
Muscle Structure Influences Utrophin Expression in Mice
Duchenne muscular dystrophy (DMD) is a severe muscle wasting disorder caused by mutations in the dystrophin gene. Utrophin is structurally similar to dystrophin and improving its expression can prevent skeletal muscle necrosis in the mdx mouse model of DMD. Consequently, improving utrophin expression is a primary therapeutic target for treating DMD. While the downstream mechanisms that influence utrophin expression and stability are well described, the upstream mechanisms are less clear. Here, we found that perturbing the highly ordered structure of striated muscle by genetically deleting desmin from mdx mice increased utrophin expression to levels that prevented skeletal muscle necrosis. Thus, the mdx:desmin double knockout mice may prove valuable in determining the upstream mechanisms that influence utrophin expression to develop a therapy for DMD.
Published in the journal:
. PLoS Genet 10(6): e32767. doi:10.1371/journal.pgen.1004431
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004431
Summary
Duchenne muscular dystrophy (DMD) is a severe muscle wasting disorder caused by mutations in the dystrophin gene. Utrophin is structurally similar to dystrophin and improving its expression can prevent skeletal muscle necrosis in the mdx mouse model of DMD. Consequently, improving utrophin expression is a primary therapeutic target for treating DMD. While the downstream mechanisms that influence utrophin expression and stability are well described, the upstream mechanisms are less clear. Here, we found that perturbing the highly ordered structure of striated muscle by genetically deleting desmin from mdx mice increased utrophin expression to levels that prevented skeletal muscle necrosis. Thus, the mdx:desmin double knockout mice may prove valuable in determining the upstream mechanisms that influence utrophin expression to develop a therapy for DMD.
Introduction
Duchenne muscular dystrophy (DMD) is an X-linked muscle disorder that affects approximately 1∶4000 boys [1]. DMD is caused by mutations in the large 2.2 Mb dystrophin gene [2], [3]. The dystrophin protein functions as a large molecular spring that connects the skeletal muscle cytoskeleton to the transmembrane dystrophin glycoprotein complex (DGC) [4]–[9]. The lack of dystrophin in DMD is accompanied by a significant reduction in the expression of the DGC leaving the membrane highly susceptible to contraction-induced injury and hypoxic stress [10]–[18]. DMD patients develop severe cardiorespiratory distress and generally live into their third decade with the help of palliative care.
The absence of dystrophin leads to various molecular and cellular homeostatic responses that slow the loss of skeletal muscle [19]. For instance, the dystrophin paralog, utrophin is expressed on the sarcolemma of dystrophic fibers acting to mitigate necrosis [20]–[25]. Skeletal muscle necrosis in the mdx mouse model of DMD is prevented by the expression of a full-length utrophin transgene when expressed at twice the levels of the endogenous utrophin [26]. Utrophin expression in DMD patients correlates with the severity of disease and time to wheelchair demonstrating the therapeutic potential of utrophin in humans [25], [27]–[31]. An utrophin therapy would benefit all DMD patients and circumvent a potential T-cell mediated immune response that could impair the long-term benefit of prospective dystrophin replacement strategies [32]–[34]. Accordingly, increasing the expression of utrophin is a primary target for therapy of DMD [33]. While promising utrophin-mediated therapies are being tested in clinical trials [33], [35], the mechanisms that influence utrophin expression are not fully understood.
Utrophin is normally expressed on the sarcolemma of developing and regenerating muscle fibers [21], [22], [36]. Utrophin is ultimately replaced by dystrophin in the sarcolemma of normal maturing fibers and remains concentrated at the neuromuscular and myotendinous junctions. However, low levels of utrophin can remain on the sarcolemma of dystrophin-deficient mdx mouse skeletal muscle fibers independent from muscle regeneration [37]. While various factors that influence utrophin expression and stability within the sarcolemma are well described [33], [38], [39], the upstream mechanisms are less clear. We recently discovered an increase in utrophin expression in mdx4cv mice expressing the microdystrophinΔR4–R23 transgene [40]. The polyproline site within hinge 2 of microdystrophinΔR4–R23 led to myotendinous strain injury and the formation of ringed fibers where the peripheral sarcomeres surround the central sarcomeres [40], [41]. Notably, we found a significant increase in utrophin expression within the limb muscles that contained ringed fibers, but not in the diaphragm muscles that did not contain ringed fibers [40]. Accordingly, we hypothesize that structural changes within skeletal muscle can influence utrophin expression, independent from muscle regeneration.
To examine the role of muscle structure on the pathogenesis of DMD we generated mdx:desmin double knockout (dko) mice. Desmin is an intermediate filament protein that maintains the highly ordered structure of striated muscles by connecting the sarcomeres to the sarcolemma and organelles [42]–[45]. Desmin influences the organization of dystrophin and ankyrin in a costameric lattice that connects the Z-disks of peripheral sarcomeres to the sarcolemma [46], [47]. Desmin−/− mice develop a severe dilated cardiomyopathy with a mild skeletal myopathy [45], [48]. The skeletal myopathy is associated with misaligned sarcomeres and changes to the distribution and function of mitochondria [45], [48]. Here, we report that a ∼2.5-fold increase in utrophin expression in dko skeletal muscle fibers prevented necrosis in a fiber-type specific manner.
Results
Premature death of dko mice
We initially found that desmin expression was increased in mdx4cv mouse skeletal muscles by western analysis of whole muscle lysates (Fig. 1A), confirming previous reports in mdx mice [49], [50]. To examine the role of desmin in the pathogenesis of DMD we bred mdx4cv:desmin+/− mice to generate the dko pups (N = 5, F>4). The dko pups were born in the expected Mendelian ratios [71 (25%) +/+; 144 (51%) +/−; 67 (24%) −/−]. We examined only the male mice for this study, as DMD patients are males. The dko mice developed a mild kyphosis (Fig. 1B). The genotype was confirmed by immunohistological analyses of dystrophin and desmin expression in skeletal muscle (Fig. 1C). The dko mice gained less body mass than the wild-type (24%), desmin−/− (18%), and mdx4cv controls (36%; P<0.001 one way ANOVA; Fig. 1D). The desmin−/− and dko mice were euthanized when they lost body mass and/or exhibited labored breathing and reduced mobility consistent with cardiorespiratory failure. Kaplan-Meyer survival analyses demonstrated a significantly reduced lifespan in the dko mice with a median survival of 76 days for males compared to a median survival of 609 days for the desmin−/− males (Fig. 1E, P<0.001). The average lifespan for mdx4cv males is 21.5 months [51]. We chose a time point of 11 weeks for the experiments in this study, unless otherwise stated. Approximately a quarter of the dko mice (22%) developed malocclusion, which contributed to the reduced body mass and increased mortality rate particularly in mice younger than 8 weeks of age. The malocclusion was treated with trimming the teeth every week and feeding the mice crushed food pellets mixed with hydrated gel. Malocclusion consistently presented in dko mice through various backcrosses suggesting that this was likely a phenotype of the dko mice and not a separate genetic defect. Furthermore, none of the wild-type, desmin−/− or mdx4cv mice developed malocclusion during the course of this study. The dko mice that developed malocclusion were included for body mass and survival analysis, but not for further analyses.

Profound reduction in dystrophic histopathology in dko mice
We next examined the gross dystrophic histopathology in various limb and respiratory muscles. Wild-type mice had few central nuclei (<1%), and no detectable calcified or necrotic fibers (Fig. 2A-D). Desmin−/− mice had a mild skeletal myopathy with a low level of central nuclei (∼5%) and rare necrotic fibers (Fig. 2AD), but no calcification was evident (Fig. 2A,C), as previously described [45], [48], [52]. The mdx4cv skeletal muscles were highly dystrophic with predominantly centrally nucleated fibers (Fig. 2A,B). Of the different limb and respiratory muscles we examined, only the mdx4cv diaphragms consistently contained calcified fibers (Fig. 2A,C), whereas all mdx4cv muscles contained patches of necrotic fibers (Fig. 2A,D). The proportion of dko limb and respiratory skeletal muscles with central nuclei was significantly reduced when compared to the mdx4cv muscles (Fig. 2A,B). None of the dko skeletal muscle fibers were calcified and there were 96% fewer necrotic fibers than the mdx4cv gastrocnemius muscles (P<0.001; Fig. 2A,C,D). Inflammation was also reduced in the dko gastrocnemius muscle with a 93% reduction in macrophages (P<0.01; Fig 2A,E) and an 82% reduction in CD3 positive T-lymphocytes (P<0.001; Fig. 2A,F) when compared to the mdx4cv controls. Thus, multiple indices of dystrophic histopathology in the mdx4cv mice were improved by the absence of desmin.

Increased expression of utrophin prevented necrosis of dko skeletal muscle fibers
We next examined whether the dystrophic pathology in the dko muscles was improved by an increase in utrophin expression. We examined the gastrocnemius muscle because of its distinct fiber-type distribution. Utrophin was restricted to the neuromuscular junctions in mature (11 week) wild-type and desmin−/− skeletal muscle fibers (Fig. 3A). Utrophin was expressed at low levels on the extrasynaptic sarcolemma in mdx4cv muscles (Fig. 3A), as previously described in mdx mice [21], [22], [36]. Utrophin was highly expressed in the dko extrasynaptic sarcolemma in some, but not all of the gastrocnemius muscle fibers (Fig. 3A). We next performed a titration of dko utrophin by western analyses to generate a non-linear regression to quantitate the changes in utrophin expression (Fig. S1). The significant increase in utrophin expression in mdx4cv mice compared to wild-type mice was confirmed by western analysis of total gastrocnemius muscle lysates (Fig. 3B; P<0.001). Importantly, we found a 2.54-fold increase in utrophin expression in the dko when compared with the mdx4cv controls (Fig. 3B; P<0.001). Because not all myofibers express utrophin in the dko we next quantitated the level of utrophin fluorescence intensity on the sarcolemma. We quantitated utrophin fluorescence in the wild-type sarcolemma as the negative control and the wild-type neuromuscular synapse as the peak of detection to ensure our quantitation is not beyond the limits of detection. The fluorescence intensity of utrophin was significantly increased in mdx4cv muscles compared to wild-type muscles (P<0.001; Fig. 3C). The utrophin fluorescence intensity increased by 2.86-fold in the dko sarcolemma when compared to the mdx4cv (P<0.001). To test whether this increase in fluorescence intensity in the dko reached therapeutic levels, we compared mdx:utrophin double knockout muscles treated with microutrophinΔR4–R21 using the same gastrocnemius muscles from our previous study [53], which demonstrated that microutrophinΔR4–R21 prevented skeletal muscle necrosis. We found that the sarcolemmal fluorescence intensity of utrophin was increased by 22% in the dko muscles when compared to the mdx:utrophin double knockout muscles expressing microutrophinΔR4–R21 (P<0.01). We found no change in utrophin mRNA in the gastrocnemius muscles of wild-type, desmin−/−, mdx4cv and dko mice, when measured by qPCR (Fig. 3D). Upregulation of utrophin was associated with a reduction in necrosis and regeneration in the dko, as only 9% of the fibers with extrasynaptic utrophin had central nuclei compared with 46% central nuclei in fibers without extrasynaptic utrophin (P<0.001; Fig. 3E). Thus, an increase in utrophin expression in a fraction of the dko muscle fibers prevented cycles of necrosis and regeneration.

Utrophin expression on the sarcolemma of maturing 1a, 2a and 2d/x fiber types
Utrophin expression is found on the sarcolemma of all developing wild-type muscle fibers and subsequently becomes restricted to the neuromuscular junctions [21], [54]. The prevention of skeletal muscle necrosis in the dko mice implied that the developmental loss of utrophin expression from the extrasynaptic sarcolemma did not occur. Furthermore, the expression of utrophin on the extrasynaptic sarcolemma of a portion of dko fibers suggests that utrophin may be expressed in certain muscle fiber types. To test this, we compared the expression of the utrophin A isoform relative to muscle fiber types at 3 weeks of age (Fig. 4). We found that utrophin was near absent from the extrasynaptic sarcolemma of wild-type gastrocnemius muscles by 3 weeks of age (Fig. 4). We found utrophin in the cytoplasm of a portion of the wild-type fast 2b fibers (Fig. 4). Furthermore, antibodies to the utrophin A isoform labeled blood vessels in wild-type muscles at 3 weeks of age (Fig. 4), but not at 11 weeks of age (Fig. 3), which was similar to the immunohistochemical staining pattern of the utrophin A isoform in humans [55]. Utrophin expression was absent from the extrasynaptic sarcolemma in most fast 2b fibers in desmin−/−, mdx4cv and dko muscles (Fig. 4). However, utrophin remained at low levels on the sarcolemma of 1a, 2a and 2d/x fiber types in desmin−/− and mdx4cv gastrocnemius muscles. The reduced utrophin expression in the extrasynaptic sarcolemma of mdx4cv muscles coincided with the appearance of patches of necrotic fibers (Fig. 4). In contrast, utrophin prevented skeletal muscle necrosis in the dko muscles by remaining on the extrasynaptic sarcolemma of maturing 1a, 2a and 2d/x fiber-types (Fig. 4). We next performed a titration of utrophin by western analyses of the 3-week-old dko muscles to generate a non-linear regression to quantitate the changes in utrophin expression (Fig. S2). We found a 29.6% increase in utrophin in the mdx4cv muscles compared to wild-type controls (Fig. 4B; P<0.05). Utrophin in the dko was increased by a further 60.9% compared to the mdx4cv muscles (P<0.001). Similar to 11 weeks of age (Fig. 3D), we found no change in the relative amounts of mRNA at 3 weeks of age when comparing all genotypes (Fig. 4C). Thus, utrophin expression was increased in the dko in a fiber-type specific manner to prevent necrosis.

Utrophin protects the sarcolemma of 1a, 2a and 2d/x dko skeletal muscle fiber types
To examine whether utrophin prevented necrosis by maintaining the integrity of the muscle membrane, we systemically delivered 200 µl of 1% (w/v) Evan's blue dye (EBD) into the mdx4cv and dko mice and looked for permeable skeletal muscle fibers (Fig. 5A). We found large patches of skeletal muscle fibers in the mdx4cv mice that were permeable to EBD (Fig. 5A), as previously described [40]. Utrophin was selectively expressed in the dko 1a, 2a and 2d/x fiber types and prevented the infiltration of EBD into these fibers (Fig. 5A). This correlated with an ∼80% reduction in centrally nucleated 1a, 2a and 2d/x fiber types in the dko compared to the corresponding mdx4cv muscles (P<0.001; Fig. 5B). Only the fast 2b fibers in the dko were permeable to EBD, which correlated with an ∼5 fold increase in centrally nucleated 2b fibers when compared with the other fiber-types in the dko (P<0.001; Fig. 5B). The total number of permeable fibers in the dko gastrocnemius muscles was ∼91% less than the mdx4cv muscles (Fig. 5C; P<0.001). Thus, utrophin prevented necrosis in the dko 1a, 2a and 2d/x fiber types by maintaining the integrity of the membrane.

We found a distinct separation of the fast 2b fibers from the 1a, 2a and 2d/x fiber types in the dko gastrocnemius muscles suggestive of a fiber-type switch in the dko muscles (Fig. 5A). We examined the fiber-type proportions in the smaller soleus muscle that contains all fiber-types in wild-type C57Bl/6mice. Analysis of fiber-type proportions in the soleus muscles at 11 weeks of age revealed a significant shift from the 2a fibers in the wild-type toward the slow 1a fibers in the desmin−/−, mdx4cv and dko muscles (P<0.001; Fig. S3). However, we found no significant change in fiber-type proportions when comparing between the desmin−/−, mdx4cv and dko muscles (Fig. S3). Thus, the skeletal muscle fiber-types were redistributed in the dko muscles, but we found no evidence of a fiber-type switch.
The increase in utrophin on the dko sarcolemma (Fig. 3C) may have resulted from reduced surface area of the 1a, 2a and 2d/x fibers compared with the corresponding mdx4cv muscles. However, the fiber area of 1a, 2a and 2d/x fiber types within the gastrocnemius muscles was unchanged when comparing wild-type, desmin−/−, mdx4cv and dko muscles (Fig. 5D). The fast 2b fibers in the desmin−/− and mdx4cv gastrocnemius were hypertrophic when compared to wild-type muscles (Fig. 5D). In contrast, the fast 2b fibers in the dko muscles were selectively atrophic. The desmin−/− muscles contained some smaller caliber fibers that increased the overall variability in muscle fiber area. The muscle fiber areas were highly variable in the mdx4cv muscles. Thus, the increase in utrophin expression on the dko sarcolemma did not result from changes in the average area of 1a, 2a and 2d/x fiber types.
Utrophin-independent mechanisms influence dystrophic pathology in the dko muscles
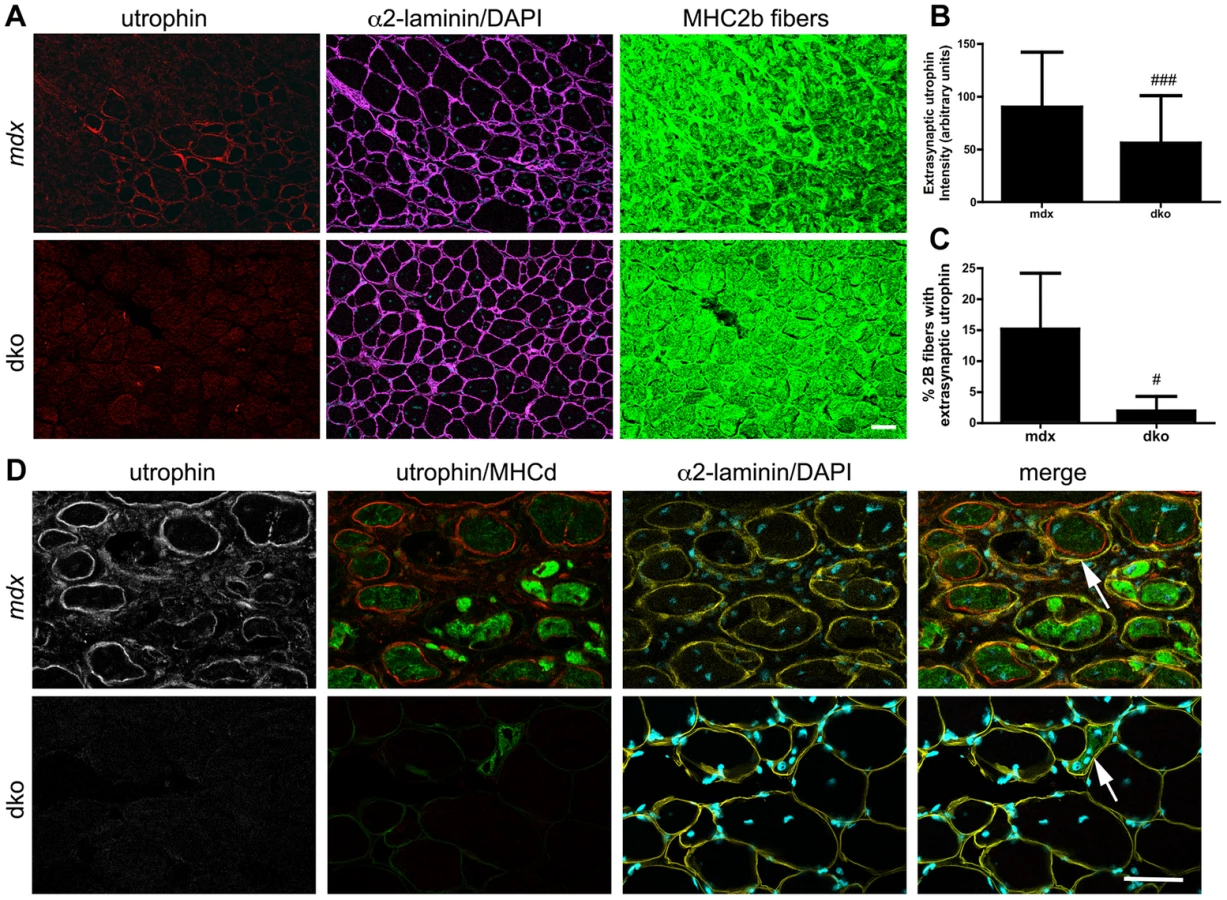
We also found a 36% reduction in the proportion of centrally nucleated fast 2b fibers in the dko when compared to the mdx4cv fast 2b fibers (P<0.01; Fig. 5B), which was consistent with the low level of central nuclei in utrophin negative fibers in the dko (46%) compared to all mdx4cv control fibers (76%) (Fig. 3E). To directly test whether utrophin-independent mechanisms were influencing the dystrophic pathology we performed a more detailed examination of the most superficial region of the gastrocnemius muscles that contained a near pure population of fast 2b fibers (Fig. 6). We found a significant reduction in the extrasynaptic utrophin expression on the fast 2b fibers in the dko compared with mdx4cv muscles (Fig. 6A,B; P<0.001). Moreover, there was a significant reduction in the number of fast 2b fibers expressing extrasynaptic utrophin in the dko when compared to the mdx4cv fast 2b fibers (Fig. 6A,C; P<0.05). Utrophin was expressed on the extrasynaptic sarcolemma in groups of regenerating mdx4cv 2b fibers as the myofibers expanded toward the basal lamina shell (Fig. 6D). Utrophin expression was maintained on the mdx4cv sarcolemma as the muscles matured and developmental myosin heavy chain dissipated (Fig. 6D). In contrast, examination of four dko gastrocnemius muscles revealed that the regenerating 2b fibers were directly enveloped by the basal lamina rather than utrophin (Fig. 6D). Together, these results demonstrate that utrophin expression was reduced in the extrasynaptic sarcolemma of dko fast 2b fibers. Thus, utrophin-independent mechanisms were also mitigating the dystrophic pathology of dko muscles.
Regenerative potential of skeletal muscles
The regenerative capacity of skeletal muscles depleted of desmin is profoundly impaired in cell culture [56], [57]. However, muscle generation in desmin−/− skeletal muscles in vivo is apparently normal [58]. Desmin−/− muscles injured with cardiotoxin can lead to persistent expression of developmental myosin heavy chain [59]. We found that regenerating fibers in uninjured gastrocnemius muscles were rare (up to 2 fibers) in the wild-type and desmin−/− mice (Fig. 7A,B). The mdx4cv muscles contained patches of regenerating fibers (Fig. 7). However, the dko muscles contained 47% fewer regenerating fibers than the mdx4cv muscles (P<0.01; Fig. 7A,B). To examine whether the regenerative capacity of muscles was impaired in the dko we delivered notexin to injure the gastrocnemius muscles and examined the muscles 4 and 6 days post injury. We found that regenerating fibers were expressing developmental myosin in wild-type, desmin−/−, mdx4cv and dko treated muscles at 4 days post injury (Fig. 7). At 6 days post injury we found that half (2 out of 4) of the injured wild-type muscles expressed developmental myosin (Fig. 7). Neither the desmin−/−, mdx4cv or dko muscles expressed developmental myosin 6 days post notexin injury (Fig. 7). We found no other overt changes in the regenerative capacity of the muscles when comparing the different strains of mice (Fig. 7). Thus, the improved dystrophic pathology in the dko muscles did not result from overt changes to the regenerative capacity of the skeletal muscles.

Utrophin concentrated β-dystroglycan in the sarcolemma, but not the nNOS, α-dystrobrevin and α1-syntrophin (NODS) complex in the dko mice
We next examined whether the significant increase in utrophin expression in the dko muscles restored the expression of β-dystroglycan and the NODS complex to the sarcolemma. Adjacent sections of gastrocnemius muscles revealed that β-dystroglycan and members of the NODS complex were concentrated within the sarcolemma of wild-type and desmin-/- skeletal muscles (Fig. 8A). The expression of β-dystroglycan and the NODS complex were increased in the desmin-/- mice (Fig. 8B,C), as previously described [60]. The expression of β-dystroglycan and the NODS complex at the sarcolemma of mdx4cv skeletal muscles were significantly diminished (Fig. 8), as previously described [40], [61] (Fig. 8). The increase in utrophin expression in the dko sarcolemma was accompanied by the increased concentration of β-dystroglycan (Fig. 8A). Immunoblots of whole muscle lysates revealed no significant difference in β-dystroglycan expression when comparing the wild-type or the mdx4cv controls with the dko (Fig. 8B,C). However, the expression of the NODS complex on the sarcolemma of dko muscles was not restored (Fig. 8).

Desmin can interact with α-dystrobrevin in the NODS complex indirectly through synemin, syncoilin and dysbindin [46]. Therefore, we examined whether desmin expression influenced the restoration of the NODS complex (Fig. S4). We found that utrophin was expressed on the sarcolemma of 4-week-old mdx4cv soleus muscles with minimal expression of the NODS complex (Fig. S4). Thus, the lack of the NODS complex on the sarcolemma of dko skeletal muscle fibers did not result from the absence of desmin.
Structural/functional changes in dko skeletal muscles
We next examined whether diaphragm function in the dko was influenced by structural defects within and around the muscles. We measured the specific contractile force of diaphragm strips in vitro. We found that the specific force production of the desmin−/− diaphragm was similar to wild-type at 11 weeks of age (Fig. 9A). In contrast, the specific force production of both mdx4cv and dko diaphragms were significantly diminished (Fig. 9A; P<0.001). Detailed histological analyses of the mdx4cv and dko diaphragms revealed that utrophin colocalized with α-sarcomeric actin in a costameric lattice (Fig. 9B). However, the alignment of α-sarcomeric actin in the dko was severely perturbed similar to the rectilinear pattern of utrophin (Fig. 9B). Electron microscopy analyses revealed that the sarcomeres aligned in wild-type muscles, but this alignment was impaired in desmin−/− muscles (Fig. 9C), as previously described [44], [45]. The alignment of sarcomeres in mdx4cv myofibers was similar to wild-type (Fig. 9C). However, the alignment of sarcomeres in the dko was severely impaired within and between individual muscle fibers (Fig. 9C). Gross histological analyses of the diaphragm revealed a 1.83-fold increase in the deposition of collagen in desmin−/− compared to wild-type (P<0.05; Fig. 9D,E). The mdx4cv diaphragms were significantly larger and contained proportionally more collagen than wild-type (4.05-fold increase; P<0.001) and desmin−/− controls (2.21-fold increase; P<0.001; Fig. 9D,E). The dko diaphragm was similar in size to the wild-type and desmin−/− controls (Fig. 9D), but contained proportionately similar amounts of collagen as the mdx4cv diaphragm (28% in the dko compared to 29% in mdx4cv; Fig. 9D,E). Together, these results demonstrate that the impaired respiratory function in the dko mice resulted, at least in part, from the impaired alignment of sarcomeres and deposition of collagen between the myofibers in the diaphragm.

Discussion
Increasing utrophin expression is a promising target for treatment of DMD [33]. While the downstream signaling pathways that influence utrophin expression are well described [33], [38], [39], the upstream mechanisms are less clear. Here, we found that perturbing the highly ordered structure of striated muscle by genetically deleting desmin from mdx4cv mice increased utrophin expression to levels that prevented skeletal muscle necrosis. We report a ∼2.5-fold increase in utrophin expression in the dko sarcolemma of 1a, 2a and 2d/x fiber types, which prevented necrosis by maintaining the integrity of the sarcolemma. Understanding the structural mechanisms that influence utrophin expression in the dko skeletal muscles may contribute to development of a therapy for DMD.
Potential mechanisms that influence utrophin expression in the dko muscles
We found that the onset of necrosis in the mdx4cv gastrocnemius muscles was coincident with the loss of utrophin expression from the maturing fibers (Fig. 4), as previously described [22], [36]. MyoD initiates skeletal muscle differentiation and maturation by activating many skeletal muscle genes and suppressing others [62]. MyoD activates the transcription of miR-206, which targets the utrophin mRNA for degradation leading to the loss of utrophin expression from the sarcolemma and its replacement by dystrophin [63]. Analysis of C2C12 cells suggests that several other miRNAs may also repress the expression of utrophin [64]. The loss of utrophin expression from the sarcolemma of maturing fibers was delayed in desmin−/− muscles and prevented in the dko muscles. It will be interesting to test whether desmin can influence the expression, trafficking, or function of miRNA's that knock-down utrophin expression.
An alternate possibility is that an early pulse in utrophin transcription [65] increased utrophin expression to levels that could overcome the knockdown effects of the miRNA's. Muscle contraction can change the shape of nuclei [66], which can change gene expression [67]–[69]. Desmin interacts with myonuclei via plectin and lamin A/C [70]–[72]. The myonuclei in the desmin−/− muscles remain oval shaped in response to muscle contraction [66]. This could potentially lead to the persistence of a developmental gene expression program that underlies the increased utrophin expression in the dko.
Utrophin is normally expressed at low levels on the sarcolemma of the slower oxidative fibers in wild-type mice [73]. Inducing the oxidative myogenic program can alleviate the dystrophic pathology in mdx mice by stimulating utrophin expression. For instance, activation of PGC1α [74]–[76], calcineurin A/NFAT [77]–[80], GA binding protein [74], Ca2+/calmodulin [81], AMP activated protein kinase [82], and the transcriptional activator PPARβ/δ [83] can each induce the slow oxidative program in mdx muscle and increase utrophin expression. Metabolic changes to the muscle can also influence utrophin expression [84]. While we found no significant change in fiber-types when comparing mdx, desmin−/− and dko soleus muscles (Fig. S3), we did find utrophin expression on the extrasynaptic sarcolemma of 1a, 2a and 2d/x fiber-types, but not in the fast 2b fibers. Thus, our results are consistent with the activation of the slower oxidative myogenic pathways that can induce utrophin expression.
The absence of desmin in stressed muscle is associated with a shift in the expression of muscle proteins to those found in slow-twitch fibers [85], [86]. These changes may be mediated in part by changes in the activity of calcineurin linked to alter myoplasmic Ca2+ levels, which could result from a loss of local protein kinase A (PKA) signaling linked to the loss of desmin. The copolymerization of desmin with synemin in the intermediate filament reticulum contributes to synemin's localization around Z-disks [87], [88]. As synemin is an A kinase anchor protein (AKAP) [89] the absence of desmin in the dko is likely to alter local PKA activity associated with the sarcomere. Calcium homeostasis is likely to be affected locally as PKA can regulate many channels and transporters essential for normal excitation-contraction coupling [90]–[93]. However, our finding that the mRNA levels for utrophin do not change in extracts of dko gastrocnemius muscle, compared to age-matched wild-type, desmin−/− and mdx4cv muscles, argue against this mechanism.
While there are various signaling pathways that can activate utrophin transcription in mdx mice, we found no changes in utrophin mRNA in the dko total gastrocnemius muscle lysates when compared to the mdx4cv, desmin−/− or wild-type muscles. The persistence of utrophin on the dko sarcolemma of maturing skeletal muscle fibers is consistent with increased utrophin stability and post-transcriptional mechanisms. Proteins experimentally over-expressed within the mdx extrasynaptic sarcolemma such as sarcospan [94], [95], cytotoxic T cell GalNac transferase [96] and biglycan [35] can stabilize utrophin to prevent skeletal muscle necrosis. RhoA, a small GTPase also increases utrophin expression without apparently influencing transcription [97]. Stabilizing RNA, a known function for the type III intermediate filament protein vimentin [98], is another potential mechanism that can increase utrophin expression without changing transcription [99], [100]. Desmin may also influence protein degradation pathways by trafficking lysosomes through the muscle via its interaction with myospryn [101], [102].
The lack of NODS expression may impair the therapeutic efficacy of utrophin in the dko
Increasing utrophin expression by increasing utrophin transcription or stabilization can restore the expression of the DGC to the sarcolemma [53], [96], [103]–[105], except for nNOS [106]. We found that utrophin was able to concentrate β-dystroglycan to the sarcolemma in the dko 1a, 2a and 2d/x fiber types. However, the expression of the NODS sub-complex was not restored in the dko muscles. nNOS influences blood flow to the skeletal muscles and can lead to hypoxic stress injury post-exercise [12]. However, the long-term effects of the lack of nNOS are difficult to predict considering Becker muscular dystrophy patients expressing truncated dystrophins can have a mild phenotype without restoring nNOS to the sarcolemma [12], [107]. The low level of α-dystrobrevin on the sarcolemma may have contributed to the low level of central nuclei in the dko mice, as α-dystrobrevin−/− mice have a mild dystrophy and residual expression of α2-dystrobrevin mitigates the dystrophic pathology in mdx muscles [13]. While α1-syntrophin is an important adapter protein that is required for the localization of nNOS and aquaporin to the sarcolemma of striated muscle [108], [109], its role in the pathogenesis of DMD is unclear. The low level of the NODS complex in the dko muscles did not result from the lack of desmin (Fig. S4). Thus, the low level of central nuclei (∼9%) in the dko muscle fibers with extrasynaptic utrophin likely resulted from the lack of desmin in combination with the reduced expression of the NODS complex from the extrasynaptic sarcolemma.
Utrophin-independent mechanisms influence dystrophic pathology
We found that the dystrophic pathology in the fast 2b fibers was also improved in the dko despite a significant reduction in extrasynaptic utrophin expression when compared with mdx4cv fast 2b fibers. Most striking was the fact that utrophin expression was reduced in the extrasynaptic sarcolemma of regenerating fast 2b fibers in the dko. However, we found no overt change in the regenerative capacity of the muscle stem cells in the dko gastrocnemius muscles injured with notexin. In contrast, Agbulut and colleagues found that desmin−/− muscles injured with cardiotoxin displayed persistent expression of developmental myosin, small caliber fibers and the infiltration of adipocytes [59]. Here, we found no evidence of increased adipocytes in the desmin−/− or dko muscles. Therefore, the discrepancy between our studies may have resulted from the different myotoxins. In any case, we found a significant reduction in the number of necrotic fibers in the dko supporting a mechanism that prevents dystrophy rather than influencing regeneration. Desmin is also likely to play a structural role in linking the contractile apparatus to the sarcolemma [47], [52], [101] and in regulating the passive mechanical properties of skeletal muscle [66], [110]. We found that utrophin could form costameric striations with α-sarcomeric actin in dko mice, but the rectilinear pattern was severely impaired. The exacerbated loss of sarcomere alignment in dko diaphragms suggests the absence of desmin and potentially the NODS complex could weaken the sarcomeric connections to the membrane. However, it is important to note that the specific force production of mdx4cv and dko diaphragms was comparable. The mdx4cv mice have a compensatory hypertrophy that can potentially maintain peak force production [111]. However, the dko diaphragms lack this cellular hypertrophy suggesting that the impaired diaphragm function could contribute to the respiratory distress and shortened lifespan. Considering the dko mice die prematurely from apparent cardiorespiratory failure, it is possible that reduced mobility in the cage could mitigate contraction-induced injury to the muscles. We are currently investigating whether desmin influences contraction-induced injury to the sarcolemma in mdx4cv muscles.
Conclusion
In conclusion, we report a significant increase in utrophin expression in dko skeletal muscles that prevented necrosis in a fiber-type specific manner. The fact that utrophin expression was elevated ∼2.5-fold on the dko sarcolemma when compared with mdx4cv muscles is of considerable interest for developing treatments for DMD [26]. Clearly, deleting desmin is not a therapeutic option for DMD as the dko mice die from apparent cardiorespiratory distress, but understanding the upstream mechanisms that influence utrophin expression may lead to novel treatment strategies for DMD. Furthermore, an utrophin-mediated therapy developed from the dko mice would treat all muscle fiber-types in the human as humans lack the fast 2b fiber types. Considering desmin functions to maintain the highly ordered structure of striated muscles [44], [45], it is likely that utrophin expression in the dko is initiated by changes to muscle structure/signaling relationships. We also found that utrophin-independent mechanisms were improving the dystrophic pathology in dko fast 2b fibers, which will be of interest for understanding the pathophysiology of DMD. Thus, the dko mice may provide new insights into the regulation of utrophin expression that are relevant for the treatment of DMD.
Materials and Methods
Mice and ethics statement
We utilized C57Bl/6 wild-type mice, desmin−/− mice, mdx4cv mice and mdx:desmin dko mice. All experiments were in accordance with the Institute of Animal Care and Use Committee of the University of Washington. The desmin−/− mice were a kind gift from Professor Yassemi Capetanaki. We generated the dko mice by first backcrossing the desmin−/− mice from the FVB strain to the wild-type C57Bl/6 strain for five generations (N5). The resulting desmin−/− mice on the C57Bl/6 strain were then inbred for at least four generations to obtain desmin−/− controls (>F4) or they were crossed with the mdx4cv strain on the C57Bl/6 background and inbred for at least four generations to obtain the dko mice (>F4). Therefore, the mice generated for this study were B6.FVB-Desmin and B6.FVB-Desmin-mdx4cv incipient congenic with ∼96.9% homozygosity with the C57Bl/6 background. We genotyped the mice using standard PCR for desmin and performed sequence analysis of the mdx4cv genomic DNA to avoid potential false positives as previously described [112]. The desmin−/− and dko mice were sacrificed if they lost body mass or exhibited signs of cardiorespiratory distress. Kaplan-Meyer survival analysis was performed with 16 desmin−/− male mice and 13 dko male mice.
Diaphragm function
The diaphragm physiology was performed as previously described [113]. Briefly, the diaphragm from wild-type (n = 6), desmin−/− (n = 5), mdx4cv (n = 5) and dko (n = 5) was placed in oxygenated KREBS (2 mM Ca2+, 24 mM NaHCO3, 137 mM NaCl, 5 mM KCl, 1 mM MgSO4, 1 mM NaH2PO4, D-Glucose). Strips of the diaphragm were dissected and the optimum length and peak tetanic contractile force was measured over 350 ms. Because the diaphragm strips vary in size, a direct comparison of peak contractile force is not plausible. After contraction, the diaphragm strip is weighed and specific force was calculated as peak tetanic force production × length × density (1.04) × pennation (1 for the diaphragm)/muscle mass.
Costamere analysis
Costamere analysis was performed as previously described [52]. Briefly, the mice were anaesthetized with 2,2,2-tribromoethanol (Sigma) and perfused with 2% paraformaldehyde (Electron microscopy sciences). The muscles were incubated in 2% paraformaldehyde for 2 hours at 4°C, then washed 3 times with 1× PBS, and incubated in 10% sucrose for 1 hour at 4°C, and then 20% sucrose overnight at 4°C. The muscles were then placed in cryovials and flash frozen in liquid N2. The frozen samples were placed on a frozen chuck with OCT and 40 µm thick sections were cut using a cryostat. The sections were immunostained with 1∶800 utrophin A polyclonal antibody (kind gift from Stanley Froehner) and 1∶500 α-sarcomeric actin monoclonal antibody (SIGMA). The thick sections were imaged using a Leica SP5 confocal microscope.
Electron microscopy
The electron microscopy was performed on longitudinal sections of diaphragm muscle as previously described [114].
Histology
Muscles were frozen directly in OCT cooled in 2-methylbutane in liquid N2. Ten micrometer transverse sections of skeletal muscles were stained with hematoxylin and eosin, alizarin red and Sirius red using manufacturer protocols (Electron Microscopy Sciences; Hatfeild, PA). The Sirius red staining of collagen was measured using the manufacturers protocols in Image J analyses software. Transverse frozen sections were also immunostained as previously described [40]. Briefly, the sections were incubated in blocking buffer (1% BSA, 0.05% Triton X-100 in 1× phosphate buffered saline (PBS)) for 30 minutes and immunostained with antibodies to desmin (1∶50; DAKO Corp), N-terminal dystrophin antibody (1∶800), utrophin (1∶800), α-dystrobrevin 1 (1 : 500), α-dystrobrevin 2 (1∶1000), α1-syntrophin (1∶500; the latter four antibodies were kind gifts from Stanley C. Froehner), β-dystroglycan (1∶100; Transduction Laboratories), MHCd (1∶40; Novocastra), α2-laminin (1∶800; Sigma) or nNOS (Zymed; 1∶100) for 1 hour. The sections were washed 3 times in 1× PBS for 10 minutes each and incubated in Alexa-488, Alexa-555, Alexa-594 or Alexa-647 secondary antibodies for 30 minutes (1∶800; Invitrogen). To label necrotic fibers we immunostained the muscles with mouse IgG1 antibodies conjugated to Alexa 488 (1∶800; Invitrogen). For labeling of acetylcholine receptors we incubated the sections in α-bungarotoxin conjugated to TRITC for 1 hour (1∶800; Invitrogen). The sections were washed 3 times for 10 minutes each and coverslipped with ProLong Gold mounting medium containing DAPI (Invitrogen). Muscle fiber typing was performed using conjugated monoclonal antibodies as previously described [115]. Sections were imaged with either a Leica SP5 confocal (Fig. 1, 3, 6), Nikon eclipse E1000 (Fig. 2, 7, 8) or an Olympus SZX16 dissection fluorescent microscope (Fig. 4, 5).
Quantitation of utrophin staining of muscle sections
Quantitation of maximal sarcolemmal utrophin fluorescence intensity was performed as previously described for dystrophin [116]. Briefly, gastrocnemius muscle sections and images were processed identically for quantitation. We utilized the FIJI analyses software to quantitate maximal fluorescence intensity. The utrophin fluorescence intensity on the wild-type sarcolemmal was used as a negative control and the utrophin fluorescence intensity at the wild-type synapse was used as the peak of detection. We drew a line across the images to ensure unbiased quantitation and measured the peak fluorescent intensity that coincided with extrasynaptic sarcolemma staining. The sarcolemmal utrophin fluorescence intensity from mdx4cv, dko and microutrophinΔR4–R21 treated mdx∶utrophin double knockout muscles all fell within these limits. The mean +/ − S.D. fluorescence intensity from n = 4 mice from 92 wild-type, 99 desmin−/−, 100 mdx4cv, 112 dko, and 77 microutrophinΔR4–R21 treated mdx:utrophin double knockout myofibers were compared.
Evans blue dye
The mdx4cv and dko mice (n = 4) were administered 200 µl of 0.22 µm filter sterilized 1% (w/v) EBD solution in HBSS intravenously by retro-orbital injection. Mice were sacrificed 3 hours after EBD administration. The gastrocnemius muscles were frozen in OCT in 2-methylbutane in liquid N2. Ten micrometer sections were cut and stained for utrophin (1∶800; kind gift from Stanley Froehner). Utrophin was labeled with Alexa-488 goat anti-rabbit secondary antibody (Invitrogen). The sections were viewed and imaged using the Olympus SZX16 dissection fluorescent microscope.
Muscle fiber regeneration
The gastrocnemius muscles of wild-type, desmin−/−, mdx4cv and dko (n = 8) were administered 30 µl of 1 µg/ml notexin in PBS at 11 weeks of age. The mice were sacrificed 4 days (n = 4) and 6 days (n = 4) post-injury. The gastrocnemius muscles were frozen in OCT. Ten micrometer sections were immunostained with α2-laminin (1∶800; Sigma) and developmental myosin heavy chain (1∶40; Novocastra) and directly compared to adjacent sections stained with hematoxylin and eosin. Considering monoclonal antibodies can label necrotic fibers, we defined regenerating fibers as those fibers that expressed developmental myosin heavy chain and contained centrally located nuclei.
Immunoblotting
Western blots were performed on whole muscle lysates as previously described [40]. Briefly, the gastrocnemius muscles of 3 and 11-week-old wild-type, desmin−/−, mdx4cv and dko (n = 6) were ground in liquid N2 and homogenized in extract buffer (50 mM Tris-HCl, 150 mM NaCl, 0.2% SDS, 24 mM Na Deoxycholate, 1% NP40, 47.6 mM Na Fluoride, 200 mM Na Orthovanadate, Roche). Protein concentration of whole muscle was determined by Coomassie Plus Bradford Assay (Pierce). Equal amounts of protein (10 µg) were resolved on a 4–12% SDS polyacrylamide gel. The blots were incubated in utrophin (1∶1000; kind gift from Stanley C. Froehner) overnight at 4°C. The α-sarcomeric actin primary antibody (1∶500; Sigma) was used as a loading control as its expression was unchanged when comparing the different strains of mice, as previously described for wild-type versus mdx4cv [40], [117]. We also loaded 20 µg of total protein to compare the expression of desmin, β-dystroglycan (1∶100; BD Transduction laboratories), α1-syntrophin (1∶500; kind gift from Stanley C. Froehner), pan α-dystrobrevin (1∶1000; BD Transduction laboratories) primary antibodies. The primary antibodies were detected with IgG HRP secondary antibodies (1∶6000; Jackson ImmunoResearch Labs). The blots were developed with ECL plus (Pierce) and scanned with the Storm 860 imaging system (Amersham Biosciences). The band intensity was measured using Image J software (NIH). The relative amount of utrophin in each blot was determined using a non-linear regression generated by a titration of utrophin from the dko from 1.25 µg up to 20 µg of total loaded protein and examined using the PRISM statistics software (Figures S1, S2; n = 4 for wild-type and desmin−/− and n = 8 for mdx4cv and dko samples).
Real time PCR
To isolate the RNA, approximately 20 µg of gastrocnemius muscle previously ground by mortar and pestle in liquid N2 was used to extract total RNA following manufacturers instructions (TRI Reagent, Molecular Research Center). We used gastrocnemius muscles from 11 week old (Fig. 3D) or 3 week old mice (Fig. 4C). The pelleted RNA was suspended in 50 µl nuclease free elution solution (Ambion, Austin, TX). Five µg of total RNA was treated with Turbo DNA-free (Ambion, Austin, TX) in order to remove trace amounts of contaminating DNA. The DNAase Treated RNA (0.5 µg) was diluted to 8 µl with nuclease free water followed by use of the SuperScript™ III First-Strand Synthesis kit (Invitrogen, Carlsbad, CA) to generate cDNA. Subsequently 2 µl of the cDNA was used for qPCR with utrophin primer-probe sets. The mouse utrophin primers sequences were: Forward 5′ - ACCAGCTGGACCGATGGA-3′, Reverse 5′ - CTCGTCCCAGTCGAAGAGATCT-3′, Probe 5′-6FAM - CGTTCAACGCCGTGCTCCACC-3′-BHQa1-Q. As a reference gene the oligonucleotide set was used to target the mouse Ywhaz gene sequence (Tyrosine 3-monooxygenase; [118]): Forward 5′ - GCTGGTGATGACAAGAAAGGAAT-3′, Reverse 5′ - GGTGTGTCGGCTGCATCTC-3′, Probe 5′-6FAM - TGGACCAGTCACAGCAAGCATACCAAGA-3′-BHQa1-Q.
Statistics
The data were compared using a one-way ANOVA with a Tukey post-test that compares all data sets with a Student's t-test. The relative amounts of utrophin in western analyses were determined using a non-linear regression generated from a titration of utrophin in the dko gastrocnemius muscles (from 1.25 µg–20 µg of total added protein). All data analyses were performed using the PRISM software.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. MendellJR, ShillingC, LeslieND, FlaniganKM, al-DahhakR, et al. (2012) Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol 71 : 304–313.
2. HoffmanEP, BrownRHJr, KunkelLM (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51 : 919–928.
3. KoenigM, HoffmanEP, BertelsonCJ, MonacoAP, FeenerC, et al. (1987) Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 50 : 509–517.
4. BhasinN, LawR, LiaoG, SaferD, EllmerJ, et al. (2005) Molecular extensibility of mini-dystrophins and a dystrophin rod construct. J Mol Biol 352 : 795–806.
5. MirzaA, SagathevanM, SahniN, ChoiL, MenhartN (2010) A biophysical map of the dystrophin rod. Biochim Biophys Acta 1804 : 1796–1809.
6. HendersonDM, LeeA, ErvastiJM (2010) Disease-causing missense mutations in actin binding domain 1 of dystrophin induce thermodynamic instability and protein aggregation. Proc Natl Acad Sci U S A 107 : 9632–9637.
7. ErvastiJM, SonnemannKJ (2008) Biology of the striated muscle dystrophin-glycoprotein complex. Int Rev Cytol 265 : 191–225.
8. ErvastiJM, CampbellKP (1991) Membrane organization of the dystrophin-glycoprotein complex. Cell 66 : 1121–1131.
9. KoenigM, KunkelLM (1990) Detailed analysis of the repeat domain of dystrophin reveals four potential hinge segments that may confer flexibility. J Biol Chem 265 : 4560–4566.
10. ErvastiJM, OhlendieckK, KahlSD, GaverMG, CampbellKP (1990) Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 345 : 315–319.
11. ErvastiJM (2007) Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta 1772 : 108–117.
12. KobayashiYM, RaderEP, CrawfordRW, IyengarNK, ThedensDR, et al. (2008) Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature 456 : 511–515.
13. GradyRM, GrangeRW, LauKS, MaimoneMM, NicholMC, et al. (1999) Role for alpha-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nat Cell Biol 1 : 215–220.
14. BrooksSV (1998) Rapid recovery following contraction-induced injury to in situ skeletal muscles in mdx mice. J Muscle Res Cell Motil 19 : 179–187.
15. DellorussoC, CrawfordRW, ChamberlainJS, BrooksSV (2001) Tibialis anterior muscles in mdx mice are highly susceptible to contraction-induced injury. J Muscle Res Cell Motil 22 : 467–475.
16. FaulknerJA, NgR, DavisCS, LiS, ChamberlainJS (2008) Diaphragm muscle strip preparation for evaluation of gene therapies in mdx mice. Clin Exp Pharmacol Physiol 35 : 725–729.
17. MoensP, BaatsenPH, MarechalG (1993) Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J Muscle Res Cell Motil 14 : 446–451.
18. PetrofBJ, ShragerJB, StedmanHH, KellyAM, SweeneyHL (1993) Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A 90 : 3710–3714.
19. BanksGB, ChamberlainJS (2008) The value of mammalian models for duchenne muscular dystrophy in developing therapeutic strategies. Curr Top Dev Biol 84 : 431–453.
20. LoveDR, HillDF, DicksonG, SpurrNK, BythBC, et al. (1989) An autosomal transcript in skeletal muscle with homology to dystrophin. Nature 339 : 55–58.
21. KhuranaTS, WatkinsSC, ChafeyP, ChellyJ, TomeFM, et al. (1991) Immunolocalization and developmental expression of dystrophin related protein in skeletal muscle. Neuromuscul Disord 1 : 185–194.
22. PonsF, NicholsonLV, RobertA, VoitT, LegerJJ (1993) Dystrophin and dystrophin-related protein (utrophin) distribution in normal and dystrophin-deficient skeletal muscles. Neuromuscul Disord 3 : 507–514.
23. DeconinckAE, RafaelJA, SkinnerJA, BrownSC, PotterAC, et al. (1997) Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 90 : 717–727.
24. GradyRM, TengH, NicholMC, CunninghamJC, WilkinsonRS, et al. (1997) Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: a model for Duchenne muscular dystrophy. Cell 90 : 729–738.
25. ChevronMP, EchenneB, DemailleJ (1994) Absence of dystrophin and utrophin in a boy with severe muscular dystrophy. N Engl J Med 331 : 1162–1163.
26. TinsleyJ, DeconinckN, FisherR, KahnD, PhelpsS, et al. (1998) Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat Med 4 : 1441–1444.
27. KarpatiG, CarpenterS, MorrisGE, DaviesKE, GuerinC, et al. (1993) Localization and quantitation of the chromosome 6-encoded dystrophin-related protein in normal and pathological human muscle. J Neuropathol Exp Neurol 52 : 119–128.
28. MizunoY, NonakaI, HiraiS, OzawaE (1993) Reciprocal expression of dystrophin and utrophin in muscles of Duchenne muscular dystrophy patients, female DMD-carriers and control subjects. J Neurol Sci 119 : 43–52.
29. KleopaKA, DrousiotouA, MavrikiouE, OrmistonA, KyriakidesT (2006) Naturally occurring utrophin correlates with disease severity in Duchenne muscular dystrophy. Hum Mol Genet 15 : 1623–1628.
30. VainzofM, Passos-BuenoMR, ManN, ZatzM (1995) Absence of correlation between utrophin localization and quantity and the clinical severity in Duchenne/Becker dystrophies. Am J Med Genet 58 : 305–309.
31. TaylorJ, MuntoniF, DubowitzV, SewryCA (1997) The abnormal expression of utrophin in Duchenne and Becker muscular dystrophy is age related. Neuropathol Appl Neurobiol 23 : 399–405.
32. FlaniganKM, CampbellK, ViolletL, WangW, GomezAM, et al. (2013) Anti-dystrophin T cell responses in Duchenne muscular dystrophy: prevalence and a glucocorticoid treatment effect. Hum Gene Ther 24 : 797–806.
33. FaircloughRJ, WoodMJ, DaviesKE (2013) Therapy for Duchenne muscular dystrophy: renewed optimism from genetic approaches. Nat Rev Genet 14 : 373–378.
34. MuellerC, ChulayJD, TrapnellBC, HumphriesM, CareyB, et al. (2013) Human Treg responses allow sustained recombinant adeno-associated virus-mediated transgene expression. J Clin Invest 123 : 5310–5318.
35. AmentaAR, YilmazA, BogdanovichS, McKechnieBA, AbediM, et al. (2011) Biglycan recruits utrophin to the sarcolemma and counters dystrophic pathology in mdx mice. Proc Natl Acad Sci U S A 108 : 762–767.
36. HelliwellTR, ManNT, MorrisGE, DaviesKE (1992) The dystrophin-related protein, utrophin, is expressed on the sarcolemma of regenerating human skeletal muscle fibres in dystrophies and inflammatory myopathies. Neuromuscul Disord 2 : 177–184.
37. WeirAP, MorganJE, DaviesKE (2004) A-utrophin up-regulation in mdx skeletal muscle is independent of regeneration. Neuromuscul Disord 14 : 19–23.
38. LjubicicV, BurtM, JasminBJ (2013) The therapeutic potential of skeletal muscle plasticity in Duchenne muscular dystrophy: phenotypic modifiers as pharmacologic targets. FASEB J 28 : 548–568.
39. MarshallJL, KwokY, McMorranBJ, BaumLG, Crosbie-WatsonRH (2013) The potential of sarcospan in adhesion complex replacement therapeutics for the treatment of muscular dystrophy. FEBS J 280 : 4210–4229.
40. BanksGB, CombsAC, ChamberlainJR, ChamberlainJS (2008) Molecular and cellular adaptations to chronic myotendinous strain injury in mdx mice expressing a truncated dystrophin. Hum Mol Genet 17 : 3975–3986.
41. BanksGB, JudgeLM, AllenJM, ChamberlainJS (2010) The polyproline site in hinge 2 influences the functional capacity of truncated dystrophins. PLoS Genet 6: e1000958.
42. LazaridesE, HubbardBD (1976) Immunological characterization of the subunit of the 100 A filaments from muscle cells. Proc Natl Acad Sci U S A 73 : 4344–4348.
43. LazaridesE, BalzerDRJr (1978) Specificity of desmin to avian and mammalian muscle cells. Cell 14 : 429–438.
44. LiZ, MericskayM, AgbulutO, Butler-BrowneG, CarlssonL, et al. (1997) Desmin is essential for the tensile strength and integrity of myofibrils but not for myogenic commitment, differentiation, and fusion of skeletal muscle. J Cell Biol 139 : 129–144.
45. MilnerDJ, WeitzerG, TranD, BradleyA, CapetanakiY (1996) Disruption of muscle architecture and myocardial degeneration in mice lacking desmin. J Cell Biol 134 : 1255–1270.
46. ErvastiJM (2003) Costameres: the Achilles' heel of Herculean muscle. J Biol Chem 278 : 13591–13594.
47. O'NeillA, WilliamsMW, ResneckWG, MilnerDJ, CapetanakiY, et al. (2002) Sarcolemmal organization in skeletal muscle lacking desmin: evidence for cytokeratins associated with the membrane skeleton at costameres. Mol Biol Cell 13 : 2347–2359.
48. LiZ, Colucci-GuyonE, Pincon-RaymondM, MericskayM, PourninS, et al. (1996) Cardiovascular lesions and skeletal myopathy in mice lacking desmin. Dev Biol 175 : 362–366.
49. WildingJR, SchneiderJE, SangAE, DaviesKE, NeubauerS, et al. (2005) Dystrophin - and MLP-deficient mouse hearts: marked differences in morphology and function, but similar accumulation of cytoskeletal proteins. FASEB J 19 : 79–81.
50. LewisC, OhlendieckK (2010) Proteomic profiling of naturally protected extraocular muscles from the dystrophin-deficient mdx mouse. Biochem Biophys Res Commun 396 : 1024–1029.
51. ChamberlainJS, MetzgerJ, ReyesM, TownsendD, FaulknerJA (2007) Dystrophin-deficient mdx mice display a reduced life span and are susceptible to spontaneous rhabdomyosarcoma. FASEB J 21 : 2195–2204.
52. LoveringRM, O'NeillA, MurielJM, ProsserBL, StrongJ, et al. (2011) Physiology, structure, and susceptibility to injury of skeletal muscle in mice lacking keratin 19-based and desmin-based intermediate filaments. Am J Physiol Cell Physiol 300: C803–813.
53. OdomGL, GregorevicP, AllenJM, FinnE, ChamberlainJS (2008) Microutrophin delivery through rAAV6 increases lifespan and improves muscle function in dystrophic dystrophin/utrophin-deficient mice. Mol Ther 16 : 1539–1545.
54. TakemitsuM, IshiuraS, KogaR, KamakuraK, ArahataK, et al. (1991) Dystrophin-related protein in the fetal and denervated skeletal muscles of normal and mdx mice. Biochem Biophys Res Commun 180 : 1179–1186.
55. SewryCA, NowakKJ, EhmsenJT, DaviesKE (2005) A and B utrophin in human muscle and sarcolemmal A-utrophin associated with tumours. Neuromuscul Disord 15 : 779–785.
56. LiH, CapetanakiY (1994) An E box in the desmin promoter cooperates with the E box and MEF-2 sites of a distal enhancer to direct muscle-specific transcription. EMBO J 13 : 3580–3589.
57. WeitzerG, MilnerDJ, KimJU, BradleyA, CapetanakiY (1995) Cytoskeletal control of myogenesis: a desmin null mutation blocks the myogenic pathway during embryonic stem cell differentiation. Dev Biol 172 : 422–439.
58. CapetanakiY, MilnerDJ, WeitzerG (1997) Desmin in muscle formation and maintenance: knockouts and consequences. Cell Struct Funct 22 : 103–116.
59. AgbulutO, LiZ, PerieS, LudoskyMA, PaulinD, et al. (2001) Lack of desmin results in abortive muscle regeneration and modifications in synaptic structure. Cell Motil Cytoskeleton 49 : 51–66.
60. KoniecznyP, FuchsP, ReipertS, KunzWS, ZeoldA, et al. (2008) Myofiber integrity depends on desmin network targeting to Z-disks and costameres via distinct plectin isoforms. J Cell Biol 181 : 667–681.
61. OhlendieckK, CampbellKP (1991) Dystrophin-associated proteins are greatly reduced in skeletal muscle from mdx mice. J Cell Biol 115 : 1685–1694.
62. FongAP, TapscottSJ (2013) Skeletal muscle programming and re-programming. Curr Opin Genet Dev 23 : 568–573.
63. RosenbergMI, GeorgesSA, AsawachaicharnA, AnalauE, TapscottSJ (2006) MyoD inhibits Fstl1 and Utrn expression by inducing transcription of miR-206. J Cell Biol 175 : 77–85.
64. BasuU, LozynskaO, MoorwoodC, PatelG, WiltonSD, et al. (2011) Translational regulation of utrophin by miRNAs. PLoS One 6: e29376.
65. GramoliniAO, JasminBJ (1999) Expression of the utrophin gene during myogenic differentiation. Nucleic Acids Res 27 : 3603–3609.
66. ShahSB, DavisJ, WeislederN, KostavassiliI, McCullochAD, et al. (2004) Structural and functional roles of desmin in mouse skeletal muscle during passive deformation. Biophys J 86 : 2993–3008.
67. MarklundN, BareyreFM, RoyoNC, ThompsonHJ, MirAK, et al. (2007) Cognitive outcome following brain injury and treatment with an inhibitor of Nogo-A in association with an attenuated downregulation of hippocampal growth-associated protein-43 expression. J Neurosurg 107 : 844–853.
68. VerganiL, GrattarolaM, NicoliniC (2004) Modifications of chromatin structure and gene expression following induced alterations of cellular shape. Int J Biochem Cell Biol 36 : 1447–1461.
69. ChenCS, MrksichM, HuangS, WhitesidesGM, IngberDE (1997) Geometric control of cell life and death. Science 276 : 1425–1428.
70. NikolovaV, LeimenaC, McMahonAC, TanJC, ChandarS, et al. (2004) Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. J Clin Invest 113 : 357–369.
71. FavreB, SchneiderY, LingasamyP, BouameurJE, BegreN, et al. (2011) Plectin interacts with the rod domain of type III intermediate filament proteins desmin and vimentin. Eur J Cell Biol 90 : 390–400.
72. KoniecznyP, WicheG (2008) Muscular integrity—a matter of interlinking distinct structures via plectin. Adv Exp Med Biol 642 : 165–175.
73. GramoliniAO, BelangerG, ThompsonJM, ChakkalakalJV, JasminBJ (2001) Increased expression of utrophin in a slow vs. a fast muscle involves posttranscriptional events. Am J Physiol Cell Physiol 281: C1300–1309.
74. AngusLM, ChakkalakalJV, MejatA, EiblJK, BelangerG, et al. (2005) Calcineurin-NFAT signaling, together with GABP and peroxisome PGC-1{alpha}, drives utrophin gene expression at the neuromuscular junction. Am J Physiol Cell Physiol 289: C908–917.
75. HollingerK, Gardan-SalmonD, SantanaC, RiceD, SnellaE, et al. (2013) Rescue of dystrophic skeletal muscle by PGC-1alpha involves restored expression of dystrophin-associated protein complex components and satellite cell signaling. Am J Physiol Regul Integr Comp Physiol 305: R13–23.
76. SelsbyJT, MorineKJ, PendrakK, BartonER, SweeneyHL (2012) Rescue of dystrophic skeletal muscle by PGC-1alpha involves a fast to slow fiber type shift in the mdx mouse. PLoS One 7: e30063.
77. ChakkalakalJV, StocksleyMA, HarrisonMA, AngusLM, Deschenes-FurryJ, et al. (2003) Expression of utrophin A mRNA correlates with the oxidative capacity of skeletal muscle fiber types and is regulated by calcineurin/NFAT signaling. Proc Natl Acad Sci U S A 100 : 7791–7796.
78. ChakkalakalJV, HarrisonMA, CarbonettoS, ChinE, MichelRN, et al. (2004) Stimulation of calcineurin signaling attenuates the dystrophic pathology in mdx mice. Hum Mol Genet 13 : 379–388.
79. St-PierreSJ, ChakkalakalJV, KolodziejczykSM, KnudsonJC, JasminBJ, et al. (2004) Glucocorticoid treatment alleviates dystrophic myofiber pathology by activation of the calcineurin/NF-AT pathway. FASEB J 18 : 1937–1939.
80. StupkaN, SchertzerJD, Bassel-DubyR, OlsonEN, LynchGS (2007) Calcineurin-A alpha activation enhances the structure and function of regenerating muscles after myotoxic injury. Am J Physiol Regul Integr Comp Physiol 293: R686–694.
81. MichelRN, ChinER, ChakkalakalJV, EiblJK, JasminBJ (2007) Ca2+/calmodulin-based signalling in the regulation of the muscle fibre phenotype and its therapeutic potential via modulation of utrophin A and myostatin expression. Appl Physiol Nutr Metab 32 : 921–929.
82. LjubicicV, MiuraP, BurtM, BoudreaultL, KhogaliS, et al. (2011) Chronic AMPK activation evokes the slow, oxidative myogenic program and triggers beneficial adaptations in mdx mouse skeletal muscle. Hum Mol Genet 20 : 3478–3493.
83. MiuraP, ChakkalakalJV, BoudreaultL, BelangerG, HebertRL, et al. (2009) Pharmacological activation of PPARbeta/delta stimulates utrophin A expression in skeletal muscle fibers and restores sarcolemmal integrity in mature mdx mice. Hum Mol Genet 18 : 4640–4649.
84. MiuraP, AndrewsM, HolcikM, JasminBJ (2008) IRES-mediated translation of utrophin A is enhanced by glucocorticoid treatment in skeletal muscle cells. PLoS One 3: e2309.
85. MeyerGA, LieberRL (2012) Skeletal muscle fibrosis develops in response to desmin deletion. Am J Physiol Cell Physiol 302: C1609–1620.
86. MeyerGA, SchenkS, LieberRL (2013) Role of the cytoskeleton in muscle transcriptional responses to altered use. Physiol Genomics 45 : 321–331.
87. PriceMG, LazaridesE (1983) Expression of intermediate filament-associated proteins paranemin and synemin in chicken development. J Cell Biol 97 : 1860–1874.
88. BellinRM, SernettSW, BeckerB, IpW, HuiattTW, et al. (1999) Molecular characteristics and interactions of the intermediate filament protein synemin. Interactions with alpha-actinin may anchor synemin-containing heterofilaments. J Biol Chem 274 : 29493–29499.
89. RussellMA, LundLM, HaberR, McKeeganK, CianciolaN, et al. (2006) The intermediate filament protein, synemin, is an AKAP in the heart. Arch Biochem Biophys 456 : 204–215.
90. OzawaT (2010) Modulation of ryanodine receptor Ca2+ channels (Review). Mol Med Rep 3 : 199–204.
91. DanilaCI, HamiltonSL (2004) Phosphorylation of ryanodine receptors. Biol Res 37 : 521–525.
92. FullerMD, EmrickMA, SadilekM, ScheuerT, CatterallWA (2010) Molecular mechanism of calcium channel regulation in the fight-or-flight response. Sci Signal 3: ra70.
93. GramoliniAO, TrivieriMG, OuditGY, KislingerT, LiW, et al. (2006) Cardiac-specific overexpression of sarcolipin in phospholamban null mice impairs myocyte function that is restored by phosphorylation. Proc Natl Acad Sci U S A 103 : 2446–2451.
94. PeterAK, MarshallJL, CrosbieRH (2008) Sarcospan reduces dystrophic pathology: stabilization of the utrophin-glycoprotein complex. J Cell Biol 183 : 419–427.
95. MarshallJL, HolmbergJ, ChouE, OcampoAC, OhJ, et al. (2012) Sarcospan-dependent Akt activation is required for utrophin expression and muscle regeneration. J Cell Biol 197 : 1009–1027.
96. NguyenHH, JayasinhaV, XiaB, HoyteK, MartinPT (2002) Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc Natl Acad Sci U S A 99 : 5616–5621.
97. Gauthier-RouviereC, Bonet-KerracheA (2009) RhoA leads to up-regulation and relocalization of utrophin in muscle fibers. Biochem Biophys Res Commun 384 : 322–328.
98. ChallaAA, StefanovicB (2011) A novel role of vimentin filaments: binding and stabilization of collagen mRNAs. Mol Cell Biol 31 : 3773–3789.
99. MoorwoodC, SoniN, PatelG, WiltonSD, KhuranaTS (2013) A cell-based high-throughput screening assay for posttranscriptional utrophin upregulation. J Biomol Screen 18 : 400–406.
100. ChakkalakalJV, MiuraP, BelangerG, MichelRN, JasminBJ (2008) Modulation of utrophin A mRNA stability in fast versus slow muscles via an AU-rich element and calcineurin signaling. Nucleic Acids Res 36 : 826–838.
101. CapetanakiY, BlochRJ, KouloumentaA, MavroidisM, PsarrasS (2007) Muscle intermediate filaments and their links to membranes and membranous organelles. Exp Cell Res 313 : 2063–2076.
102. LiW, ZhangQ, OisoN, NovakEK, GautamR, et al. (2003) Hermansky-Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1). Nat Genet 35 : 84–89.
103. RafaelJA, TinsleyJM, PotterAC, DeconinckAE, DaviesKE (1998) Skeletal muscle-specific expression of a utrophin transgene rescues utrophin-dystrophin deficient mice. Nat Genet 19 : 79–82.
104. SonnemannKJ, Heun-JohnsonH, TurnerAJ, BaltgalvisKA, LoweDA, et al. (2009) Functional substitution by TAT-utrophin in dystrophin-deficient mice. PLoS Med 6: e1000083.
105. MercadoML, AmentaAR, HagiwaraH, RafiiMS, LechnerBE, et al. (2006) Biglycan regulates the expression and sarcolemmal localization of dystrobrevin, syntrophin, and nNOS. FASEB J 20 : 1724–1726.
106. LiD, BarejaA, JudgeL, YueY, LaiY, et al. (2010) Sarcolemmal nNOS anchoring reveals a qualitative difference between dystrophin and utrophin. J Cell Sci 123 : 2008–2013.
107. EnglandSB, NicholsonLV, JohnsonMA, ForrestSM, LoveDR, et al. (1990) Very mild muscular dystrophy associated with the deletion of 46% of dystrophin. Nature 343 : 180–182.
108. AlbrechtDE, FroehnerSC (2002) Syntrophins and dystrobrevins: defining the dystrophin scaffold at synapses. Neurosignals 11 : 123–129.
109. AdamsME, MuellerHA, FroehnerSC (2001) In vivo requirement of the alpha-syntrophin PDZ domain for the sarcolemmal localization of nNOS and aquaporin-4. J Cell Biol 155 : 113–122.
110. ShahSB, LoveJM, O'NeillA, LoveringRM, BlochRJ (2012) Influences of desmin and keratin 19 on passive biomechanical properties of mouse skeletal muscle. J Biomed Biotechnol 2012 : 704061.
111. Kornegay JN, Childers MK, Bogan DJ, Bogan JR, Nghiem P, et al.. (2012) The paradox of muscle hypertrophy in muscular dystrophy. Phys Med Rehabil Clin N Am 23 : 149–172, xii.
112. BanksGB, CombsAC, ChamberlainJS (2010) Sequencing protocols to genotype mdx, mdx(4cv), and mdx(5cv) mice. Muscle Nerve 42 : 268–270.
113. GregorevicP, AllenJM, MinamiE, BlankinshipMJ, HaraguchiM, et al. (2006) rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat Med 12 : 787–789.
114. BanksGB, ChamberlainJS, FroehnerSC (2009) Truncated dystrophins can influence neuromuscular synapse structure. Mol Cell Neurosci 40 : 433–441.
115. GregorevicP, MeznarichNA, BlankinshipMJ, CrawfordRW, ChamberlainJS (2008) Fluorophore-labeled myosin-specific antibodies simplify muscle-fiber phenotyping. Muscle Nerve 37 : 104–106.
116. BanksGB, GregorevicP, AllenJM, FinnEE, ChamberlainJS (2007) Functional capacity of dystrophins carrying deletions in the N-terminal actin-binding domain. Hum Mol Genet 16 : 2105–2113.
117. HanftLM, RybakovaIN, PatelJR, Rafael-FortneyJA, ErvastiJM (2006) Cytoplasmic gamma-actin contributes to a compensatory remodeling response in dystrophin-deficient muscle. Proc Natl Acad Sci U S A 103 : 5385–5390.
118. GubernC, HurtadoO, RodriguezR, MoralesJR, RomeraVG, et al. (2009) Validation of housekeeping genes for quantitative real-time PCR in in-vivo and in-vitro models of cerebral ischaemia. BMC Mol Biol 10 : 57.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Early Back-to-Africa Migration into the Horn of Africa
- PINK1-Mediated Phosphorylation of Parkin Boosts Parkin Activity in
- OsHUS1 Facilitates Accurate Meiotic Recombination in Rice
- Ancient DNA Analysis of 8000 B.C. Near Eastern Farmers Supports an Early Neolithic Pioneer Maritime Colonization of Mainland Europe through Cyprus and the Aegean Islands
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy