGettiganPA
2007 Clustal W and Clustal X version 2.0. Bioinformatics 23 2947 2948
22. SuzekBE
ErmolaevaMD
SchreiberM
SalzbergSL
2001 A probabilistic method for identifying start codons in bacterial genomes. Bioinformatics 17 1123 1130
23. SmithMG
GianoulisTA
PukatzkiS
MekalanosJJ
OrnstonLN
2007 New insights into Acinetobacter baumannii pathogenesis revealed by high-density pyrosequencing and transposon mutagenesis. Genes Dev 21 601 614
24. DelcherAL
BratkeKA
PowersEC
SalzbergSL
2007 Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 23 673 679
25. WooPC
LeungPK
TsoiHW
YuenKY
2001 Cloning and characterisation of malE in Burkholderia pseudomallei. J Med Microbiol 50 330 338
26. WinstanleyC
HalesBA
HartCA
1999 Evidence for the presence in Burkholderia pseudomallei of a type III secretion system-associated gene cluster. J Med Microbiol 48 649 656
27. KyteJ
DoolittleRF
1982 A simple method for displaying the hydropathic character of a protein. J Mol Biol 157 105 132
28. KrauseL
McHardyAC
NattkemperTW
PuhlerA
StoyeJ
2007 GISMO–gene identification using a support vector machine for ORF classification. Nucleic Acids Res 35 540 549
29. SerresMH
GopalS
NahumLA
LiangP
GaasterlandT
2001 A functional update of the Escherichia coli K-12 genome. Genome Biol 2 RESEARCH0035
30. LefebureT
StanhopeMJ
2007 Evolution of the core and pan-genome of Streptococcus: positive selection, recombination, and genome composition. Genome Biol 8 R71
31. TettelinH
MasignaniV
CieslewiczMJ
DonatiC
MediniD
2005 Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci U S A 102 13950 13955
32. NiermanWC
DeShazerD
KimHS
TettelinH
NelsonKE
2004 Structural flexibility in the Burkholderia mallei genome. Proc Natl Acad Sci U S A 101 14246 14251
33. KimHS
SchellMA
YuY
UlrichRL
SarriaSH
2005 Bacterial genome adaptation to niches: divergence of the potential virulence genes in three Burkholderia species of different survival strategies. BMC Genomics 6 174
34. YuY
KimHS
ChuaHH
LinCH
SimSH
2006 Genomic patterns of pathogen evolution revealed by comparison of Burkholderia pseudomallei, the causative agent of melioidosis, to avirulent Burkholderia thailandensis. BMC Microbiol 6 46
35. PearsonT
GiffardP
Beckstrom-SternbergS
AuerbachR
HornstraH
2009 Phylogeographic reconstruction of a bacterial species with high levels of lateral gene transfer. BMC Biol 7 78
36. TuanyokA
AuerbachRK
BrettinTS
BruceDC
MunkAC
2007 A horizontal gene transfer event defines two distinct groups within Burkholderia pseudomallei that have dissimilar geographic distributions. J Bacteriol 189 9044 9049
37. ChenSL
HungCS
XuJ
ReigstadCS
MagriniV
2006 Identification of genes subject to positive selection in uropathogenic strains of Escherichia coli: a comparative genomics approach. Proc Natl Acad Sci U S A 103 5977 5982
38. AnisimovaM
BielawskiJP
YangZ
2001 Accuracy and power of the likelihood ratio test in detecting adaptive molecular evolution. Mol Biol Evol 18 1585 1592
39. YangZ
2007 PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 24 1586 1591
40. KorberB
2000 HIV Signature and Sequence Variation Analysis. Dordrecht, Netherlands Kluwer Academic Publishers 55 72 Computational Analysis of HIV Molecular Sequences, Chapter 4 Allen G. Rodrigo and Gerald H. Learn, eds
41. SawyerS
1989 Statistical tests for detecting gene conversion. Mol Biol Evol 6 526 538
42. BruenTC
PhilippeH
BryantD
2006 A simple and robust statistical test for detecting the presence of recombination. Genetics 172 2665 2681
43. DidelotX
FalushD
2007 Inference of bacterial microevolution using multilocus sequence data. Genetics 175 1251 1266
44. CraigL
PiqueME
TainerJA
2004 Type IV pilus structure and bacterial pathogenicity. Nat Rev Microbiol 2 363 378
45. Essex-LoprestiAE
BoddeyJA
ThomasR
SmithMP
HartleyMG
2005 A type IV pilin, PilA, Contributes To Adherence of Burkholderia pseudomallei and virulence in vivo. Infect Immun 73 1260 1264
46. LiuB
KooGC
YapEH
ChuaKL
GanYH
2002 Model of differential susceptibility to mucosal Burkholderia pseudomallei infection. Infect Immun 70 504 511
47. AktoriesK
BarbieriJT
2005 Bacterial cytotoxins: targeting eukaryotic switches. Nat Rev Microbiol 3 397 410
48. HallA
1998 Rho GTPases and the actin cytoskeleton. Science 279 509 514
49. StevensMP
FriebelA
TaylorLA
WoodMW
BrownPJ
2003 A Burkholderia pseudomallei type III secreted protein, BopE, facilitates bacterial invasion of epithelial cells and exhibits guanine nucleotide exchange factor activity. J Bacteriol 185 4992 4996
50. De BuckE
LammertynE
AnneJ
2008 The importance of the twin-arginine translocation pathway for bacterial virulence. Trends Microbiol 16 442 453
51. HuxtableRJ
1992 Physiological actions of taurine. Physiol Rev 72 101 163
52. SaitouN
NeiM
1987 The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4 406 425
53. PerriereG
GouyM
1996 WWW-query: an on-line retrieval system for biological sequence banks. Biochimie 78 364 369
54. EichhornE
van der PloegJR
KerteszMA
LeisingerT
1997 Characterization of alpha-ketoglutarate-dependent taurine dioxygenase from Escherichia coli. J Biol Chem 272 23031 23036
55. van der PloegJR
WeissMA
SallerE
NashimotoH
SaitoN
1996 Identification of sulfate starvation-regulated genes in Escherichia coli: a gene cluster involved in the utilization of taurine as a sulfur source. J Bacteriol 178 5438 5446
56. ChuaKL
ChanYY
GanYH
2003 Flagella are virulence determinants of Burkholderia pseudomallei. Infect Immun 71 1622 1629
57. TuanyokA
KimHS
NiermanWC
YuY
DunbarJ
2005 Genome-wide expression analysis of iron regulation in Burkholderia pseudomallei and Burkholderia mallei using DNA microarrays. FEMS Microbiol Lett 252 327 335
58. BrownSA
PalmerKL
WhiteleyM
2008 Revisiting the host as a growth medium. Nat Rev Microbiol 6 657 666
59. SpragueLD
NeubauerH
2004 Melioidosis in animals: a review on epizootiology, diagnosis and clinical presentation. J Vet Med B Infect Dis Vet Public Health 51 305 320
60. PeacockSJ
SchweizerHP
DanceDA
SmithTL
GeeJE
2008 Management of accidental laboratory exposure to Burkholderia pseudomallei and B. mallei. Emerg Infect Dis 14 e2
61. DarlingAE
TreangenTJ
MesseguerX
PernaNT
2007 Analyzing patterns of microbial evolution using the mauve genome alignment system. Methods Mol Biol 396 135 152
62. ChenF
MackeyAJ
StoeckertCJJr
RoosDS
2006 OrthoMCL-DB: querying a comprehensive multi-species collection of ortholog groups. Nucleic Acids Res 34 D363 368
63. AltschulSF
GishW
MillerW
MyersEW
LipmanDJ
1990 Basic local alignment search tool. J Mol Biol 215 403 410
64. SuyamaM
TorrentsD
BorkP
2006 PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res 34 W609 612
65. WongWS
YangZ
GoldmanN
NielsenR
2004 Accuracy and power of statistical methods for detecting adaptive evolution in protein coding sequences and for identifying positively selected sites. Genetics 168 1041 1051
66. BoddeyJA
FleggCP
DayCJ
BeachamIR
PeakIR
2006 Temperature-regulated microcolony formation by Burkholderia pseudomallei requires pilA and enhances association with cultured human cells. Infect Immun 74 5374 5381
Štítky
Hygiena a epidemiologie Infekční lékařství LaboratořČlánek vyšel v časopise
PLOS Pathogens
2010 Číslo 4
- Parazitičtí červi v terapii Crohnovy choroby a dalších zánětlivých autoimunitních onemocnění
- Vakcíny proti klíšťové encefalitidě
- Kdy je nejlepší očkovat
- Možné vedlejší účinky očkování
- Imunogenita vakcín
Nejčtenější v tomto čísle
- The Effect of Vaccination on the Evolution and Population Dynamics of Avian Paramyxovirus-1
- Reconstitution of SARS-Coronavirus mRNA Cap Methylation
- Increased Monocyte Turnover from Bone Marrow Correlates with Severity of SIV Encephalitis and CD163 Levels in Plasma
- Deficiencies in Jasmonate-Mediated Plant Defense Reveal Quantitative Variation in Pathogenesis
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy