
Aberrant Autolysosomal Regulation Is Linked to The Induction of Embryonic Senescence: Differential Roles of Beclin 1 and p53 in Vertebrate Spns1 Deficiency
Spinster homolog 1 (Spns1) in vertebrates, as well as Spinster (Spin) in Drosophila, is a hypothetical lysosomal H+-carbohydrate transporter, which functions at a late stage of autophagy. The Spin/Spns1 defect induces aberrant autolysosome formation that leads to embryonic senescence and accelerated aging symptoms, while the molecular mechanisms of the pathogenesis are unknown in vivo. Using zebrafish, we show that Beclin 1 suppression ameliorates Spns1 loss-mediated senescence as well as autolysosomal impairment, whereas p53 deficit unexpectedly exacerbates these characteristics. We demonstrate that basal p53 activity has a certain protective role(s) against the Spns1 defect via suppressing autophagosome-lysosome fusion, while p53 activated by ultraviolet radiation amplifies the Spns1 deficit. In addition, we found that excessive lysosomal biogenesis and prolonged suboptimal acidification, modulated by v-ATPase, could be the primary reason for the appearance on the hallmarks of Spns1 deficiency. Our findings thus suggest that Spns1 is critically involved in lysosomal acidification and trafficking during autophagy, and differentially acts in a pathway with Beclin 1 and p53 in the regulation of senescence.
Published in the journal:
. PLoS Genet 10(6): e32767. doi:10.1371/journal.pgen.1004409
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1004409
Summary
Spinster homolog 1 (Spns1) in vertebrates, as well as Spinster (Spin) in Drosophila, is a hypothetical lysosomal H+-carbohydrate transporter, which functions at a late stage of autophagy. The Spin/Spns1 defect induces aberrant autolysosome formation that leads to embryonic senescence and accelerated aging symptoms, while the molecular mechanisms of the pathogenesis are unknown in vivo. Using zebrafish, we show that Beclin 1 suppression ameliorates Spns1 loss-mediated senescence as well as autolysosomal impairment, whereas p53 deficit unexpectedly exacerbates these characteristics. We demonstrate that basal p53 activity has a certain protective role(s) against the Spns1 defect via suppressing autophagosome-lysosome fusion, while p53 activated by ultraviolet radiation amplifies the Spns1 deficit. In addition, we found that excessive lysosomal biogenesis and prolonged suboptimal acidification, modulated by v-ATPase, could be the primary reason for the appearance on the hallmarks of Spns1 deficiency. Our findings thus suggest that Spns1 is critically involved in lysosomal acidification and trafficking during autophagy, and differentially acts in a pathway with Beclin 1 and p53 in the regulation of senescence.
Introduction
Autophagy is an evolutionarily conserved intracellular catabolic process whereby cytoplasmic proteins and organelles are engulfed into autophagosomes and subsequently degraded in autolysosomes, following fusion with lysosomes. Biologically significant roles of autophagy have been illuminated in a variety of physiological and pathophysiological conditions, such as occurs during the adaptation to nutrient starvation, the clearance of damaged proteins and cell organelles, development, cell survival and death, tumor progression and suppression, elimination of pathogens, and aging [1]. It has also been suggested that autophagy can have a beneficial effect on longevity in many lower organisms from yeast to flies, although a clear role in lifespan extension still remains elusive in vertebrates [2]. Furthermore, several interventions that promote longevity, including caloric restriction and chemical treatment with rapamycin, have exploited their impact through autophagy [3].
Zebrafish is an ideal organism to study the entire developmental process ex utero and are easily accessible for both experimental and genetic manipulations. Therefore, the zebrafish model system has become a popular platform to explore the mechanisms of human diseases [4]. Recently in our laboratory, we screened mutagenized zebrafish embryos for the altered expression of senescence-associated β-galactosidase (SA-β-gal), which is a versatile senescence biomarker widely used in both cellular senescence and organismal aging studies [5], [6], [7]. SA-β-gal has also been utilized for various detection of embryonic/larval senescence in our studies and those of others [8], [9], [10], [11]. We successfully validated the use of embryonic SA-β-gal production as a valuable screening tool by analyzing over 500 zebrafish mutants [12]. Of our identified mutants, the highest SA-β-gal activity was found to be associated with an insertion in the gene denoted “not really started” (nrs) (currently denoted as zebrafish spinster homolog 1, spns1), which is a homolog of Drosophila spinster, a gene that regulates aging and lifespan in flies [13]. Zebrafish harboring a homozygous mutation in the spns1 gene revealed embryonic/larval lethality, associated with yolk opaqueness and senescence [12], [14]. Adult zebrafish with a heterozygous deletion of spns1 show accelerated signs of aging, including an increased accumulation of the “aging pigment” lipofuscin in the muscle and liver, and have shortened lifespan [12]. Spinster has been implicated in a lysosomal storage function in flies [13], [15], and Spns1 deficiency leads to impaired autophagic termination and lysosome reformation problems in the mammalian cell culture system [16]. However, it remains unknown how Spns1 physiologically and pathophysiologically has an impact on autophagic homeostasis in conjunction with senescence in higher organisms in vivo, where we lack an appropriate vertebrate model system except for zebrafish.
Beclin 1, an autophagic regulator, is essential for early embryonic development, and is a haploinsufficient tumor suppressor [17]. During starvation of cultured cells, the accumulation of large and long-lasting autolysosomes caused by Spns1 deficiency is attenuated by concurrent beclin 1 knockdown, suggesting dependence on autophagy induction and progression [16]. p53, the most extensively characterized tumor suppressor, is a master regulator with pleiotropic effects on genomic stability, cell cycle, proliferation, cell death, tumorigenesis, stress response, senescence and energy metabolism, and is also involved in autophagic regulation [18]. p53 had been exclusively considered as a positive regulator of autophagy [19], but was recently found also to act as an autophagic inhibitor [20], [21]. Thus, the role of p53 in autophagy regulation requires further study since it may underlie key aspects of metabolism, aging, and cancer biology.
We examined the impact of Spns1 impairment on the autophagic process and on the induction of embryonic senescence in zebrafish, in order to clarify how autolysosomal processing is linked to these two tumor suppressors, Beclin 1 and p53. In this study, we found that inhibition of Beclin 1 can attenuate the yolk opacity and senescence caused by the Spns1 defect, whereas deficiency of “basal” p53 augments them (“basal” meaning in the absence of extrinsic genotoxic stress, e.g., ultraviolet light). Conversely, p53 “activated” by DNA damage apparently induced autophagy and apoptosis, intensifying the Spns1-deficient phenotype. Moreover, a chemical and genetic blockage of lysosomal acidification by inhibition of vacuolar-type H+-ATPase (v-ATPase) prevented the appearance of the hallmarks of Spns1 deficiency irrespective of the p53 state, while at the same time preventing autophagosome-lysosome fusion. Our findings thus suggest that Spns1 is critically involved in lysosomal acidification and trafficking during autophagy, and acts in the same pathway as Beclin 1 and p53 in the regulation of senescence.
Results
Accumulation of cytoplasmic membranous inclusions and LC3 puncta in spns1-mutant fish
Spin/Spns1 has been implicated in the regulation of autophagic lysosomal homeostasis in mammalian cells and flies [15], [16]. In fact, in zebrafish, electron microscopy revealed that compared with the wild-type control, spns1-mutant larvae accumulated cytoplasmic membranous inclusions corresponding to late endosomal, autophagic, and lysosomal structures in the hypodermal and retinal epithelial cells (Figure S1A). To verify that the autophagic process of spns1-deficient (spns1hi891/hi891) vertebrates is fundamentally disturbed, we generated EGFP-tagged microtubule-associated protein 1 light chain 3 (LC3) transgenic zebrafish with the spns1-mutant background. In the resulting EGFP-LC3-transgenic spns1-mutant [Tg(CMV:EGFP-LC3); spns1hi891/hi891] fish line, grossly enhanced EGFP intensity was observed throughout the body in comparison with the original Tg(CMV:EGFP-LC3) line [22], [23] (Figure 1A). In addition, intracellular localization of EGFP-LC3 was detectable as aggregated puncta in periderm or basal epidermal cells of the skin (above the eye on the head or in the caudal fin) and epithelial cells of several other organs including yolk sac, retina, and liver (Figure 1B), suggesting excessive autophagosome and/or autolysosome accumulation.

To gain additional information concerning the site of action of Spns1, we examined LC3 conversion as a hallmark of autophagy induction in whole zebrafish embryos by immunoblotting to distinguish the autophagosome-associated phosphatidylethanolamine-conjugated LC3-II from the cytosolic LC3-I form by showing the increased mobility of LC3-II. In spns1 mutants, both endogenous LC3-II and exogenous EGFP-LC3-II were detected at higher levels (Figure 1C).
Extending our analysis to a second animal model, we also examined autophagy activity in Caenorhabditis elegans containing a loss-of function mutation in the gene homologous to spin-1 (C13C4.5) [24]. Similar to our results in zebrafish, the C. elegans spin-1 mutation conferred augmented autophagic induction, as demonstrated by the increased expression and cytoplasmic aggregation of the EGFP::LGG-1 reporter gene product (LGG-1 is the ortholog of LC3) in seam cells of mutant animals (Figure S1B and C). We found the spin-1 mutant worms were more sensitive to starvation-induced death (Figure S1D), consistent with defective autophagy. In addition, decrease of Spns1 in heterozygous zebrafish as well as loss of Spin-1 in homozygous worms resulted in significant reductions in their adult lifespan (Figure S1E and F). These data suggest that across these different species, the defects in the spns1/spin-1 gene induce autophagic abnormality with excessive autophagosomes and/or autolysosomes, potentially leading to the accumulation of undegraded macromolecules and organelles in cells of mutant animals, which subsequently have a shortened life expectancy.
Lysosomal, but not mitochondrial, abnormalities in the pathogenesis of spns1 mutants
Spin/Spns1 is a multi-pass transmembrane protein localized in late endosomes and lysosomes [15], [25]. In mammalian cells, however, Spns1 has been reported to occasionally localize to mitochondria [26]. To elucidate a potential relationship between lysosomal and mitochondrial biogenesis with the pathogenesis induced by the Spns1-defective animals in vivo, we performed double staining of these two organelles by using LysoTracker (red) and MitoTracker (green) probes. In whole animal images, we found prominent increases of LysoTracker intensity in spns1-mutant fish, whereas no significant difference was detected by MitoTracker staining (Figure S2A). By further utilizing Tg(CMV:EGFP-LC3);spns1hi891/hi891 animals, concurrent LysoTracker staining revealed significant numbers of intracellular yellow (both green - and red-positive) puncta. Since the EGFP green signal is normally lost by quenching in acidic compartments such as the lysosome [27], this finding suggests the existence of insufficiently acidic autolysosomes (Figure 1D and E). In contrast, staining with a mitochondrial superoxide indicator, MitoSOX, revealed no critical abnormality of superoxide generated in the mitochondria (Figure S2B). These results suggest that Spns1 deficiency fundamentally leads to impaired lysosomal and/or autolysosomal acidification, but not to any significant modulation of mitochondrial biogenesis and oxidative stress.
Formation of enlarged mal-acidic cellular deposits caused by the Spns1 defect
Autophagosomes subsequently fuse with lysosomes to degrade their contents. The Spns1 defect causes excessively enlarged undegraded deposits of autolysosomal compartments in cells [16]. The inability of spns1 mutants to degrade protein aggregates, despite the apparent induction of autophagosomes, prompted us to ask whether Spns1 is required for degradation of autophagic cargos by ensuring proper acidification in autolysosomes. To address this question, we generated EGFP-LC3;mCherry-LC3 double-transgenic zebrafish [Tg(CMV:EGFP-LC3;mCherry-LC3); spns1hi891/hi891] to determine the acidification efficiency. As EGFP fluorescence is lost in acidic compartments, but mCherry red fluorescence is not, the coexpression of EGFP-LC3 and mCherry-LC3 can label insufficiently acidified autolysosomes as well as non-acidic autophagosomes to produce yellow fluorescence (positive for both green EGFP and red mCherry), whereas acidic autolysosomes would only show a red fluorescent signal.
To first validate that the EGFP signal was decreased or lost by quenching in acidic autolysosomes of wild-type animals, we utilized two lysosomal protease inhibitors, pepstatin A, an inhibitor of cathepsins D and E, and E-64-d, an inhibitor of cathepsins B, H and L. Because these inhibitors can target the proteases without altering autolysosomal acidity, we anticipated that the EGFP signal would only be reduced in truly acidic vesicles. In wild-type animals, as expected, only the large punctate signals of EGFP-LC3 were faded, whereas neither the LysoTracker nor mCherry-LC3 signals were affected (Figure S2C and D). On the other hand, as shown in Figure 1F, once spns1 morpholino antisense oligonucleotide (MO) was injected into the GFP - and mCherry-LC3-double transgenic fish embryos to knockdown the gene expression, we observed a prominent increase in the number of yellow-fluorescent enlarged intracellular vesicles as compared with those in standard control MO-injected animals, consistent with the accumulation of insufficiently acidified autolysosomes. The EGFP-LC3-positive vesicles in the spns1 mutants were further confirmed to be autolysosomes by the co-expression of a mCherry-tagged lysosomal membrane marker, lysosomal-associated membrane protein 1 (Lamp1) (Figure 1G). mCherry-LC3-positive enlarged vesicular aggregations that accumulated in the spns1-mutant fish were suppressed by expression of EGFP-tagged Spns1 vector (Spns1 WT) but not by that of an empty EGFP vector or an EGFP-tagged mutant Spns1 vector (Spns1 E153K; presumably disrupted for the transporter activity) [15], [16] (Figure 1H).
In addition, the vast majority of EGFP-LC3-positive vesicles in spns1 mutants were found to be still positive for a fluorogenic lysosomal substrate DQ Red BSA at the earlier phenotypic stages (∼60 hours post fertilization; hpf) (Figure S2E). DQ Red BSA fluoresces upon lysosomal degradation due to dequenching; the released peptide fragments are brightly fluorescent. Thus, the autolysosomes of spns1-mutant fish appeared to still contain hydrolytic activity at least in early autolysosomes, indicating that the primary reason for the retained EGFP-LC3 signal is probably due to suboptimal acidity at later stages. Therefore, the observed increase in both EGFP-LC3 and mCherry-LC3 double-positive yellow fluorescent intracellular vesicles in spns1-mutant fish could be attributed to ineffective or insufficient acidification (“mal-acidification”) at the late autolysosomal stage.
Rescue of the Spns1 deficit in zebrafish by suppression of Beclin 1
Based on a recent report of an autophagy-dependent effect of spns1 knockdown in a mammalian cell culture [16] and our current observations described above in the zebrafish model, we assumed that inhibition of the early stages of autophagy by blocking the class III phosphatidylinositol 3-kinase (PtdIns3K) complex containing Vps34/Pik3c3 and Beclin 1 would reduce aggregated LC3 puncta in cells of spns1 mutants and ameliorate yolk opaque abnormalities induced by the Spns1 deficiency. We therefore designed a splice-block morpholino antisense oligonucleotide (MO) targeting the zebrafish beclin 1 (becn1; zbeclin 1) gene at the 5′ end of exon 4 (Figure 2A). RT-PCR and DNA sequencing results showed this splice-block MO (beclin 1 MO) generated a loss of exon 4 and a premature stop codon, resulting in a truncated protein lacking the entire Bcl2 homology domain 3 (BH3 domain) (Figure 2B). The phenotype induced by the knockdown of beclin 1 by the MO during early development was not particularly evident at the gross morphology level apart from some minor developmental retardation at 24 hpf, without any obvious abnormality later on (Figure S3A). In contrast, the concurrent suppression of both spns1 and beclin 1 by MO targeting strikingly diminished the yolk opaqueness seen with the spns1 morphants and produced an increased number of viable larvae that survive beyond 72 hpf (Figure 2C–E). We also performed beclin 1 MO injections into spns1-mutant embryos, and reproducibly confirmed the ameliorated yolk phenotype through 3 dpf (Figure S3B), but mutant animals subsequently relapsed into deterioration, presumably due to the persistent impact of the Spns1 mutation and/or transient activity of the beclin 1 MO. These results indicate that suppression of the early stage of autophagy by beclin 1 knockdown can offset the deleterious effect of Spns1 deficiency that occurs at the late stage of autophagy.

We next examined whether the enlarged aggregations of LC3 in spns1 morphants and mutants can be restored by Beclin 1 knockdown. spns1 MO and/or beclin 1 MO were introduced into Tg(CMV:EGFP-LC3) fish embryos and resultant specimens were observed by confocal microscopy at the cellular level. The appearance of punctate vesicle-like intracellular aggregates and deposits observed in spns1 morphants was diminished by the beclin 1 knockdown (Figure 3A). LC3 has several functional homologs, including gamma-aminobutyric acid A (GABA)-receptor associated protein (GABARAP) and GABARAPL2/GATE-16. It has been reported that both LC3 and GABARAP are indispensable for the autophagic process in mammalian cells [21]. The restorative effect of beclin 1 knockdown was also demonstrated in spns1-depleted Tg(CMV:EGFP-GABARAP;mCherry-LC3) fish. The concomitant microinjection of spns1 MO and beclin 1 MO showed consistently similar outcomes in terms of the obvious reduction of both EGFP-GABARAP and mCherry-LC3 puncta (Figure 3B), as observed with the EGFP-LC3 puncta (Figure 3A).
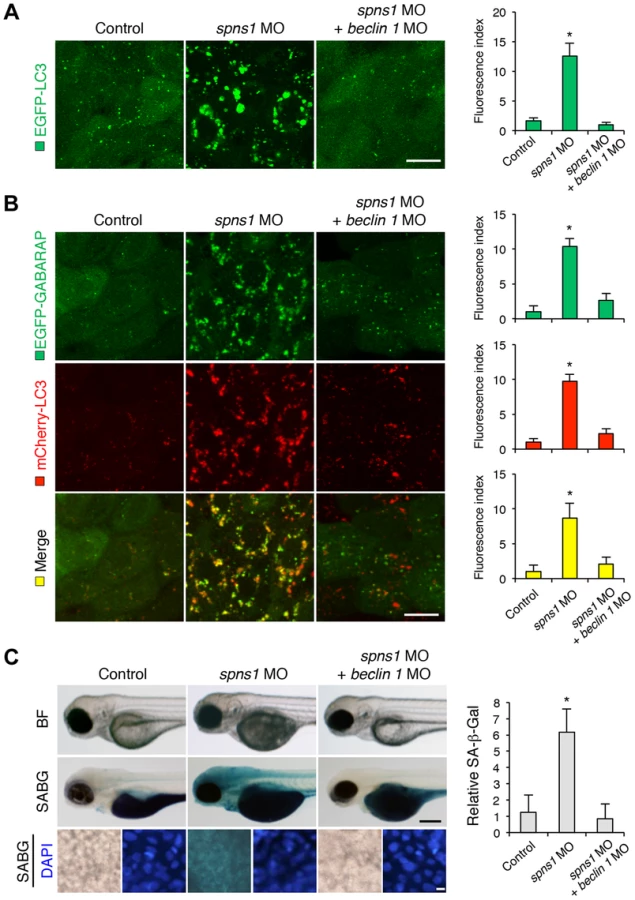
Another hallmark of spns1-mutant fish is the striking induction of senescence-associated β-galactosidase (SA-β-gal), which is an endogenous lysosomal β-D-galactosidase detectable at pH 6.0 [12], [28]. Previously we demonstrated that an embryonic (or larval) senescence phenotype caused by specific gene mutations (or MO-mediated knockdowns) and also by stress is readily detectable via SA-β-gal staining of zebrafish embryos and larvae [12]. Additionally, we also tested another lysosomal hydrolase/glycosidase, α-L-fucosidase (α-fuc) that has been reported in mammalian cells as a novel sensitive biomarker, senescence-associated α-fuc (SA-α-fuc) [29]. We found that higher activity of SA-α-fuc, as well as of SA-β-gal, was detected in spns1-mutant fish, compared with wild-type control fish, with SA-β-gal being the more sensitive assay (Figure S3C; see also Text S1 and S2). We therefore examined the effect of beclin 1 MO by staining with SA-β-gal in both spns1 morphants and mutants. Consistent with the restored yolk clarity and reduced LC3 puncta observed with beclin 1 knockdown in conditions of Spns1 deficiency, the beclin 1 MO markedly decreased the intensity of SA-β-gal at 3.5 dpf (Figure 3C and Figure S3B), whereas control injections (water and standard control MO) did not significantly affect the SA-β-gal activity in spns1 morphant and mutant animals (Figure 3C and Figure S3B). These results suggest that the aberrant SA-β-gal activity in spns1-defective animals coincides with autophagic initiation and its progression, and is accompanied by an increase in autolysosomes at the late autophagy stage.
While the excessive accumulation of autophagosomes and autolysosomes was observed in spns1-deficient animals, we anticipated that induction of apoptosis may be accompanied or preceded by the autophagic abnormality. We found, however, that such apoptotic induction was undetectable in spns1 mutants and morphants (Figure S4 and Figure S5; see also Text S1 and S2). Acridine orange (AO) staining, which can correspond to the detection of acidified compartments [30], [31] as well as of apoptotic and necrotic cell death [32], [33], showed positive signals co-stained by LysoTracker in spns1 mutants (Figure S4). However, when we performed a TUNEL assay for detecting DNA fragmentation associated with apoptosis, we found no staining upon knockdown of spns1 (Figure S5), while the positive control of ultraviolet light (UV) irradiation produced a TUNEL-positive signal, as reported previously [34]. The UV irradiation-mediated apoptosis was slightly but not significantly inhibited by beclin 1 knockdown (Figure S6a), which can fully suppress autophagy induced by UV (Figure S6B), suggesting that Beclin 1 plays a critical role in initiating autophagy, but is potentially dispensable for the induction of UV-mediated apoptosis in zebrafish embryos.
Impact of the p53 status on Spns1 deficiency in zebrafish
It has been reported that cells deficient in Beclin 1 exhibit an elevated DNA damage response [35], along with an increase in reactive oxygen species (ROS) [36]. In addition, a reduction of p53 by proteasomal degradation has been documented under the condition of beclin 1 knockdown [37]. The stress-responsive function of p53 still remains poorly understood with regard to how it is linked to autophagy impairment. In fact, although activated nuclear p53 can induce autophagy [38], it has also been reported that a removal of basal cytosolic p53 can stimulate autophagy [20]. We wondered which state of p53, if either, is involved in the Spns1 impairment. Moreover, since p53 activation is ordinarily thought to induce cellular senescence, which is also the case for zebrafish embryonic senescence [33], we suspected that the suppression of senescence by Beclin 1 depletion might be due to an intrinsic reduction in p53 [37]. We therefore investigated the requirement of p53 in the Spns1 deficiency-mediated senescence in zebrafish embryos under various experimental conditions through the genetic manipulations described below.
First, we examined the potential contribution of Spns1 and p53 separately in spns1 and p53 mutant fish backgrounds. We tested spns1 MO and p53 MO in p53 mutants and spns1 mutants, respectively (Figure S7). Unexpectedly, either p53 mutation or knockdown enhanced, rather than suppressed, the senescence phenotype under the Spns1-defective conditions. This increased SA-β-gal activity that is induced by p53 suppression was further demonstrated by coinjection of p53 MO and spns1 MO into normal wild-type animals to rule out any background influence in the mutants (Figure 4A, upper panels and B).

Next, we performed coinjections of p53 and spns1 MOs into Tg(CMV:EGFP-LC3) fish to concurrently monitor the autophagic process with EGFP-positive LC3 aggregate formation, in addition to subsequent senescence induction (Figure 4A, middle and lower panels, and C). Upon transient knockdown, although the total EGFP fluorescence became brighter, the number of EGFP-LC3 puncta were only slightly increased by p53 MO, compared with the control injected fish (Figure 4C, columns 1 and 2). On the one hand, enhanced LC3 puncta induction was observed when both MOs were coinjected, as similarly seen in the case of spns1 MO injection only (Figure 4C, columns 3 and 4), suggesting that autophagy induction associated with Spns1 depletion does not require p53. On the other hand, there were more cumulative LysoTracker-positive aggregates (dysfunctional autolysosomes) colocalized with LC3 by the double knockdown than spns1 knockdown alone, as EGFP-LC3 and LysoTracker double-positive yellow puncta were obviously increased by the p53 suppression in spns1 morphants (Figure 4A, middle panels, and C, columns 11 and 12). Moreover, the enhancing effect of p53 knockdown on senescence in spns1 morphants was obviously seen (Figure 4A, upper panels, and Figure S7).
We further generated spns1-mutant fish harboring a p53 mutation (tp53zdf1/zdf1), spns1hi891/hi891;tp53zdf1/zdf1, and confirmed that there was no requirement of normal p53 inheritance for the induction of embryonic senescence resulting from Spns1 deficiency, but instead there was an enhancement of SA-β-gal activity caused by the constitutive loss of wild-type p53 (Figure 4D and E). To further obtain robust hallmarks of senescence in zebrafish embryos, we examined the expression of other markers and/or mediators of senescence in spns1-defective animals. Quantitative RT-PCR (qPCR) as well as semi-qPCR in individual embryos demonstrated that the expression of p21waf1/cip1 and plasminogen activator inhibitor-1 (pai-1), which are known downstream targets of the p53 pathway [39], were upregulated in spns1 morphants and mutants (Figure 4F, and Figure S13; see also Text S1 and S2). Senescence marker protein-30 (smp-30) was downregulated in spns1-deficient animals compared with the corresponding controls. While the induction of p21waf1/cip1 as well as bax was apparently regulated in a p53-dependent manner, both the pai-1 induction and the smp-30 reduction in spns1 mutants were not influenced by the p53 defect.
We extended the analysis by monitoring the conversion of LC3-I into LC3-II among normal wild-type, tp53zdf1/zdf1, spns1hi891/hi89, and spns1hi891/hi891;tp53zdf1/zdf1 fish through 4 dpf. Autophagy was minimally induced in tp53zdf1/zdf1 fish based on detection of LC3-II conversion by immunoblotting, while the total amount of LC3 (LC3-I plus -II) was increased (Figure 4G and H). In spns1hi891/hi891;tp53zdf1/zdf1 fish, the LC3-II conversion/accumulation was similar to that seen in spns1-mutant fish (Figure 4G and H). These results suggest that either decrease or loss of basal p53 enhances the Spns1 impairment, potentially by augmenting autophagy progression (but not initiation) and/or lysosomal biogenesis (i.e., subsequent autolysosomal formation and maturation).
We then proceeded to assess the epistasis among spns1, beclin 1 and p53. We first confirmed that Beclin 1 suppression had no significant impact on p53 morphants or tp53zdf1/zdf1 fish (Figure 4H and I, columns 1, 3, 5, and 7, and Figure S8). Conversely, p53 depletion prevented the ability of beclin 1 MO to suppress the appearance of the yolk opaqueness and senescence phenotypes of spns1 mutants (Figure 4H and I, and Figure S8). Moreover, the beclin 1 knockdown significantly suppressed the SA-β-gal activity in spns1hi891/hi891;tp53zdf1/zdf1 fish to a similar extent as seen in standard control MO-injected spns1hi891/hi891;tp53+/+ fish (Figure 4H and I, columns 2 and 8, and Figure S8). However, the reduction of the SA-β-gal activity was more obvious in beclin 1 MO-injected spns1hi891/hi891;tp53+/+ fish than in spns1hi891/hi891;tp53zdf1/zdf1 fish (Figure 4H and I, columns 6 and 8, and Figure S8). Thus, basal p53 activity has a certain protective role(s) in preventing the deleterious phenotypes caused by genetic ablation of the spns1 gene, by competing with Beclin 1-mediated autophagy progression.
Although basal p53 can contribute to attenuating the Spns1 deficiency conceivably through suppressing autophagic progression and lysosomal biogenesis, we also wondered whether “activated p53” in response to DNA damage (e.g., UV) has any impact on the Spns1 deficiency, based on the result that the UV irradiation activates and/or enhances autophagy in zebrafish embryos (Figure S6B). As anticipated, apoptosis was similarly induced in spns1 mutants, compared with wild-type animals after UV treatment, whereas such apoptotic induction was almost undetectable under the p53 mutant condition (Figure S9A; see also Text S1 and S2). The UV exposure apparently augmented both autophagic progress (i.e., GFP-LC3 puncta formation) and lysosomal biogenesis (i.e., LysoTracker-stained puncta) in spns1 mutants only when functional p53 was present (Figure S9B and C).
A DNA damage response can be visualized as persistent foci of damaged nuclear DNA and its interacting proteins such as the phosphorylated histone variant, γH2AX [40]. DNA damage induced by UV treatment and the subsequent cell-cycle arrest in S phase were demonstrated by an increase of γH2AX intensity and a decrease of 5-bromo-2-deoxyuridine (BrdU) incorporation, respectively (Figure S10; see also Text S1 and S2). spns1 mutants had a negligible difference in γH2AX levels but had a significant reduction of BrdU incorporation, irrespective of the p53 state (Figure S10), which is indicative of a slowdown of cell proliferation without apparent DNA damage. The immunostaining of a mitotic marker, phosphorylated histone H3 (pH 3) also showed a significant reduction in tp53+/+-spns1-mutant animals, even without UV treatment. There was a similar tendency of pH 3 reduction in non-irradiated spns1;tp53-double mutants, but it was not statistically significant (Figure S11). Embryonic SA-β-gal activity was consistently increased by the UV stimulation in both wild-type and spns1-mutant animals in the presence of p53 (Figure S12).
Finally, we extended our analysis to examine the expression profiles of p21waf1/cip1, pai-1, and smp-30 as potential markers and/or mediators of senescence in spns1-defective animals (Figure 4F and Figure S13). beclin 1 morphants did not show any significantly detectable changes in expression of these genes (Figure S14A). Importantly, however, the suppression of beclin 1 significantly counteracted the impact of the spns1 depletion by restoring expression of the pai-1 and smp-30 genes (Figure S14A). As described above, even in the absence of p53, the altered regulation of these two critical senescence markers was still detectable in spns1-deficient animals (Figure S14B), indicating that p53-independent regulation may be responsible for the expression of these genes. In contrast, the induction of p21waf1/cip1, bax, and mdm2 genes in the spns1-defective condition was apparently p53 dependent and UV responsive, as confirmed by the level of their expression in p53 mutants (Figure S14B). It is also important to note that expression of ink4ab (the zebrafish homolog of both p15ink4b and p16ink4a) was induced by UV treatment but not by Spns1 loss (Figure S15). Taken together, the upregulation of pai-1 and p21, and the downregulation of smp-30 in spns1-defective fish embryos are consistent with the induction of senescence characteristics in aging organisms [40], [41], [42], [43], [44], [45].
Chemical modulation and monitoring of the autolysosomal acidification in spns1-mutant fish
Chemical genetic approaches are emerging in the zebrafish model system, and increasing numbers of chemical compounds are currently available for examining autophagic regulation [46], [47], [48]. We determined the effects of several chemical compounds and selective modulators of autophagy on Spns1 deficiency to see if any chemical(s) ameliorates or exacerbates the Spns1 phenotypes of zebrafish embryos. Of the chemicals tested, bafilomycin A1 (BafA) and other proton pump inhibitors (PPIs) such as the acid reducer omeprazole stood out due to their apparent inhibitory effect on overall phenotypic deterioration in spns1 animals (Figure 5A, B and Figure S16). BafA is a specific inhibitor of vacuolar-type H+-ATPase (v-ATPase), and inhibits lysosomal acidification, slowing or blocking degradation of LC3-II within autolysosomes as well as inhibiting the fusion between autophagosomes and lysosomes [49], [50], and subsequently it also enhances EGFP-LC3 puncta accumulation corresponding to mammalian autophagosomes [27]. Consistently, we found that BafA significantly increased the formation of cellular LC3 puncta as well as their gross EGFP intensity in wild-type zebrafish (Figure 5C–F). Intriguingly, both the progression of yolk opacity and SA-β-gal activity in spns1 mutants during the time period of 36–60 hpf were entirely suppressed by BafA treatment (Figure 5A and B). While EGFP-LC3 puncta signals in BafA-treated spns1 mutants did not appear significantly different compared with those in untreated counterparts, LysoTracker-positive compartments in cells were reduced by BafA treatment (Figure 5E and F), similar to the result seen with whole animal staining (Figure 5C and D). This is likely due to ‘prior’ alkalinization in lysosomes/autolysosomes and reduction of their biogenesis (Figure 5E and F). Importantly, these effects of BafA on spns1-mutant animals were essentially unaltered under the p53-depleted condition. Thus, BafA-induced pre-alkalinization might compensate for vulnerability of the spns1 mutants lacking basal p53 activity (Figure 5A).

BafA specifically binds to subunit c of the v-ATPase and thereby inhibits its enzymatic and proton-pump activity, but it has been shown that the concentration used and the duration of treatment with this drug are fairly critical to observe this effect [49]. In addition, BafA may have other off-target effects [51]. Therefore, we specifically knocked down the v-ATPase subunit gene atp6v0c by using a MO, whose effectiveness had already been demonstrated [52]. We obtained comparably consistent outcomes for the ameliorative effect of atp6v0c knockdown on the Spns1 deficiency (Figure S17A and B). In addition, we found that three other PPIs (omeprazole, lansoprazole, and pantoprazole), which can all interfere with the v-ATPase [53], [54], could also suppress the senescence phenotype in spns1 mutants (Figure S17C).
We further utilized LysoSensor dye to monitor acidification levels in lysosomes and autolysosomes, to verify that possible pre-alkalinization by BafA treatment or direct atp6v0c knockdown can efficiently suppress the spns1-mutant phenotypes. In contrast to the LysoTracker probes, which exhibit fluorescence that is largely independent of the pH level, the LysoSensor reagents can show a pH-dependent increase in fluorescence intensity upon acidification [55]. The neutral pH-sensitive LysoSensor 153 fluoresces optimally at pH 7.5, while the acidic pH-sensitive LysoSensor 189 fluoresces optimally at pH 5.2. When these probes (green) were used in combination with LysoTracker (red), we found a much stronger signal with LysoSensor 153 than with LysoSensor 189 in spns1-mutant animals (Figure S18A and B), which was also quite obvious at the cellular level (Figure 5G and H). By contrast, treatment of wild-type animals with lysosomal protease inhibitors, pepstatin A and E-64-d, which allows the retention of intact autolysosomal/lysosomal acidity while preventing autolysosomal maturation and turnover, showed highly acidic vesicles stained by LysoSensor 189, rather than by LysoSensor 153 (Figure 5G and H). Lysosomal compartments in spns1 mutants may still retain some weak acidification allowing lysosomal biogenesis and subsequent autophagosome-lysosome fusion, as short-term treatment (for 1 h) with BafA can completely abolish the acidic compartments stained by LysoSensor and significantly reduce the LysoTracker-positive signals (Figure S18C and D).
Finally, we examined the colocalization of EGFP-LC3 puncta and lysosomes in wild-type fish in the presence of BafA or pepstatin A and E-64-d, compared to that in the spns1hi891/hi891 fish (Figure S19A and B). In wild-type animals, BafA caused the accumulation of EGFP-LC3 and colocalization of EGFP-LC3-mCherry-LC3 signals (Figure S19C), but no accumulation of LysoTracker, indicating a block in fusion of autophagosomes with lysosomes (Figure S19B). Inhibition of lysosomal hydrolase activity with pepstatin A and E-64-d resulted in accumulation of lysosomes (red) and autolysosomes (yellow by overlapping EGFP-LC3 and LysoTracker) (Figure S19B). In contrast, the spns1hi891/hi891 fish (Figure S19A) and their cells (Figure S19B and C) displayed both an accumulation of autolysosomes (yellow by overlapping EGFP-LC3 and LysoTracker) and autophagosomes (yellow by overlapping of EGFP-LC3 and mCherry-LC3) without any inhibitors, again indicating defects in both fusion of autophagosomes with lysosomes and autolysosomal maturation. Collectively, these results demonstrate that the appearance of deleterious changes in spns1 animals is due to aberrant autophagic progression caused by impaired suboptimal acidification in malformed autolysosomes, and that p53 may also be involved in the process of both lysosomal and autolysosomal pathogenesis in Spin1 deficiency.
Discussion
We demonstrated that loss of Spns1 leads to defects in autophagic and lysosomal homeostasis in zebrafish, and the tumor suppressors Beclin 1 and p53 are differentially involved in autophagy and senescence pathways regulated by Spns1. A reduction of Beclin 1 as an autophagy regulator can attenuate the Spns1 defect, whereas a decrease/loss of basal p53 activity, as well as activated p53 by DNA damage, augments it and exacerbates the deleterious phenotype in zebrafish. If both Spns1 and p53 were abrogated, the Beclin 1 reduction was no longer effective in suppressing the spns1-mutant phenotypes sufficiently, whereas v-ATPase reduction was robust enough to suppress the phenotypes regardless of p53 state.
Importantly, we have successfully generated valuable zebrafish tools by crossing the fluorescent protein-tagged LC3 - and GABARAP-transgenic lines with the spns1-mutant line to monitor real-time alterations of autophagic abnormalities in vivo. Vertebrates have approximately seven Atg8 homologs [56], and the best studied of these is LC3. GABARAP shows many similarities with LC3, but its conjugation is only mildly affected by starvation, and under certain conditions conjugation may be activated independent of target of rapamycin (TOR) inactivation [57], [58]. We have found, however, an indistinguishable behavior between LC3 and GABARAP in the transgenic animals harboring either spns1 mutation or depletion, although it has been suggested that LC3 and GABARAP differentially act in autophagosome biogenesis [59].
The evolutionarily conserved autophagy gene beclin 1 (vps30/atg6) is frequently inactivated at one locus in several cancers [60], [61]. Studies in mice have also demonstrated that beclin 1 is a haploinsufficient tumor suppressor [17], [62]. It has been demonstrated that Spns1-loss-associated EGFP-LC3 puncta accumulation in cells, which reflects autophagic progression by autophagosome formation, is suppressed by the depletion of Beclin 1, Atg7, or Ulk1, as well as by treatment with a PtdIns3K inhibitor, 3-methyladenine [16]. Consistently, we also demonstrated that beclin 1 morphants were resistant to forming LC3 puncta induced by Spns1 deficiency in zebrafish. However, once both spns1 and p53 were depleted, the beclin 1 knockdown was no longer effective enough to suppress the punctate accumulation of LC3 as well as the mutant hallmarks of both yolk opaqueness and embryonic senescence characteristic of Spns1 deficiency in zebrafish.
p53 is one of the most commonly mutated tumor suppressor genes found in many types of human cancers [63]. We found that the loss of basal p53 compromises the ability to properly adjust autolysosomal formation, and exacerbated the spns1 deficiency, while beclin 1 knockdown can ameliorate it by suppressing the early stage of autophagy. p53 has been linked to the regulation of autophagy, but the exact nature of its role still remains controversial. On the one hand, onocogenic and genotoxic stress events result in p53 stabilization and activation, which can stimulate autophagy through both transcription-independent mechanisms (e.g., AMP-activated protein kinase; AMPK activation and TOR inhibition) and transcription-dependent mechanisms (e.g., transcriptional upregulations of PTEN, tuberous sclerosis complex 1/TSC1 and damage-regulated autophagy modulator/DRAM1) [64]. On the other hand, it has been reported that genetic or chemical inhibition of basal cytoplasmic p53, or proteasomal depletion of p53 during starvation and/or endoplasmic reticulum stress, activates autophagy through transcription-independent mechanisms involving AMPK activation and TOR inhibition [20]. Loss of p53 may lead to homeostatic imbalance in cells, such as induction of bioenergetic compromise, increased ROS, and/or defective cell-cycle checkpoints, which can lead to autophagy induction. Thus, p53 depletion may promote or enhance autophagy indirectly as a result of imbalanced metabolic stress conditions. This therefore suggests that p53 maintains cellular homeostasis by adjusting the rate of autophagy in a context-dependent manner, as circumstances require.
Intriguingly, Spns1-loss-induced embryonic senescence (SLIES) represents an atypical senescence response that is distinct from p53-induced senescence and can be suppressed by autophagy inhibition mediated through beclin 1 knockdown (Figure 6). Since the Beclin 1 suppression may lead to reduction in the level of p53 [37], and then might subsequently prevent SLIES, we intensively examined the effect of p53 depletion on SLIES. To our initial surprise, SLIES cannot be suppressed by the loss of p53 at all, but is rather enhanced. This seems to contradict the conventional role of p53 as an inducer of cellular senescence in various contexts including the zebrafish model [33], [65]. However, given recent evidence of a certain anti-aging mechanism by p53 in mice and p53-mediated suppression of senescence in cells [66], [67], it might not be surprising that p53 can also act both as a suppressor of senescence and of autophagy in some contexts, although the exact molecular mechanism remains elusive at this point. In addition, there remains a p53-independent cellular senescence mechanism that still depends on its authentic downstream target, p21Waf1/Cip1/Cdkn1a, among others, such as Arf and p27Kip1/Cdkn1b triggered by Skp2 inactivation [68]. Moreover, a recent report indicated that p21Waf1/Cip1/Cdkn1a also has a tissue-selective and context-dependent modulation of senescence in BubR1 progeroid mice [69]. In addition, most recently, SA-β-gal-positive “developmental senescence” observed in mice, which shares some, but not all, regulatory pathways detectable in adults, was shown to involve the activation of p21Waf1/Cip1/Cdkn1a in the absence of a DNA damage response and any alteration in p53, p16Ink4a, or p19Arf [70], [71]. Interestingly, we found that in spns1-deficient fish embryos, the upregulation of p21 and pai-1 expression and the downregulation of smp-30 expression were detected without a DNA damage response. Further investigation and elucidation of their functional roles as senescence mediators or attenuators will be required to determine how they are responsible for SLIES.

p53 is also well known for its pro-apoptotic cell death-inducing activities, but it can conversely possess pro-survival effects, particularly under mild stress conditions [72], [73], [74]. In zebrafish embryos, however, we determined that SLIES occurs regardless of p53-mediated impacts on apoptotic cell death and/or the cell-cycle checkpoint response as well. Thus, spns1-mutant animals show a new type of developmental senescence that can be triggered by autophagic initiation followed by autolysosomal fusion in the absence of the authentic senescence moderator p53, while basal p53 and activated p53 can play contrasting roles; attenuation in SLIES and mediation in apoptosis, respectively.
“Activated p53” is not specifically involved in the spns1-ablated condition but can generally induce and/or augment the deleterious condition caused by the DNA damage response and apoptosis. In contrast, “basal p53” may have an antagonistic effect on lysosomal biogenesis (or autolysosomal maturation) rather than on the autophagic progress in the absence of Spns1. Alternatively, the p53 status may rather influence endosomal-lysosomal homeostasis where Spns1 is primarily involved. It should be noted that p53 may be involved in lysosomal stabilization [75], as well as in various metabolic changes and the regulation of energy metabolism including aerobic glycolysis (the Warburg effect) in which the lysosome is also engaged for degradation [38].
Our preliminary observation suggests that SLIES and the yolk opaqueness hallmarks of spns1 embryos are only mildly affected by chemical (e.g., rapamycin)-mediated autophagy induction. This may be a reflection of the consistent outcome of already attenuated TOR (re)activation due to impaired autophagic lysosome reformation by Spns1 deficiency, as has been demonstrated in mammalian cells and flies [16], [76]. We are also wondering if basal p53 depletion may have any effect on autophagy enhancing activity independent of or different from the rapamycin-sensitive TOR pathway.
Of note, rather than simple depletion of wild-type p53, the p53 mutant (tp53zdf1/zdf1) fish used here retain an accumulation of the mutant p53 protein (p53M214K) [77], which corresponds to the human p53M246K mutant protein whose function is completely abolished [78]. A recent study suggests that this mutant p53 protein is degraded through chaperone-mediated autophagy (CMA) in a lysosome-dependent fashion [79]. Thus, the regulation of the stability of mutant p53 differs from that of wild-type p53. There is a possibility of activating the CMA pathway by inhibiting (macro)autophagy, to specifically promote the degradation of mutant p53, under a nutrient-starved condition. Therefore, it is also important to examine any involvement of the Spns1 function in the protein stability of mutant p53, whether the Spns1 defect selectively activates the CMA pathway for the removal of mutant p53 or not.
Altogether, our present results support the notion that the interruption of the intrinsic nutrient supply through autophagy, supposedly from yolk in zebrafish embryos and larvae [80], may lead to profound energetic exhaustion under the aberrant autolysosomal condition resulting from Spns1 deficiency, and this effect is dependent on the p53 state.
Since BafA can inhibit the import of H+ through the v-ATPase into the lysosome lumen, while the Spns1 defect presumably prohibits the symport of H+/sugar by loss of its function, it was anticipated that BafA might at least temporarily rescue the spns1-mutant animals, by restoring the balanced acidity condition of autolysosomes and/or lysosomes, as well as subsequent autophagosome-lysosome fusion. In fact, we found that BafA effectively inhibited the progression of both yolk opacity and embryonic senescence that appeared in spns1 mutants. Moreover, a direct depletion of the v-ATPase subunit c (a direct target of BafA) by MO recapitulated the restorative effect on the mutant animals. Importantly, the lysosomal pH of spns1 mutants was found to be less acidic, suggesting that protons may pass through the membrane via other H+-coupled transporters and/or channels such as lysosomal amino acid transporter 1 (LYAAT-1/SLC36A1) [81], chloride channel 7 (CLCN7) [82], and cystic fibrosis transmembrane conductance regulator (CFTR) [83].
It should be noted that SA-β-gal is acid β-D-galactosidase, a lysosomal glycoside hydrolase (glycosidase), which catalyses the hydrolysis of β-galactosides into monosaccharides [28], and its substrates also include various glycoproteins, gangliosides (glycosphingolipids), lactose, and lactosylceramidases [84], [85]. The aberrant increase of the in situ SA-β-gal activity induced by Spns1 deficiency indicates that such a glycosidase product itself can be preserved in autolysosomes and/or lysosomes, but may not function properly in vivo without an essential permease(s) to transport degradation products that need to be delivered into the cytoplasm as energy sources.
The extent to which our current observations of Spns1 functions during early development pertain to actual aging and age-related disease situations remains to be rigorously determined under both physiological and pathological conditions in animals. However, an increase in the abundance of the lysosomal hydrolases is presumably linked to the increased lysosomal biogenesis observed in senescent cells. Indeed, cumulative evidence suggests that an increased number of lysosomes and elevated lysosomal activity have been associated with replicative senescence [85]. Thus, the current finding suggests that temporal suppression of autophagy through Beclin 1 and/or v-ATPase by approved therapeutics (e.g., omeprazole) may be an effective therapeutic approach in the prevention of autophagic impairments similar to the Spns1 deficiency (Figure 6). Similar intervention has been demonstrated successfully in a mouse model of Pompe disease, a lysosomal glycogen storage disorder [86].
Materials and Methods
Zebrafish maintenance and ethics
Zebrafish (AB and casper strains) were maintained under a 14 : 10 h light/dark cycle and fed living brine shrimp twice daily. Brine shrimp were presented using 1 mL pipettes (about 0.75 mL brine shrimp per 20 fish). Flake food was also given every other day in proportion to the number of fish in the tanks. A continuously cycling aquarium system was used to maintain water quality (Aquatic Habitats Inc.). Zebrafish embryos were collected from pairwise matings of adults and raised at 28.5°C. The embryos to be used in the experiments were then staged by hours post fertilization (hpf) at 28.5°C and also by morphological appearance for experiments [87]. All animal experiments were approved by and conducted in accordance with the guidelines established by the Institutional Animal Care and Use Committee (IACUC) at The Scripps Research Institute, IACUC approval number 09-0009.
Confocal microscopy and imaging
Zebrafish embryos (in the case of the AB fish line) were transferred into 0.003% (w/v) 1-phenyl-2-thiourea (PTU) prior to 24 hpf to prevent pigmentation. Embryos or larvae were then mounted live in water containing 0.16 mg/ml tricaine (Sigma, A5040) for imaging. Images were taken using the FluoView 1000 confocal laser scanning microscope system (Olympus) with a 60× objective lens). Since EGFP - or mCherry-LC3 and EGFP-GABARAP showed both a uniform cytosolic signal and more intense spots, threshold values were set to reduce the cytosolic signal and identify the more intense dots. The same threshold value was applied for all samples in the indicated experiments. The extent of colocalization between LysoTracker and LysoSensor signals and EGFP - or mCherry-LC3 and EGFP-GABARAP dots was quantified in three independent visual fields from three independent embryos. All values are represented as mean ± standard deviation (S.D.). Mounted animals were photographed using each specific fluorescent signal by confocal laser microscopy. Fluorescence intensities were quantified using Adobe Photoshop over a color range that was chosen according to 25 additive color selections of regions that showed visually positive signals. For analyses of cells within the zebrafish embryos, these regions were selected in each actual embryo only and not in the yolk. Following pixel selection, a fuzziness setting of 64 was used, and the chosen pixel number was determined using the image histogram calculation.
Morpholino oligonucleotides
Morpholino oligonucleotides (MOs) were designed and synthesized by Gene Tools, LLC (Philomath, OR). The sequence of the beclin 1 MO is 5′-CATCCTGCAAAACACAAATGGCTTA-3′, which overlaps the intron-exon boundary at the 5′-splice junction of exon 4 in the zebrafish beclin 1 gene. The sequence of the standard control MO is 5′-CCTCTTACCTCAGTTACAATTTATA-3′. MOs were resuspended in sterile water at a concentration of 1 mM as stock solutions. For microinjection into embryos, the stock solutions (1 mM) were diluted to 125, 250, 500, and 750 µM. A 10 nl volume of each MO solution was injected into the yolk during the one-cell stage. All other MO sequences have been reported previously [8], [12], [52], [88], except Inverse-sequence p53 MO (inv. p53 MO); 5′-GTTAAGAACGTTTCGTTACCGCG3′.
MitoTracker, LysoTracker, LysoSensor and DQ Red BSA staining
The vital mitochondrial and lysosomal dyes MitoTracker Green FM (Invitrogen; molecular probes, M7514), LysoTracker Red DND-99 (Invitrogen; molecular probes, L7528), LysoSensor Green DND-189 (Invitrogen; molecular probes, L7535) and LysoSensor Green DND-153 (Invitrogen; molecular probes, L7534) were diluted to final concentrations of 1 µM, 10 µM, 1 µM and 1 µM, respectively, in E3 medium (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4), and pre-warmed to 28.5°C. Each dye was then added to an equal volume of fresh water on embryos and incubated at 28.5°C in the dark for 30 min to 1 h. Embryos were then rinsed four times in fresh E3 medium before imaging. DQ Red BSA (Invitrogen; molecular probes, D12050) was diluted to a final concentration of 0.5 mg/ml in E3 medium, directly injected into the yolk sac at 72–84 hpf, and subjected animals were incubated for 4 h prior to observation by microscopy.
Transmission electron microscopy
Zebrafish larvae were fixed in 4% paraformaldehyde, 2.5% glutaraldehyde, 0.02% picric acid, 0.1 M Na cacodylate buffer, washed and fixed in 1% osmium tetroxide in 0.1 M Na cacodylate buffer. They were subsequently treated with 0.5% tannic acid followed by 1% sodium sulfate. The pellets were treated with propylene oxide and embedded in Epon/Araldite. Thin sections (70 nm) of the pelleted samples were cut on a Reichert Ultracut E (Leica, Deerfield, IL) using a diamond knife (Diatome, Electron Microscopy Sciences, Hatfield, PA), mounted on parlodion-coated copper slot grids and stained in uranyl acetate and lead citrate. Sections were examined on a Philips CM100 transmission electron microscope (FEI, Hillsbrough, OR). Images were documented and measurements were taken using a Megaview III CCD camera (Olympus Soft Imaging Solutions, Lakewood CO). Transverse sections were obtained through the trunk muscle region, the yolk and the eye region.
RNA isolation and RT-PCR analysis for zebrafish beclin 1
RT-PCR analysis of a single zebrafish embryo was performed to determine the effects of the splice-block MO for the zebrafish beclin 1 gene. Total RNA was extracted from 24–48 hpf embryos injected with control MO, beclin 1 MO, or beclin 1 plus spns1 MO, using TRIzol reagent according to the manufacturer's protocol (Invitrogen). cDNA was synthesized using M-MLV reverse transcriptase (Promega), followed by PCR with ExTaq (Takara). For semi-quantitative analysis, the linear amplification ranges were then determined for each of the primer sets. PCR primers used to amplify the fragments of the zebrafish beclin 1 gene were designed using a web-based primer design tool, PrimerQuest (Integrated DNA Technology, Inc.) (zbeclin 1 EX3 forward primer; 5′-CAAACAAGATGGCGTGGCTCGAAA-3′, zbeclin 1 EX4 forward primer; 5′-GTGGAACTATGGAGAACTTGAGTCGCA-3′, and zbeclin1 EX7 reverse primer; 5′-TCCAACTCCAGCTGCTGTCTCTT-3′). The amplified products were validated by sequencing. As controls for these PCR analyses, ef1α and β-actin were examined. The forward and reverse primers used to amplify ef1α were 5′-ACCACCGGCCATCTGATCTACAAA-3′ and 5′-ACGGATGTCCTT GACAGACACGTT-3′, respectively, and for β-actin were 5′-CCCAGACATCAGGGAGTGAT-3′ and 5′-CACCGATCCAGACGGAGTAT-3′, respectively. For amplification by PCR, the initial denaturing step at 94°C for 5 min was followed by 18–25 amplification cycles of 30 sec at 94°C; 30 sec at 60°C; 60 sec at 72°C, and a final extension period of 10 min at 72°C. Amplified products were separated on a 1.5% agarose gel stained with ethidium bromide and the bands were visualized and recorded using a Multi Image Light Cabinet (Cell Bioscience). Other PCR primers, parameters and conditions are summarized in Supplemental Table S1 and S2.
SA-β-gal assay and quantification
Zebrafish embryos and larvae at 48–72 dpf were washed three times in phosphate buffered saline (PBS) and fixed overnight in 4% paraformaldehyde with PBS at 4°C. After fixation, the samples were washed three times in PBS, pH 7.5, twice again in PBS, pH 6.0 at 4°C, and then incubated at 37°C (in the absence of CO2) for 12–16 h with SA-β-gal staining solution (5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 2 mM MgCl2 in PBS at pH 6.0). All animals were photographed under the same conditions using reflected light with a macro microscope, AZ100 (Nikon). SA-β-gal activity in each animal was quantified using a selection tool in Adobe Photoshop software for a color range that was chosen using 25 additive color selections of regions that showed visual SA-β-gal staining. For analyses of embryos, these regions were selected in each embryo proper only and not in the yolk in order to exclude variability in the initial yolk volume and yolk consumption levels over time. Since the yolk stains much more intense for SA-β-gal at all stages of development than any other embryonic tissues in general, it was desirable to eliminate this as a source of variability. Following pixel selection, a fuzziness setting of 14 was used, and the chosen pixel number was determined using the image histogram calculation.
Immunoblotting
Embryos were dechorionated, deyolked and homogenized in RIPA buffer. Protein concentrations of embryo lysates were determined using the bicinchoninic acid (BCA) protein assay. The lysates were mixed with equal volumes of 2× SDS sample buffer, heated at 95°C for 2 min, and resolved on 12.5% or 15% gels. After transfer, the polyvinylidene difluoride membranes were incubated with primary antibodies [anti-LC3A/B (Cell Signaling Technology, Inc., #4108), anti-β-actin (Cell Signaling Technology, #4967), or anti-GFP (Life Technologies, A11122) antibody], diluted in TBST overnight at 4°C. After washing, the blot was then incubated with a secondary anti-rabbit horseradish peroxidase-conjugated antibody (Cell Signaling Technology, #7074) at room temperature for 1 h and visualized using an ECL kit (Perkin Elmer) in accordance with the manufacturer's instructions.
Generating transgenic zebrafish
To generate transgenic zebrafish expressing mCherry-tagged LC3, the corresponding expression construct pminiTol2-mCherry-LC3 was generated using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) in accordance with the manufacturer's instructions. pT3TS-Tol2 was linearized by XbaI and transcribed with T3 RNA polymerase using the Ambion mMESSAGE mMACHINE kit (Ambion, AM1348) to produce Tol2 transposase mRNA. Approximately 5 nl of the mixture of plasmid DNA (100 ng/µl) (pminiTol2-mCherry-LC3) and 50 pg of Tol2 transposase mRNA (100 ng/µl) were coinjected into newly fertilized embryos at the one-cell stage to produce transgenic fish. Injected embryos were raised to adulthood and out-crossed to wild-type fish to identify germline-transmitted transgenic founders (F0) as described previously [22]. Positive founders were determined by screening F1 embryos for visible mCherry expression. The mCherry-positive offspring were then allowed to grow to maturity for further experiments.
Chemical treatments
Bafilomycin A1 (BafA) (LC Laboratories, B-1080), omeprazole, lansoprazole, and pantoprazole (Sigma, O104, L8533, and P0021, respectively) treatment was performed from 36 through 48 hpf or 48 through 60 hpf in E3 medium at 28.5°C in 12 - or 6-well plates. The chemicals dissolved in DMSO were added to the embryo water (E3 medium) at the final concentrations of 200 nM for BafA and 25 µM for lansoprazole, omeprazole and pantoprazole. Pepstatin A (Fisher BioReagent, BP26715) and E-64-d (Enzo Life Sciences, BML-PI107) treatment was administrated from 60 through 72 hpf for 12 h in E3 medium at 28.5°C in 12 - or 6-well plates. These reagents were both dissolved in DMSO and added to the embryo water (E3 medium) at the final concentration of 5 µg/ml.
Quantitative analysis and statistics
Data processing and statistical analyses were performed using Statistical Package for the Social Sciences (SPSS) version 14.0. This software was used to generate each of the graphs shown in the text to perform statistical tests where appropriate.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. HeC, KlionskyDJ (2009) Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43 : 67–93.
2. VellaiT, Takacs-VellaiK, SassM, KlionskyDJ (2009) The regulation of aging: does autophagy underlie longevity? Trends Cell Biol 19 : 487–494.
3. ChoiAM, RyterSW, LevineB (2013) Autophagy in human health and disease. N Engl J Med 368 : 651–662.
4. DooleyK, ZonLI (2000) Zebrafish: a model system for the study of human disease. Curr Opin Genet Dev 10 : 252–256.
5. DimriGP, LeeX, BasileG, AcostaM, ScottG, et al. (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 92 : 9363–9367.
6. Debacq-ChainiauxF, ErusalimskyJD, CampisiJ, ToussaintO (2009) Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc 4 : 1798–1806.
7. KishiS, UchiyamaJ, BaughmanAM, GotoT, LinMC, et al. (2003) The zebrafish as a vertebrate model of functional aging and very gradual senescence. Exp Gerontol 38 : 777–786.
8. KoshimizuE, ImamuraS, QiJ, ToureJ, ValdezDMJr, et al. (2011) Embryonic senescence and laminopathies in a progeroid zebrafish model. PLoS One 6: e17688.
9. CaoL, LiW, KimS, BrodieSG, DengCX (2003) Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev 17 : 201–213.
10. KeyesWM, WuY, VogelH, GuoX, LoweSW, et al. (2005) p63 deficiency activates a program of cellular senescence and leads to accelerated aging. Genes Dev 19 : 1986–1999.
11. CaoL, XuX, BuntingSF, LiuJ, WangRH, et al. (2009) A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell 35 : 534–541.
12. KishiS, BaylissPE, UchiyamaJ, KoshimizuE, QiJ, et al. (2008) The identification of zebrafish mutants showing alterations in senescence-associated biomarkers. PLoS Genet 4: e1000152.
13. NakanoY, FujitaniK, KuriharaJ, RaganJ, Usui-AokiK, et al. (2001) Mutations in the novel membrane protein spinster interfere with programmed cell death and cause neural degeneration in Drosophila melanogaster. Mol Cell Biol 21 : 3775–3788.
14. YoungRM, MartyS, NakanoY, WangH, YamamotoD, et al. (2002) Zebrafish yolk-specific not really started (nrs) gene is a vertebrate homolog of the Drosophila spinster gene and is essential for embryogenesis. Dev Dyn 223 : 298–305.
15. DermautB, NorgaKK, KaniaA, VerstrekenP, PanH, et al. (2005) Aberrant lysosomal carbohydrate storage accompanies endocytic defects and neurodegeneration in Drosophila benchwarmer. J Cell Biol 170 : 127–139.
16. RongY, McPheeCK, DengS, HuangL, ChenL, et al. (2011) Spinster is required for autophagic lysosome reformation and mTOR reactivation following starvation. Proc Natl Acad Sci U S A 108 : 7826–7831.
17. YueZ, JinS, YangC, LevineAJ, HeintzN (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 100 : 15077–15082.
18. VousdenKH, PrivesC (2009) Blinded by the Light: The Growing Complexity of p53. Cell 137 : 413–431.
19. FengZ, ZhangH, LevineAJ, JinS (2005) The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A 102 : 8204–8209.
20. TasdemirE, MaiuriMC, GalluzziL, VitaleI, Djavaheri-MergnyM, et al. (2008) Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 10 : 676–687.
21. Scherz-ShouvalR, WeidbergH, GonenC, WilderS, ElazarZ, et al. (2010) p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc Natl Acad Sci U S A 107 : 18511–18516.
22. HeC, BartholomewCR, ZhouW, KlionskyDJ (2009) Assaying autophagic activity in transgenic GFP-Lc3 and GFP-Gabarap zebrafish embryos. Autophagy 5 : 520–526.
23. HeC, KlionskyDJ (2010) Analyzing autophagy in zebrafish. Autophagy 6 : 642–644.
24. HanM, ChangH, ZhangP, ChenT, ZhaoY, et al. (2013) C13C4.5/Spinster, an evolutionarily conserved protein that regulates fertility in C. elegans through a lysosome-mediated lipid metabolism process. Protein Cell 4 : 364–372.
25. SweeneyST, DavisGW (2002) Unrestricted synaptic growth in spinster-a late endosomal protein implicated in TGF-beta-mediated synaptic growth regulation. Neuron 36 : 403–416.
26. YanagisawaH, MiyashitaT, NakanoY, YamamotoD (2003) HSpin1, a transmembrane protein interacting with Bcl-2/Bcl-xL, induces a caspase-independent autophagic cell death. Cell Death Differ 10 : 798–807.
27. NiHM, BockusA, WozniakAL, JonesK, WeinmanS, et al. (2011) Dissecting the dynamic turnover of GFP-LC3 in the autolysosome. Autophagy 7 : 188–204.
28. LeeBY, HanJA, ImJS, MorroneA, JohungK, et al. (2006) Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 5 : 187–195.
29. HildebrandDG, LehleS, BorstA, HaferkampS, EssmannF, et al. (2013) alpha-Fucosidase as a novel convenient biomarker for cellular senescence. Cell Cycle 12 : 1922–1927.
30. UmataT, MoriyamaY, FutaiM, MekadaE (1990) The cytotoxic action of diphtheria toxin and its degradation in intact Vero cells are inhibited by bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase. J Biol Chem 265 : 21940–21945.
31. YoshimoriT, YamamotoA, MoriyamaY, FutaiM, TashiroY (1991) Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem 266 : 17707–17712.
32. SidiS, SandaT, KennedyRD, HagenAT, JetteCA, et al. (2008) Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell 133 : 864–877.
33. SajishM, ZhouQ, KishiS, ValdezDMJr, KapoorM, et al. (2012) Trp-tRNA synthetase bridges DNA-PKcs to PARP-1 to link IFN-gamma and p53 signaling. Nat Chem Biol 8 : 547–554.
34. LangheinrichU, HennenE, StottG, VacunG (2002) Zebrafish as a model organism for the identification and characterization of drugs and genes affecting p53 signaling. Curr Biol 12 : 2023–2028.
35. Karantza-WadsworthV, PatelS, KravchukO, ChenG, MathewR, et al. (2007) Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes Dev 21 : 1621–1635.
36. MathewR, KarpCM, BeaudoinB, VuongN, ChenG, et al. (2009) Autophagy suppresses tumorigenesis through elimination of p62. Cell 137 : 1062–1075.
37. LiuJ, XiaH, KimM, XuL, LiY, et al. (2011) Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell 147 : 223–234.
38. VousdenKH, RyanKM (2009) p53 and metabolism. Nat Rev Cancer 9 : 691–700.
39. KortleverRM, HigginsPJ, BernardsR (2006) Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol 8 : 877–884.
40. KuilmanT, MichaloglouC, MooiWJ, PeeperDS (2010) The essence of senescence. Genes Dev 24 : 2463–2479.
41. RodierF, CampisiJ (2011) Four faces of cellular senescence. J Cell Biol 192 : 547–556.
42. FujisawaK, TeraiS, HiroseY, TakamiT, YamamotoN, et al. (2011) Senescence marker protein 30 (SMP30)/regucalcin (RGN) expression decreases with aging, acute liver injuries and tumors in zebrafish. Biochem Biophys Res Commun 414 : 331–336.
43. FujitaT, UchidaK, MaruyamaN (1992) Purification of senescence marker protein-30 (SMP30) and its androgen-independent decrease with age in the rat liver. Biochim Biophys Acta 1116 : 122–128.
44. KondoY, InaiY, SatoY, HandaS, KuboS, et al. (2006) Senescence marker protein 30 functions as gluconolactonase in L-ascorbic acid biosynthesis, and its knockout mice are prone to scurvy. Proc Natl Acad Sci U S A 103 : 5723–5728.
45. IshigamiA, KondoY, NanbaR, OhsawaT, HandaS, et al. (2004) SMP30 deficiency in mice causes an accumulation of neutral lipids and phospholipids in the liver and shortens the life span. Biochem Biophys Res Commun 315 : 575–580.
46. KlionskyDJ, AbdallaFC, AbeliovichH, AbrahamRT, Acevedo-ArozenaA, et al. (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8 : 445–544.
47. RubinszteinDC, CodognoP, LevineB (2012) Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 11 : 709–730.
48. FlemingA, RubinszteinDC (2011) Zebrafish as a model to understand autophagy and its role in neurological disease. Biochim Biophys Acta 1812 : 520–526.
49. KlionskyDJ, ElazarZ, SeglenPO, RubinszteinDC (2008) Does bafilomycin A1 block the fusion of autophagosomes with lysosomes? Autophagy 4 : 849–950.
50. YamamotoA, TagawaY, YoshimoriT, MoriyamaY, MasakiR, et al. (1998) Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 23 : 33–42.
51. ShackaJJ, KlockeBJ, RothKA (2006) Autophagy, bafilomycin and cell death: the “a-B-cs” of plecomacrolide-induced neuroprotection. Autophagy 2 : 228–230.
52. PickartMA, SivasubbuS, NielsenAL, ShriramS, KingRA, et al. (2004) Functional genomics tools for the analysis of zebrafish pigment. Pigment Cell Res 17 : 461–470.
53. MoriyamaY, PatelV, UedaI, FutaiM (1993) Evidence for a common binding site for omeprazole and N-ethylmaleimide in subunit A of chromaffin granule vacuolar-type H(+)-ATPase. Biochem Biophys Res Commun 196 : 699–706.
54. LiuW, BakerSS, TrinidadJ, BurlingameAL, BakerRD, et al. (2013) Inhibition of lysosomal enzyme activities by proton pump inhibitors. J Gastroenterol 48(12): 1343–52.
55. LinHJ, HermanP, KangJS, LakowiczJR (2001) Fluorescence lifetime characterization of novel low-pH probes. Anal Biochem 294 : 118–125.
56. HemelaarJ, LelyveldVS, KesslerBM, PloeghHL (2003) A single protease, Apg4B, is specific for the autophagy-related ubiquitin-like proteins GATE-16, MAP1-LC3, GABARAP, and Apg8L. J Biol Chem 278 : 51841–51850.
57. TanidaI, WakabayashiM, KanematsuT, Minematsu-IkeguchiN, SouYS, et al. (2006) Lysosomal turnover of GABARAP-phospholipid conjugate is activated during differentiation of C2C12 cells to myotubes without inactivation of the mTor kinase-signaling pathway. Autophagy 2 : 264–271.
58. KabeyaY, MizushimaN, YamamotoA, Oshitani-OkamotoS, OhsumiY, et al. (2004) LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 117 : 2805–2812.
59. WeidbergH, ShvetsE, ShpilkaT, ShimronF, ShinderV, et al. (2010) LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 29 : 1792–1802.
60. LiangXH, JacksonS, SeamanM, BrownK, KempkesB, et al. (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402 : 672–676.
61. AitaVM, LiangXH, MurtyVV, PincusDL, YuW, et al. (1999) Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics 59 : 59–65.
62. QuX, YuJ, BhagatG, FuruyaN, HibshooshH, et al. (2003) Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 112 : 1809–1820.
63. MullerPA, VousdenKH (2013) p53 mutations in cancer. Nat Cell Biol 15 : 2–8.
64. MaiuriMC, GalluzziL, MorselliE, KeppO, MalikSA, et al. (2010) Autophagy regulation by p53. Curr Opin Cell Biol 22 : 181–185.
65. ItahanaK, DimriG, CampisiJ (2001) Regulation of cellular senescence by p53. Eur J Biochem 268 : 2784–2791.
66. DemidenkoZN, KorotchkinaLG, GudkovAV, BlagosklonnyMV (2010) Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci U S A 107 : 9660–9664.
67. MatheuA, MaraverA, KlattP, FloresI, Garcia-CaoI, et al. (2007) Delayed ageing through damage protection by the Arf/p53 pathway. Nature 448 : 375–379.
68. LinHK, ChenZ, WangG, NardellaC, LeeSW, et al. (2010) Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature 464 : 374–379.
69. BakerDJ, WeaverRL, van DeursenJM (2013) p21 both attenuates and drives senescence and aging in BubR1 progeroid mice. Cell Rep 3 : 1164–1174.
70. StorerM, MasA, Robert-MorenoA, PecoraroM, OrtellsMC, et al. (2013) Senescence Is a Developmental Mechanism that Contributes to Embryonic Growth and Patterning. Cell 155 : 1119–1130.
71. Munoz-EspinD, CanameroM, MaraverA, Gomez-LopezG, ContrerasJ, et al. (2013) Programmed Cell Senescence during Mammalian Embryonic Development. Cell 155 : 1104–1118.
72. ToledanoMB (2009) The guardian recruits cops: the p53-p21 axis delegates prosurvival duties to the Keap1-Nrf2 stress pathway. Mol Cell 34 : 637–639.
73. BensaadK, CheungEC, VousdenKH (2009) Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J 28 : 3015–3026.
74. SablinaAA, BudanovAV, IlyinskayaGV, AgapovaLS, KravchenkoJE, et al. (2005) The antioxidant function of the p53 tumor suppressor. Nat Med 11 : 1306–1313.
75. YuanXM, LiW, DalenH, LotemJ, KamaR, et al. (2002) Lysosomal destabilization in p53-induced apoptosis. Proc Natl Acad Sci U S A 99 : 6286–6291.
76. YuL, McPheeCK, ZhengL, MardonesGA, RongY, et al. (2010) Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465 : 942–946.
77. GuoL, LiewHP, CamusS, GohAM, CheeLL, et al. (2013) Ionizing radiation induces a dramatic persistence of p53 protein accumulation and DNA damage signaling in mutant p53 zebrafish. Oncogene 32 : 4009–4016.
78. BerghmansS, MurpheyRD, WienholdsE, NeubergD, KutokJL, et al. (2005) tp53 mutant zebrafish develop malignant peripheral nerve sheath tumors. Proc Natl Acad Sci U S A 102 : 407–412.
79. Vakifahmetoglu-NorbergH, KimM, XiaHG, IwanickiMP, OfengeimD, et al. (2013) Chaperone-mediated autophagy degrades mutant p53. Genes Dev 27 : 1718–1730.
80. FanX, KleinM, Flanagan-SteetHR, SteetR (2010) Selective yolk deposition and mannose phosphorylation of lysosomal glycosidases in zebrafish. J Biol Chem 285 : 32946–32953.
81. SagneC, AgulhonC, RavassardP, DarmonM, HamonM, et al. (2001) Identification and characterization of a lysosomal transporter for small neutral amino acids. Proc Natl Acad Sci U S A 98 : 7206–7211.
82. MindellJA (2012) Lysosomal acidification mechanisms. Annu Rev Physiol 74 : 69–86.
83. BaraschJ, KissB, PrinceA, SaimanL, GruenertD, et al. (1991) Defective acidification of intracellular organelles in cystic fibrosis. Nature 352 : 70–73.
84. SuzukiK, TanakaH, YamanakaT, Van DammeO (1980) The specificity of beta-galactosidase in the degradation of gangliosides. Adv Exp Med Biol 125 : 307–318.
85. KurzDJ, DecaryS, HongY, ErusalimskyJD (2000) Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci 113(Pt 20): 3613–3622.
86. RabenN, SchreinerC, BaumR, TakikitaS, XuS, et al. (2010) Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder–murine Pompe disease. Autophagy 6 : 1078–1089.
87. KimmelCB, BallardWW, KimmelSR, UllmannB, SchillingTF (1995) Stages of embryonic development of the zebrafish. Dev Dyn 203 : 253–310.
88. ImamuraS, UchiyamaJ, KoshimizuE, HanaiJ, RaftopoulouC, et al. (2008) A non-canonical function of zebrafish telomerase reverse transcriptase is required for developmental hematopoiesis. PLoS One 3: e3364.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2014 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Early Back-to-Africa Migration into the Horn of Africa
- PINK1-Mediated Phosphorylation of Parkin Boosts Parkin Activity in
- OsHUS1 Facilitates Accurate Meiotic Recombination in Rice
- Ancient DNA Analysis of 8000 B.C. Near Eastern Farmers Supports an Early Neolithic Pioneer Maritime Colonization of Mainland Europe through Cyprus and the Aegean Islands
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy