
Defective iA37 Modification of Mitochondrial and Cytosolic tRNAs Results from Pathogenic Mutations in TRIT1 and Its Substrate tRNA
Mitochondrial disorders are clinically diverse, and identifying the underlying genetic mutations is technically challenging due to the large number of mitochondrial proteins. Using high-throughput sequencing technology, we identified a disease-causing mutation in the TRIT1 gene. This gene encodes an enzyme, tRNA isopentenyltransferase, that adds an N6-isopentenyl modification to adenosine-37 (i6A37) in a small number of tRNAs, enabling them to function correctly during the synthesis of essential mitochondrial proteins. We show that this mutation leads to severe deficiency of tRNA-i6A37 in the patient's cells that can be rescued by introduction of the wild-type TRIT1 protein. A deficiency in oxidative phosphorylation, the process by which energy (ATP) is generated in the mitochondria, leads to a mitochondrial disease presentation. Introducing the mutant protein into model yeast species and measuring the resulting impairment provided further evidence of the pathogenic effect of the mutation. Additional studies investigating a previously reported pathogenic mutation in a mitochondrial tRNA gene demonstrated that a mutation in a substrate of TRIT1 can also cause a loss of the modification, providing evidence of a new mechanism causing mitochondrial disease in humans.
Published in the journal:
. PLoS Genet 10(6): e32767. doi:10.1371/journal.pgen.1004424
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004424
Summary
Mitochondrial disorders are clinically diverse, and identifying the underlying genetic mutations is technically challenging due to the large number of mitochondrial proteins. Using high-throughput sequencing technology, we identified a disease-causing mutation in the TRIT1 gene. This gene encodes an enzyme, tRNA isopentenyltransferase, that adds an N6-isopentenyl modification to adenosine-37 (i6A37) in a small number of tRNAs, enabling them to function correctly during the synthesis of essential mitochondrial proteins. We show that this mutation leads to severe deficiency of tRNA-i6A37 in the patient's cells that can be rescued by introduction of the wild-type TRIT1 protein. A deficiency in oxidative phosphorylation, the process by which energy (ATP) is generated in the mitochondria, leads to a mitochondrial disease presentation. Introducing the mutant protein into model yeast species and measuring the resulting impairment provided further evidence of the pathogenic effect of the mutation. Additional studies investigating a previously reported pathogenic mutation in a mitochondrial tRNA gene demonstrated that a mutation in a substrate of TRIT1 can also cause a loss of the modification, providing evidence of a new mechanism causing mitochondrial disease in humans.
Introduction
Mitochondrial diseases are characterised by biochemical defects in oxidative phosphorylation (OXPHOS) enzyme activity and arise as a consequence of nuclear - or mitochondrial-encoded gene mutations [1]. Generalised disorders of mitochondrial protein synthesis resulting in OXPHOS defects are increasingly reported as causing a clinically heterogeneous group of neonatal and infantile mitochondrial disease presentations associated with isolated or multi-organ involvement [2]. The advent of whole exome capture and sequencing technologies has revolutionised the molecular diagnosis of this patient group [3]; the molecular disease mechanism can implicate nuclear gene products involved in mitochondrial DNA (mtDNA) replication, synthesis and repair [4], mitochondrial aminoacyl tRNA synthetases [5], mitochondrial translation elongation and release factors [6], structural ribosomal proteins and assembly factors, and enzymes involved in mt-RNA modification [7].
Post-transcriptional modification of tRNAs is crucial for folding, stability and function in deciphering the genetic code during translation. Modifications of cytosolic-tRNAs (cy-tRNA) and mt-tRNAs occur notably at nucleotide positions 34 and 37 in the anticodon loop (ACL), serving to promote translational fidelity and efficiency by optimising codon-anticodon fit within the ribosome [8]. The homologous tRNA isopentenyltransferases (IPTases) are conserved from bacteria to humans and introduce an evolutionarily ancient modification, an isopentenyl group onto N6 of adenine at position 37 (i6A37). In bacteria, i6A37 is further modified by methylthiolation to ms2i6A37 which uses its methyl-sulphur group to stabilise the intrinsically weak A–U pairing between anticodon A36 and the first base of UNN codons [9]. However, the methylthiolation enzymes are not present in eukaryotes, leaving i6A37 without further modification.
To date, functional analysis in eukaryotes comes from studies in yeast which have shown that i6A37 promotes translational efficiency and fidelity in a codon-specific manner cognate with the i6A37-tRNAs [10]. The presence of i6A37 increases the specific activity of a tRNA for its codon about four-fold in S. pombe [10]. Prior work on orthologues including MiaA (E. coli), Tit1 (S. pombe), Mod5 (S. cerevisiae), and TRIT1 (human) has revealed specificity for subsets of cy - and mt-tRNAs that bear the single-stranded anticodon loop recognition sequence, ‘A36-A37-A38’, although this motif alone is not always sufficient ([11] and refs therein). However, it has also become clear that due to sequence variability in the tRNAs and the specificities of the transferases, different species contain different subsets of i6A37-modified tRNAs [10], [12]. Determining the subsets of specific mRNAs that are sensitive to i6A37 deficiency and how this contributes to phenotype is a contemporary challenge [10].
We used whole exome sequencing to identify a homozygous p.Arg323Gln mutation in the TRIT1 gene that segregates within a consanguineous UK-Pakistani family in which affected children present with encephalopathy and myoclonic epilepsy due to multiple OXPHOS deficiencies in skeletal muscle. We confirm that this mutation is responsible for a severe deficiency in the i6A37 content of cy - and mt-tRNAs, as it can be reversed by rescue with wild type TRIT1 in the patient's fibroblasts. We show that TRIT1 is targeted to mitochondria and provide evidence in both humans and yeast that this gene is required for efficient mitochondrial function. Furthermore, we have demonstrated that a previously-reported pathogenic A38G mutation of mt-tRNASer(UCN), causes i6A37 deficiency, strengthening the conclusion that TRIT1-related human disease can arise from mutation of either the enzyme or its tRNA substrate.
Results
Histochemical and biochemical analyses identified a generalised disorder of mitochondrial protein synthesis
We investigated a family with clinical indications of mitochondrial disease in two affected children. A skeletal muscle biopsy (subject II-3, detailed clinical report in Text S1) showed normal morphology and a mosaic pattern of cytochrome c oxidase (COX) deficiency (Figure 1A), which can be associated with mutations in nuclear genes involved with mtDNA translation and maintenance or mtDNA mutations. We also observed biochemical evidence of a mitochondrial respiratory chain deficiency involving complexes I (10% of controls) and IV (60% of controls), with apparent sparing of complex II and III activity (Figure 1B). Together these data confirmed the presence of a combined OXPHOS deficiency.
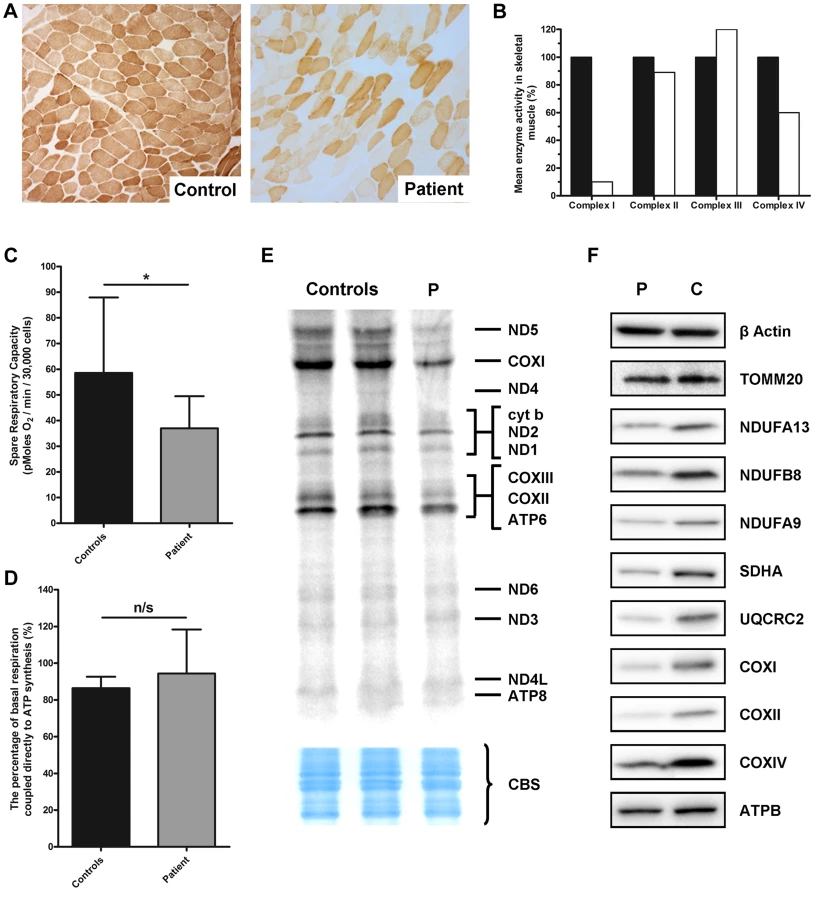
Micro-scale oxygraphy analysis provided evidence of mitochondrial respiratory dysfunction in patient fibroblasts (Figure 1C and D). Basal oxygen consumption rate (OCR) was significantly decreased (P = 0.0451) in the patient compared to controls, as was maximal OCR (P = 0.0078). The spare respiratory capacity (SRC) (Figure 1C) was significantly reduced (P = 0.0102) in patient cells whilst the coupling efficiency of ATP synthesis to respiration, a measure of proton leak, was unchanged (Figure 1D).
In vitro metabolic labelling of mitochondrial translation identified a generalised decrease in the synthesis of mtDNA-encoded proteins with particularly notable loss of ND1 and ND5 of Complex I, CYTB of Complex III and COXI, COXII and COXIII of Complex IV (Figure 1E). This was supported by immunoblotting, which revealed decreased steady-state levels of mtDNA-encoded OXPHOS components in the patient fibroblasts (Figure 1F) and a moderate decrease in SDHA protein levels which was surprising given that complex II activity in skeletal muscle was normal (Figure 1F). Levels of TOMM20 were unchanged in patient cells confirming a specific defect of OXPHOS protein synthesis rather than general loss of mitochondrial proteins.
Exome sequencing identified a mutation in TRIT1
Having excluded mtDNA rearrangements, copy number abnormalities and point mutations (Table S1), we employed whole exome sequencing of both affected siblings to elucidate a potential genetic basis of the defect. This analysis identified 3970 novel homozygous protein altering variants shared between siblings (Table S2), of which 40 were rare (Minor Allele Frequency <0.01). Based on predicted mitochondrial localisation and an autosomal recessive inheritance pattern, variant filtering identified a single candidate homozygous missense mutation shared by both affected siblings in TRIT1 (c.968G>A predicting p.Arg323Gln). This mutation was predicted to be pathogenic by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) with a score of 0.999. Targeted resequencing of the proband and familial relatives confirmed the homozygous mutation in the affected siblings and demonstrated disease segregation as both parents and an unaffected sibling were heterozygous carriers (Figure 2A). Importantly, the TRIT1 c.968G>A variant was not observed by the 1000 Genomes Project, the NHLBI Exome Sequencing Project nor a panel of 120 ethnically-matched control chromosomes (data not shown).

The p.Arg323Gln mutation occurs in exon 8 of the TRIT1 gene, which also has a putative mitochondrial targeting sequence in exon 1 and a matrin-type zinc finger domain contributed by exons 10 and 11 (Figure 2B). Mitochondrial targeting of TRIT1 is supported by prediction using the freely available online tool, MitoProt II (http://ihg.gsf.de/ihg/mitoprot.html) [13], with a confidence of 94%. Both cytosolic and mitochondrial localization is predicted by other available databases, as is also the case for its homologs Mod5, Tit1, and GRO-1. Indeed, immunoblotting of whole cell extracts and isolated mitochondrial subfractions confirmed that TRIT1 was present in the cytosolic fraction (Figure 2C, lane 2) and also detectable in proteinase K-treated mitoplasts (lane 6), consistent with the enzyme being present in the mitochondrial matrix, where tRNA molecules and the translation apparatus are active during protein synthesis. However, at this stage we cannot exclude the possibility that a significant fraction of the cytosolic portion of TRIT1 may be associated with the outer mitochondrial membrane.
The corresponding position of the affected amino acid, p.Arg323, is occupied by a basic side chain in all homologues from a range of species (Figure 2D), whereas glutamine at this position in the proband is polar but uncharged. Based on the available Mod5-tRNA co-crystal structure [14] (Figure 2E–G), the position and chemical nature of the mutation was not expected to affect mitochondrial localization, general solubility or gross structural alterations of the enzyme. In Mod5 this position is occupied by p.Lys294 whose basic side chain extends from an α-helix that lies adjacent to, but pointing away from, the catalytic site containing A37 [14]. Both this and adjacent conserved basic side chains comprise part of a series of residues on the same side of the α-helix, including Arg298 (also arginine in TRIT1) which are involved in binding the phosphate groups of nucleotides 27–29 of the AC stem (Figure 2E–G) [14]. Based on this we suspected that the mutation would not impair catalytic activity per se but might impair proper binding of the enzyme to its tRNA substrates. However, since this mutation would appear to affect only one of a series of contacts with the surface of the tRNA backbone, it was not necessarily expected to cause a severe deficiency of isopentenyltransferase activity.
The TRIT1 p.Arg323Gln mutation severely impairs i6A modification activity
Purified recombinant TRIT1 was previously used to examine i6A modification activity in vitro using an established assay employing synthetic RNA that matches the anticodon stem loop (ASL) of a substrate tRNA [11]. By this assay, the isopentenyl group of DMAPP (dimethylallyl pyrophosphate) is transferred to N6 of A37 in substrate tRNAs by the IPTase TRIT1 ([11] and references therein). His-tagged TRIT1-WT and His-tagged TRIT1-Arg323Gln were purified from E. coli in parallel and compared by gel electrophoresis (Figure 3A). The modification activity of mutant TRIT1 was negligible relative to that of wild-type TRIT1 using the standard assay (Figure 3B). We reasoned that if the mutation led to decreased affinity for its substrate, as suggested by the co-crystal structure of Mod5-tRNA, we might be able to obtain activity by increasing substrate concentration. Some activity of mutant TRIT1 could indeed be observed by increasing the concentration of the RNA substrate 4-fold, but even under these conditions it was much less active than the wild-type TRIT1 (Figure 3C). Although increasing the concentrations of enzyme and substrate further was technically-limited in these reactions, the data suggest that higher activity might be achieved with higher concentrations.

Mutant TRIT1 fails to complement activity of a codon-specific cytoplasmic translational reporter or a mitochondrial respiratory growth defect in a tit1-knockout strain of S. pombe
A previously characterized S. pombe strain with a deletion of the tit1+ gene (a homologue of TRIT1), yNB5, exhibits two distinct phenotypes [10], [11] that were examined for their sensitivity to wild-type TRIT1 and the p.Arg323Gln TRIT1 mutant. The first phenotype is manifested by a red-white colony colour assay, that monitors tRNASer(UCA)-mediated suppression of a UGA nonsense mutation in ade6-704. This in vivo assay reports on the codon-specific translational activity of the suppressor-tRNASer(UCA) to decode the ade6-704 UGA codon, which was previously shown to be highly dependent on i6A37 [10], [11]. In this assay, absence of i6A37 decreases the translational activity of the suppressor-tRNA and the cells accumulate red pigment [11]. The yNB5 strain (tit1-Δ) transformed with the empty vector is red as expected, whilst the yYH1 strain (tit1+) is white. yNB5 transformed with either wild-type TRIT1 or wild-type tit1+ are white, indicating successful complementation, whilst yNB5 transformed with mutant TRIT1 is red (Figure 3D).
The second phenotype of the yNB5 strain is slow growth in glycerol, which is a manifestation of mitochondrial respiratory dysfunction. This growth defect could be rescued by tit1+ but not by a catalytically debilitated mutant-tit1 carrying a point mutation [10]. The positive control, yYH1 (tit1+), grows well when transformed with an empty vector, but yNB5 transformed with an empty vector grows relatively poorly on glycerol. Transformation with wild-type tit1+ or wild-type TRIT1 rescued the growth defect of yNB5. However, whilst growth on glycerol after transformation with the mutant TRIT1 was slightly better than with the empty vector, rescue was less complete compared to wild-type TRIT1 (Figure 3E). This partial rescue may reflect a low level of i6A37 modification activity by the mutant TRIT1 enzyme.
Modelling of the mutation in S. cerevisiae demonstrates a respiratory deficiency
We also generated a strain of S. cerevisiae with knock-out MOD5 (a homologue of TRIT1) which was transformed with either wild-type MOD5, an empty vector, mod5K294R (humanised MOD5, which carries the Lys294Arg mutation) or mod5K294Q (mutant MOD5, which carries the Lys294Gln mutation). As observed in S. pombe, mod5-Δ yeast transformed with mutant MOD5 showed a reduced growth rate in a oxidative carbon source such as ethanol compared to mod5-Δ yeast transformed with either wild-type MOD5 or humanised MOD5 (Figure S1A). Oxidative growth defects were due to reduced respiratory activity since mod5-Δ strain transformed with an empty vector showed a significantly decreased respiration rate (P = 0.0002) in comparison to yeast transformed with wild-type MOD5, whilst mutant MOD5 failed to rescue the respiration rate of the transformed yeast as efficiently as wild-type or humanised MOD5 (Figure S1B).
The i6A37 modification is severely decreased in tRNAs from patient-derived fibroblasts carrying the p.Arg323Gln mutation
Immunoblotting of TRIT1 in fibroblasts from the proband demonstrated no significant loss of protein levels in comparison to control fibroblasts, using β-actin as a loading control (Figure 4A). A second, smaller band that was barely detectable in the control cell extract but more abundant in the patient cell extract was not always reproducible, likely reflecting nonspecific protein degradation. Notably, our evidence indicates that mitochondrial TRIT1 is the same molecular weight as the major band observed in the extracts (see Figure 2C). We next examined the in vivo levels of mitochondrial and cytosolic tRNA-i6A37 in patient and control fibroblasts using the Positive Hybridisation in the Absence of i6A (PHA6) assay [11]. As described previously, in the PHA6 assay, strong binding of the ACL probe occurs only in the absence of i6A37, whilst body probes (BP) efficiently bind the tRNA whether or not i6A37 is present and were used to indicate relative levels of the tRNAs [11]. Equal loading of the RNA was confirmed by ethidium bromide imaging of the gel (Figure 4B, upper panel) and by hybridization with appropriate BPs.

Both cy-tRNASer(UGA) and mt-tRNASer(UCN) had considerably decreased i6A37 modification in patient fibroblasts compared to control. In contrast, there was no difference in the ACL probing of mt-tRNACys, which does have a A36A37A38 target site for TRIT1 but is not modified, consistent with previous results [12]. The cy-tRNASer(UGA) appears to be fully modified in control fibroblasts (undetectable with ACL probe) consistent with prior results using HeLa cells [12] but largely unmodified in patient fibroblasts (Figure 4B). The mt-tRNASer(UCN) shows a significant, albeit decreased difference in the ACL signal between control and patient compare to cy-tRNASer(UGA). This is due in part to a significant fraction of unmodified mt-tRNASer(UCN) in the control cells, again consistent with prior results using HeLa cells [12]. This suggests that wild-type TRIT1 is only partially active on mt-tRNASer(UCN) in control fibroblasts. The more similar mt-tRNASer(UCN) ACL signals in control and patient fibroblasts is also due in part to a significantly lower amount of the overall level of mt-tRNASer(UCN) in the patient, as revealed by the mt-tRNASer(UCN) BP. We note that whilst cy-tRNASer(UGA) showed similar steady-state levels in control and patient cells, the mt-tRNASer(UCN) showed a 40% decrease in steady-state level (Figure 4B, quantification not shown but see Figure 5E), calculated using the BPs of mt-tRNASer(UCN) and mt-tRNACys as internal calibration standards [10].

Wild-type TRIT1 rescues the i6A modification in patient fibroblasts
Attempts to rescue the i6A37 hypomodification observed in patient fibroblasts were met with significant technical challenges that produced variable levels of transient transfection efficiency and ectopic TRIT1 and therefore only partial rescue of the i6A37 hypomodification (data not shown). We therefore used a retrovirus vector-based transduction approach to optimize the percentage of cells expressing ectopic TRIT1. Transduction of control and patient fibroblasts resulted in very high levels of overexpression as compared to the empty vector (Figure 5A), allowing us to examine the i6A37 content of tRNAs from patient cells (Figure 5B). Importantly, wild-type TRIT1 completely reversed the cy-tRNASer(UGA) hypomodification defect in the patient cells, whereas the empty vector did not, providing strong evidence that the native endogenous mutant TRIT1 protein is responsible for the hypomodification (Figure 5B, compare lanes 7 & 8 with 9 & 10). Given the very high overexpression of TRIT1 in these cells (Figure 5A), it was not surprising based on our mutation structure analysis, in vitro modification results and partial rescue of slow growth in glycerol by the mutant TRIT1 protein, that mutant TRIT1 was no less efficacious than wild-type TRIT1 in rescuing the i6A37 hypomodification of cy-tRNASer(UGA) in patient cells (Figure 5B).
In notable contrast to the rescue of cy-tRNASer(UGA) hypomodification, the hypomodification of mt-tRNASer(UCN) was rescued more efficiently by wild-type TRIT1 than mutant TRIT1 (Figure 5B). Moreover, restoration of i6A37 to mt-tRNASer(UCN) was specifically associated with an overall increase in the steady state levels of this tRNA as reflected by the mt-tRNASer(UCN) BP (Figure 5B; compare lanes 7 and 8 with 9 and 10). Quantification of independent triplicate sample sets revealed that whilst wild-type TRIT1 could effectively recover the i6A37 modification level of mt-tRNASer(UCN) to that observed in the control fibroblasts, ∼60% (P = 0.6286), mutant TRIT1 was significantly less efficient, at ∼20% (P = 0.0146) (Figure 5C).
Wild-type TRIT1 promotes accumulation of mt-tRNASer(UCN)
Using U5 snRNA as a loading control and mt-tRNACys and mt-tRNALeu(UUR) as non-substrate mitochondrial controls, the steady-state levels of both cy-tRNASer(UGA) (Figure 5D) and mt-tRNASer(UCN) (Figure 5E) (calculated using BPs) in transduced fibroblasts were determined. Curiously, cy-tRNASer(UGA) levels relative to U5 RNA were reproducibly found to be significantly higher in patient fibroblasts as compared to the control cells, regardless of whether the transducing vector encoded TRIT1 or not (Figure 5D). However, mt-tRNASer(UCN) levels, which were relatively lower in patient fibroblasts, were more efficiently rescued by wild-type TRIT1 (elevated relative to U5: P = 0.0162) than by mutant TRIT1 (unchanged relative to U5: P = 0.2038) or the empty vector (decreased relative to U5: P = 0.0055) when compared to control fibroblasts transduced with empty vector (Figure 5E, black bars). This quantitative trend was more significant when calibrating the mt-tRNASer(UCN) levels relative to the non-substrate control, mt-tRNACys in the patient cells (Figure 5E, grey bars).
We tried various approaches to rescue the biochemical, respiratory and molecular phenotypes in the patient fibroblasts. However due to limitations associated with the manipulation and transfection of patient and control fibroblasts, we were unable to do so with either wild-type or mutant TRIT1 despite multiple attempts (not shown). It appears that the cells had become less dependent on and/or less expressive of respiratory function with passage and handling.
The m.7480A>G mutation at position 38 in mt-tRNASer(UCN) impairs i6A37 modification
As noted in the Introduction, the tRNA substrates of all characterized isopentenyltransferases have an enzyme recognition sequence of A36A37A38 in their anticodon loops [11]. Thus we decided to further investigate a previously reported patient with mitochondrial myopathy [15] due to a pathogenic (m.7480A>G) mutation at position 38 in mt-tRNASer(UCN), a substrate of TRIT1 (Figure 6A). The PHA6 assay using a double ACL probe that matches both the wild type and mutant mt-tRNASer(UCN) (see Methods) performed on total RNA extracted from homogenised patient skeletal muscle showed reduced i6A37 modification of mt-tRNASer(UCN), to ∼14% of control levels (Figure 6B, quantification not shown). Furthermore, the steady-state level of mt-tRNASer(UCN) was also decreased by ∼30% in patient skeletal muscle compared to control (quantification not shown). Interestingly, when comparing control skeletal muscle to the previously described control fibroblasts (Figure 4B), it appears that skeletal muscle harbours relatively more mt-tRNASer(UCN) lacking the i6A modification. The in vitro modification assay was also performed on several synthetic tRNA ASLs, substantiating our finding of a deficiency in modification activity (Figure 6C). Isopentenyl modification of wild-type mt-tRNASer(UCN) (lane 3) in vitro is comparable to that observed for the positive controls, cy-tRNASec(UCA) (Lane 1) and cy-tRNASer(UGA) (Lane 2). However, the m.7480A>G mutation significantly abolishes in vitro activity of TRIT1 on mt-tRNASer(UCN) (lane 4) to the level observed in the non-substrates, mt-tRNACys (Lane 5) and mt-tRNALeu(UUR) (Lane 6). Hybridization of ASL probes to synthetic substrates modified in vitro by DMAPP was observed by the PHA6 assay to be substantially decreased compared to hybridization of the same probes to unmodified synthetic substrates (Figure 6D). Hybridization of ASL probes to the non-DMAPP substrate mt-tRNACys was unaffected by treatment with TRIT1. Notably, hybridisation of ASL probes to mutant mt-tRNASer(UCN) (m.7480A>G) was unchanged by the DMAPP modification reaction, confirming the loss of i6A37 modification in the mutant tRNA. These data validate the PHA6 assay for detection of changes in the i6A modification status of substrate mitochondrial and cytosolic tRNAs.

Discussion
Here we describe the investigation of a consanguineous kindred in which affected children presented with encephalopathy and myoclonic epilepsy associated with a disorder of mitochondrial translation. Analysis of whole exome sequencing data indicated that this was due to a recessively-inherited p.Arg323Gln mutation in TRIT1, the gene encoding the tRNA isopentenyltransferase (IPTase) responsible for i6A modification at position 37 in the anticodon loop of a subset of tRNAs [16], including mt-tRNASer(UCN), consistent with a previous report on i6A in bovine mt-tRNASer(UCN) [17].
In addition to the TRIT1 mutation-associated disorder of mitochondrial dysfunction reported here, we also demonstrated that a m.7480A>G point mutation of mt-tRNASer(UCN), previously reported as a cause of progressive mitochondrial myopathy [15], results in i6A37 hypomodification. In this case, the point mutation was in the TRIT1 sequence-specific recognition site, A36A37A38, of mt-tRNASer(UCN). The convergence of two different mechanisms, one due to a mutation in the TRIT1 enzyme and the other to a mutation in its substrate mt-tRNASer(UCN), that both cause i6A37 hypomodification of mt-tRNASer(UCN) and mitochondrial myopathy, provide strong genetic evidence of the critical importance of i6A37 in mitochondrial translation. TRIT1 should therefore be added to the increasing list of genes encoding mitochondrial tRNA-modifying enzymes, including MTU1 [18], PUS1 [19], MTO1 [20], MTFMT [21] and the various mitochondrial aminoacyl-tRNA synthetases [5], that have been associated with human disease. It is also worth noting that TRIT1 has been reported as a tumor suppressor and certain rare variant alleles are associated with poor survival from lung cancer in some ethnic groups [22], [23]; other mitochondrial-disease associated genes such as GRIM19 have also been implicated as a tumor suppressor [24]. However, it is not clear whether this is due to a relationship between cellular respiration and the mitochondrial function of TRIT1 in the lungs and/or the enzyme's cytosolic role. In addition, recent work has demonstrated an effect of Mod5 in tRNA-gene mediated gene silencing of RNA polymerase II promoters, suggesting a role for eukaryotic IPTases beyond their tRNA modification activity [25].
IPTases are conserved in sequence, structure and catalytic mechanism from bacteria to humans, particularly in the sequence surrounding the TRIT1 p.Arg323Gln mutation site. Indeed, in all of the IPTase sequences examined including E. coli MiaA, the targeted residue is either Arg or Lys. The mod5-i6A37-tRNA crystal structure shows that this residue comprises one of several basic side chains that contact the acidic backbone of the tRNA anticodon stem suggesting that the semi-conservative Arg to Gln mutation might compromise but not ablate enzyme activity. We therefore expected that any effect of the p.Arg323Gln mutation on TRIT1 activity would not be extreme and that the phenotype would be associated with a moderate decrease in tRNA i6A37 modification. However, to our surprise the mutation severely impaired the activity of the enzyme in vitro and caused a dramatic loss of i6A37 from both cy-tRNASer(UGA) and mt-tRNASer(UCN) in patient fibroblasts. It is important to note that our observation of i6A37 modification of cy-tRNASer(UGA) contrasts with a previous study that could not detect this modification in human cy-tRNASer(UGA) expressed in monkey-derived CV-1 cells [26].
In all eukaryotes examined, the IPTases modify both cytosolic and mitochondrial tRNAs and mitochondrial localization is a significant part of the biology. S. cerevisiae employs an intricate system for maintaining proper distribution of Mod5 to the mitochondria, nucleus and cytoplasm [27]–[29]. A prominent phenotype of S. pombe tit1-Δ mutants is slow growth on glycerol, a manifestation of mitochondrial respiratory dysfunction [10]. In C. elegans, the extended life span phenotype as well as deregulated development and other phenotypes of gro-1 mutants can be rescued by the mitochondrial isoform of the GRO-1 IPTase but not the nuclear and cytoplasmic isoform [30]. It is therefore noteworthy that while both cytosolic and mitochondrial tRNAs are lacking i6A37 in patient cells, the manifestations of disease clearly localize to a mitochondrial cause. Therefore, it may be important that in addition to hypomodification of mt-tRNASer(UCN), the overall levels of the mt-tRNASer(UCN) were significantly lower in patient fibroblasts. This suggests that in addition to the ∼4-fold loss of tRNA specific activity due to lack of i6A37 [10], mitochondrial translation would be even further compromised by a decrease in the absolute level of mt-tRNASer(UCN). This may contribute to a molecular basis for the apparent sensitization of the mitochondrial-associated phenotype in the patients described here.
Another informative finding was that the TRIT1-mutant could modify its substrate tRNAs with i6A37 when greatly over-expressed in the transduced patient fibroblasts. This was not completely unexpected on the basis of two experimental observations. As noted above, structure modelling suggested that the mutation would affect substrate binding but not i6A37 catalytic activity. Indeed, we observed increased transferase activity with increased substrate concentration, at least within the technical limits of the assay (Figures 3B and 3C). Second, the TRIT1 p.Arg323Gln mutant manifested partial activity to complement the slow growth in glycerol phenotype in the S. pombe tit1-Δ strain (Figure 3E). Furthermore, previous studies have shown that this phenotype remains uncomplemented by a prior characterized tit1-T12A catalytic mutant that is inactive for i6A37 modification of tRNA [10], [11]. Thus, the partial complementation of this phenotype (Figure 3E) suggests that the TRIT1 mutant retains some i6A37 activity, consistent with high activity of the nmt1+ promoter in the multi-copy expression vector.
Recent studies have concluded that although some tRNAs in human cells contain the A36A37A38 TRIT1 recognition motif they accumulate in the i6A37-unmodified form [12]. Somewhat similarly, the A36A37A38-containing tRNATrp(CCA) in S. cerevisiae remains unmodified [11]. This further suggests that the subset of i6A37-containing tRNAs may change under different conditions, due to varying concentrations of the enzyme or substrate, a situation that may occur during development and/or other physiological conditions.
Materials and Methods
Ethics statement
Written informed consent was obtained from the family in accordance with the Declaration of Helsinki and the study was approved by the Newcastle and North Tyneside 1 Ethics Committee.
Histochemical and biochemical analyses
Standard histological and histochemical analyses, including cytochrome c oxidase (COX), of a skeletal muscle biopsy were performed according to established protocols [31], on fresh-frozen skeletal muscle sections (10 µm). Mitochondrial respiratory chain complex activities were determined in skeletal muscle homogenates as previously described, and expressed relative to the activity of the matrix marker enzyme, citrate synthase [32].
Molecular genetics
Total DNA was extracted by standard procedures from all available tissues obtained with consent from familial relatives, and mtDNA rearrangements were excluded by long-range PCR. Direct sequencing of the entire mitochondrial genome was performed on homogenate skeletal muscle DNA.
Genomic DNA from the two affected siblings (II–1 and II–3) was isolated from blood (DNeasy, Qiagen, Valencia, CA); fragmented to 150–200 bp with the use of Adaptive Focused Acoustics (Covaris); end-repaired, adenylated, and ligated to adapters (Illumina Paired-End Sample Preparation Kit). Ligated libraries were hybridized with whole-exome baits that covered 27,184 genes (Agilent SureSelect Human All Exon Kit Version 2) with modifications for the SureSelect Human All Exon Kit Illumina Paired-end Sequencing Library (Version 2.0.1). Captured fragments were purified, clonally amplified and sequenced on 2 lanes of an Illumina Genome Analyser IIx using 75 bp paired-end reads.
The sequence was aligned to the human reference genome (UCSC hg19) with Burrows Wheeler Aligner (BWA) [33], then reformatted with the use of SAMtools v0.1.18 [34]. 83.1% of exon target sequence was covered by >10 reads. Single base variants were identified with Varscan v2.2 [35] and Indels were identified with Dindel v1.01 [36]. Variants were annotated using wANNOVAR [37]. Lists of on-target variants were filtered against data from the National Heart, Lung and Blood Institute (NHLBI, NIH, Bethesda, MD) Exome Sequencing Project (ESP) 6500 exomes, the 1000 Genomes project, and the exome sequences of 315 unrelated in-house control exomes to identify rare homozygous variants with a Minor allele frequency (MAF) <0.01.
Variant filtering led to a final list of 40 rare, homozygous, protein-altering variants of which 4 were mitochondrial according to the Gene-Ontology database. These genes included TRIT1, CCDC19, ARSB and SYNJ2 of which TRIT1 segregated with disease in the family. Targeted resequencing and familial segregation studies were performed by cycle sequencing using an ABI 3130xl (Applied Biosystems) system and BigDye Terminator v3.1 technology. The following primer pairs, including universal tags, were employed: forward primer, 5′-TGTAAAACGACGGCCAGTAGGGAAAATGCACACTGGAG-3′, and reverse primer, 5′-CAGGAAACAGCTATGACCTTCCCTTAGGTCAGATCCAAAA-3′. Analysis of the evolutionary conservation of the mutated amino acid across a range of homologous proteins was performed by the freely available Clustal Omega multiple sequence alignment software (http://www.ebi.ac.uk/Tools/msa/clustalo/) [38].
Culture of primary fibroblasts
Primary human fibroblast cell lines were established from the patient as well as from controls according to standard protocols and cultured at 37°C, in a humidified, 5% CO2 atmosphere. Fibroblasts were maintained as monolayers in Minimum Essential Media (MEM) (Life Technologies #21090) supplemented with FBS to 10%, 1x MEM-vitamins, 21 mM L-Glutamine, 1 mM sodium pyruvate, 1x penicillin/streptomycin, 1x non-essential amino acids, and 0.41 µM uridine.
Fibroblast transduction
TRIT1 wild-type and p.Arg323Gln mutant open reading frames were cloned into the pOP retroviral vector (Radichev et al., 2006) in frame with FLAG and HA epitope tags at the 5′ end using the XhoI and NotI sites, and sequencing was performed for confirmation. The preparation of the retroviral supernatants and the transduction of the control and patient fibroblasts were done as described [39].
Micro-scale oxygraphy analysis of live cells
Live cell respiration studies were performed by micro-scale oxygraphy using the Seahorse XFe Extracellular Flux Analyzer 24 (Seahorse Bioscience) according to manufacturer's instructions. Fibroblasts were seeded at a density of 30,000 cells/well. Mitochondrial function was assayed through the sequential addition of oligomycin (to 1.3 µM) to block the ATP synthase, 2 additions of carbonyl cyanide 4-(trifluoromethoxy)-phenylhydrazone (FCCP), a respiratory uncoupler which drives maximal respiration (to 2 µM and then to 3 µM), and antimycin (to 2.5 µM) to inhibit Complex III.
Oxygen consumption rate (OCR) and proton production rate (PPR) measurements for each well were normalized by cell number. Non-mitochondrial respiration was subtracted from all OCR values prior to analysis; spare respiratory capacity (SRC) equals maximal OCR - basal OCR, ATP coupling efficiency equals (basal OCR - oligomycin-inhibited OCR)/(basal OCR*100). Seven separate control cell lines underwent multiple testing and the means were combined to calculate control data (mean ± SD; n = 7). Patient fibroblasts were tested multiple times (n = 21). An unpaired, two-tailed Student's t-test was performed to determine the significance of differences between the data sets and P-values were considered significant at the 95% confidence interval.
SDS-PAGE and immunoblotting
Total cellular protein was extracted from patient and control fibroblasts (as well as transfected cell lines), size separated on a 10% separating gel by SDS-PAGE and transferred to a methanol-activated PVDF membrane. Immunoblotting was performed using primary antibodies to NDUFA9, NDUFB8, NDUFA13, SDHA, UQCRC2, MTCOI, MTCOII, COXIV and ATPB (all from Abcam), TRIT1 (GeneTex GTX120508) and β-actin (Sigma A5316) as a loading control and TOMM20 (Abcam) as a non-respiratory chain protein mitochondrial control. Chemiluminescent detection of the bands was achieved using the Amersham ECL Prime Kit (GE Healthcare) for signal development, following manufacturer's instructions and the membrane was viewed using the ChemiDocTMMP Imaging System (Bio-Rad).
Mitochondrial preparation and subfractionation
Subcellular fractions were prepared as described previously [40]. The same amount of protein (40 µg) from whole cell lysate, post-mitochondrial supernatant and mitochondrial subfractions was loaded onto a 12% SDS-PAGE gel, transferred to a PVDF membrane and analysed by immunoblotting using primary antibodies to TOMM20 (Santa Cruz), AIF (NEB), GDH (custom made against mature recombinant protein), NDUFA9 (Mitosciences), eIF4E (Cell Signaling). Chemiluminescent detection of the bands was achieved as described before.
Metabolic labelling of in vitro mitochondrial translation
The translation of proteins encoded by the mtDNA in patient fibroblasts was assessed by labelling with 35S-methionine/35S-cysteine (Perkin Elmer) as described previously [41]. Cytosolic translation was inhibited by co-incubation of the radioisotopes with 100 µg/ml emetine dihydrochloride. Total cell protein was extracted from both control and patient fibroblasts and 50 µg loaded onto a 15%–20% gradient gel for SDS-PAGE. Assessment of protein loading was achieved by Coomassie blue staining, and the gel was visualised by exposure to a blank PhosphorImager screen that was imaged using a Typhoon system (GE Healthcare).
In vitro modification assay
The in vitro modification activity of both wild-type and mutant TRIT1 was determined as previously described [11] using recombinantly-expressed enzyme from E. coli recovered using a Histidine tag [42]. Synthetic RNA minihelixes representing the target anticodon stem/loop (ASL) sequences of various tRNAs were used as templates for modification with 14C-labelled dimethylallyl pyrophosphate (DMAPP). In this assay, the isopentenyl group of DMAPP is transferred to A37 in substrate tRNAs by the IPTase TRIT1. The following RNA oligos were designed with an additional G-C base pair added to each ASL to stabilize the stem, and purchased from Integrated DNA Technologies (IDT): rGrUrGrCrArGrGrCrUrUrCrArArArCrCrUrGrUrArC (cy-tRNASec(UCA)), rGrArUrGrGrArCrUrUrGrArArArUrCrCrArUrC (cy-tRNASer(UGA)), rGrGrGrUrUrGrGrCrUrUrGrArArArCrCrArGrCrUrC (mt-tRNASer(UCN)), rGrGrGrUrUrGrGrCrUrUrGrArArGrCrCrArGrCrUrC (mt-tRNASer(UCN)-A7480G), rGrUrUrGrArArUrUrGrCrArArArUrUrCrGrArC (mt-tRNACys) and rGrUrArArArArCrUrUrArArArArCrUrUrUrArC (mt-tRNALeu(UUR)).
Decreased hybridization efficiency of complimentary DNA oligos due to the incorporation of the isopentenyl group in synthetic RNA minihelixes was confirmed by running an in vitro modification reaction with unlabeled DMAPP and recombinant His-TRIT1. We used the same protocol for the in vitro modification reaction as described previously [11], replacing 14C-labeled DMAPP with unlabeled DMAPP (100 nmol). For each sample, two reactions were performed. In mock-treated samples, all the components of the in vitro reaction were added except His-TRIT1. After the reaction, the RNA sample was purified by a phenol-chloroform extraction and loaded on a 15% TBE-Urea gel. The RNA was transferred to a GeneScreen Plus Hybridization Transfer Membrane (Perkin Elmer, catalog # NEF986001PK) and hybridized with 32P-labeled anticodon loop (ACL) oligos as described in the legend of Figure 6D. The sequences of DNA oligos used for this experiment are the same as used elsewhere in this paper.
The PHA6 tRNA-i6A37 detection assay
Total RNA isolation from skeletal muscle and human primary cell lines was performed using TRIzol according to manufacturer's protocol. The impact of the TRIT1 mutation on in vivo levels of the i6A37 modification in both cytosolic and mitochondrial tRNAs was assessed by the Positive Hybridisation in the Absence of i6A (PHA6) assay, which is an adaptation of high-resolution northern analysis [11]. The following anticodon loop (ACL) and body (BP) probes were used (all written as 5′-3′): mt-tRNACys ACL, TCTTCGAATTTGCAATTCAATATG and BP, AGCCCCGGCAGGTTTGAAGCT, cy-tRNASer(UGA) ACL, CCCATTGGATTT CAAGTCCAACGC, and BP, GCAGGATTCGAACCTGCGCGGG, wild-type mt-tRNASer(UCN) ACL, CAAAGCTGGTTTCAAGCCAACCCC (used for analysis of the patient carrying the TRIT1 mutation), mutant mt-tRNASer(UCN) ACL, CAAAGCTGGCTTCAAGCCAACCCC (both the wild-type and mutation-bearing probes were used together as a ‘double ACL probe’; the complement of the mutated base is underlined) and mt-tRNASer(UCN) BP, AAGGAAGGAATCGAACCCCCC, mt-tRNALeu(UUR) BP, GTTAAGAAGAGG AATTGAACCTC and U5 probe, TCCTCTCCACGGAAATCTTTA.
Phenotype rescue analyses in S. pombe
The wild-type TRIT1 or mutant TRIT1 was cloned into the pREP82X plasmid under the nmt1+ promoter before transformation into a yNB5 (tit1-Δ) strain of S. pombe. The experiments related to tRNA-mediated anti-suppression and growth deficiency in glycerol were performed as described previously [10], [11].
Phenotype rescue analyses in S. cerevisiae
The S. cerevisiae yeast strain used was BY4742 mod5-Δ (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 mod5::KanMX4) from the Euroscarf collection. The MOD5 gene was PCR-amplified with Kod HiFi Polymerase using primers MOD5CFw (gactagaaaatcgatgtgtcagg) and MOD5CSalIRv (ccgccGTCGACgcttgtcat cctccctttcc), digested with KpnI and SalI and cloned in the centromeric vector, pFL38 [43], thus obtaining the plasmid pFL38MOD5. The mod5K294R humanized and mod5K294Q mutant alleles were obtained by site-directed mutagenesis as described previously [44], on a MOD5 gene fragment obtained through amplification with the upstream forward primer MOD5MUTFw (ggagcccctgcagcttcatg) and the reverse mutagenic primer MOD5hK294RFw (cgagaacacgtcaatacgcaCGCaggcaggtaaaatggatcaag) or MOD5K294QFw (cgagaacacgtc aatacgcaCaaaggcaggtaaaatggatcaag). The amplified fragments were digested with BamHI and SalI and subcloned in BamHI-SalI-digested pFL38MOD5.
Plasmids were introduced in a BY4742 mod5Δ strain according to [45]. Growth assays were performed at 28°C in SD medium (0.69% YNB (Formedium, Norfolk, UK), without amino acids for which the strain is auxotroph) supplemented with 2% glucose (w/v) or 2% ethanol (v/v). Images of the colonies in the spots were acquired at 40X magnification with a Zenith inverted microscope through an Optikam 3 Digital Camera (Optika).
Oxygen consumption rate was measured at 30°C from suspensions of yeast cells cultured for 24 hours at 28°C in SD medium supplemented with glucose at a non-repressing concentration of 0.6% using a Clark-type oxygen electrode (Oxygraph System Hansatech Instruments England) with 1 ml of air-saturated respiration buffer (0.1 M phthalate – KOH, pH 5.0), 0.5% glucose. The reaction was started by addition of 20 mg of wet-weight cells.
Supporting Information
{kind=link}
Zdroje
1. VafaiSB, MoothaVK (2012) Mitochondrial disorders as windows into an ancient organelle. Nature 491 : 374–383.
2. CalvoSE, ComptonAG, HershmanSG, LimSC, LieberDS, et al. (2012) Molecular Diagnosis of Infantile Mitochondrial Disease with Targeted Next-Generation Sequencing. Sci Transl Med 4 : 118ra110.
3. PagliariniDJ, CalvoSE, ChangB, ShethSA, VafaiSB, et al. (2008) A mitochondrial protein compendium elucidates complex I disease biology. Cell 134 : 112–123.
4. KornblumC, NichollsTJ, HaackTB, ScholerS, PeevaV, et al. (2013) Loss-of-function mutations in MGME1 impair mtDNA replication and cause multisystemic mitochondrial disease. Nature Genet 45 : 214–219.
5. KonovalovaS, TyynismaaH (2013) Mitochondrial aminoacyl-tRNA synthetases in human disease. Mol Genet Metab 108 : 206–211.
6. AntonickaH, OstergaardE, SasarmanF, WeraarpachaiW, WibrandF, et al. (2010) Mutations in C12orf65 in Patients with Encephalomyopathy and a Mitochondrial Translation Defect. Am J Hum Genet 87 : 115–122.
7. NichollsTJ, RorbachJ, MinczukM (2013) Mitochondria: Mitochondrial RNA metabolism and human disease. Int J Biochem Cell Biol 45 : 845–849.
8. SuzukiT, NagaoA, SuzukiT (2011) Human Mitochondrial tRNAs: Biogenesis, Function, Structural Aspects, and Diseases. Annu Rev Genet 45 : 299–329.
9. JennerLB, DemeshkinaN, YusupovaG, YusupovM (2010) Structural aspects of messenger RNA reading frame maintenance by the ribosome. Nat Struct Mol Biol 17 : 555–U548.
10. LamichhaneTN, BlewettNH, CrawfordAK, CherkasovaVA, IbenJR, et al. (2013) Lack of tRNA Modification Isopentenyl-A37 Alters mRNA Decoding and Causes Metabolic Deficiencies in Fission Yeast. Mol Cell Biol 33 : 2918–2929.
11. LamichhaneTN, BlewettNH, MaraiaRJ (2011) Plasticity and diversity of tRNA anticodon determinants of substrate recognition by eukaryotic A37 isopentenyltransferases. RNA 17 : 1846–1857.
12. LamichhaneTN, MattijssenS, MaraiaRJ (2013) Human cells have a limited set of tRNA anticodon loop substrates of the tRNA isopentenyltransferase TRIT1 tumor suppressor. Mol Cell Biol 33 : 4900–4908.
13. ClarosMG, VincensP (1996) Computational Method to Predict Mitochondrially Imported Proteins and their Targeting Sequences. Eur J Biochem 241 : 779–786.
14. ZhouC, HuangRH (2008) Crystallographic snapshots of eukaryotic dimethylallyltransferase acting on tRNA: Insight into tRNA recognition and reaction mechanism. Proc Natl Acad Sci 105 : 16142–16147.
15. BidookiS, JacksonMJ, JohnsonMA, Chrzanowska-LightowlersZMA, TaylorRW, et al. (2004) Sporadic mitochondrial myopathy due to a new mutation in the mitochondrial tRNASer(UCN) gene. Neuromusc Disord 14 : 417–420.
16. GolovkoA, HjalmG, SitbonF, NicanderB (2000) Cloning of a human tRNA isopentenyl transferase. Gene 258 : 85–93.
17. YokogawaT, WatanabeY, KumazawaY, UedaT, HiraoI, et al. (1991) A novel cloverleaf structure found in mammalian mitochondrial tRNASer(UCN). Nucleic Acids Research 19 : 6101–6105.
18. ZehariaA, ShaagA, PappoO, Mager-HeckelA-M, SaadaA, et al. (2009) Acute Infantile Liver Failure Due to Mutations in the TRMU Gene. Am J Hum Genet 85 : 401–407.
19. BykhovskayaY, CasasK, MengeshaE, InbalA, Fischel-GhodsianN (2004) Missense Mutation in Pseudouridine Synthase 1 (PUS1) Causes Mitochondrial Myopathy and Sideroblastic Anemia (MLASA). Am J Hum Genet 74 : 1303–1308.
20. GhezziD, BaruffiniE, HaackTB, InvernizziF, MelchiondaL, et al. (2012) Mutations of the Mitochondrial-tRNA Modifier MTO1 Cause Hypertrophic Cardiomyopathy and Lactic Acidosis. Am J Hum Genet 90 : 1079–1087.
21. TuckerEJ, HershmanSG, KohrerC, Belcher-TimmeCA, PatelJ, et al. (2011) Mutations in MTFMT Underlie a Human Disorder of Formylation Causing Impaired Mitochondrial Translation. Cell Metab 14 : 428–434.
22. SpinolaM, FalvellaFS, GalvanA, PignatielloC, LeoniVP, et al. (2007) Ethnic differences in frequencies of gene polymorphisms in the MYCL1 region and modulation of lung cancer patients' survival. Lung Cancer 55 : 271–277.
23. SpinolaM, GalvanA, PignatielloC, ContiB, PastorinoU, et al. (2005) Identification and functional characterization of the candidate tumor suppressor gene TRIT1 in human lung cancer. Oncogene 24 : 5502–5509.
24. KalakondaS, NallarSC, JaberS, KeaySK, RorkeE, et al. (2013) Monoallelic loss of tumor suppressor GRIM-19 promotes tumorigenesis in mice. Proceedings of the National Academy of Sciences of the United States of America 110: E4213–E4222.
25. Pratt-HyattM, PaiDA, HaeuslerRA, WozniakGG, GoodPD, et al. (2013) Mod5 protein binds to tRNA gene complexes and affects local transcriptional silencing. Proc Natl Acad Sci 110: E3081–E3089.
26. CaponeJP, SharpPA, RajBhandaryUL (1985) Amber, ochre and opal suppressor tRNA genes derived from a human serine tRNA gene. EMBO J 4 : 213–221.
27. GillmanEC, SlusherLB, MartinNC, HopperAK (1991) MOD5 translation initiation sites determine n6-isopentenyladenosine modification of mitochondrial and cytoplasmic transfer-RNA. Mol Cell Biol 11 : 2382–2390.
28. TolericoLH, BenkoAL, ArisJP, StanfordDR, MartinNC, et al. (1999) Saccharomyces cerevisiae Mod5p-II contains sequences antagonistic for nuclear and cytosolic locations. Genetics 151 : 57–75.
29. BenkoAL, VaduvaG, MartinNC, HopperAK (2000) Competition between a sterol biosynthetic enzyme and tRNA modification in addition to changes in the protein synthesis machinery causes altered nonsense suppression. Proc Natl Acad Sci 97 : 61–66.
30. LemieuxJ, LakowskiB, WebbA, MengY, UbachA, et al. (2001) Regulation of physiological rates in Caenorhabditis elegans by a tRNA-modifying enzyme in the mitochondria. Genetics 159 : 147–157.
31. OldSL, JohnsonMA (1989) Methods of microphotometric assay of succinate dehydrogenase and cytochrome-c oxidase activities for use on human skeletal muscle. Histochem J 21 : 545–555.
32. Kirby DM, Thorburn DR, Turnbull DM, Taylor RW, Pon LA, et al. (2007) Biochemical Assays of Respiratory Chain Complex Activity. Methods in Cell Biology: Academic Press. pp. 93–119.
33. LiH, DurbinR (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25 : 1754–1760.
34. LillR (2009) Function and biogenesis of iron–sulphur proteins. Nature 460 : 831–838.
35. KoboldtDC, ChenK, WylieT, LarsonDE, McLellanMD, et al. (2009) VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25 : 2283–2285.
36. AlbersCA, LunterG, MacArthurDG, McVeanG, OuwehandWH, et al. (2011) Dindel: Accurate indel calls from short-read data. Genome Res 21 : 961–973.
37. ChangX, WangK (2012) wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet 49 : 433–436.
38. LarkinMA, BlackshieldsG, BrownNP, ChennaR, McGettiganPA, et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23 : 2947–2948.
39. RadichevI, KwonSW, ZhaoYM, DePamphilisML, VassilevA (2006) Genetic analysis of human ORC2 reveals specific domains that are required in vivo for assembly and nuclear localization of the origin recognition complex. Journal of Biological Chemistry 281 : 23264–23273.
40. BruniF, GramegnaP, OliveiraJMA, LightowlersRN, Chrzanowska-LightowlersZMA (2013) REXO2 Is an Oligoribonuclease Active in Human Mitochondria. Plos One 8: e64670 doi:10.1371/journal.pone.0064670
41. McKenzie M, Lazarou M, Ryan MT (2009) Analysis of respiratory chain complex assembly with radiolabelled nuclear - and mitochondrial-encoded subunits. In: Allison WS, editor.Methods in Enzymology.San Diego: Elsevier Academic Press Inc. pp. 321–339.
42. MakridesSC (1996) Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol Rev 60 : 512–538.
43. BonneaudN, OzierkalogeropoulosO, LiGY, LabouesseM, MinviellesebastiaL, et al. (1991) A Family of Low and High Copy Replicative, Integrative and Single-Stranded Saccharomyces cerevisiae Escherichia coli Shuttle Vectors. Yeast 7 : 609–615.
44. HoSN, HuntHD, HortonRM, PullenJK, PeaseLR (1989) Site-Directed Mutagenesis by Overlap Extension using the Polymerase Chain Reaction. Gene 77 : 51–59.
45. GietzRD, SchiestlRH (2007) Quick and easy yeast transformation using the LiAc/SS carrier DNA/PEG method. Nature Protoc 2 : 35–37.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Early Back-to-Africa Migration into the Horn of Africa
- PINK1-Mediated Phosphorylation of Parkin Boosts Parkin Activity in
- OsHUS1 Facilitates Accurate Meiotic Recombination in Rice
- Ancient DNA Analysis of 8000 B.C. Near Eastern Farmers Supports an Early Neolithic Pioneer Maritime Colonization of Mainland Europe through Cyprus and the Aegean Islands
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy