
High-Resolution Transcriptome Maps Reveal Strain-Specific Regulatory Features of Multiple Isolates
Campylobacter jejuni is currently the leading cause of bacterial gastroenteritis in humans. Comparison of multiple Campylobacter strains revealed a high genetic and phenotypic diversity. However, little is known about differences in transcriptome organization, gene expression, and small RNA (sRNA) repertoires. Here we present the first comparative primary transcriptome analysis based on the differential RNA–seq (dRNA–seq) of four C. jejuni isolates. Our approach includes a novel, generic method for the automated annotation of transcriptional start sites (TSS), which allowed us to provide genome-wide promoter maps in the analyzed strains. These global TSS maps are refined through the integration of a SuperGenome approach that allows for a comparative TSS annotation by mapping RNA–seq data of multiple strains into a common coordinate system derived from a whole-genome alignment. Considering the steadily increasing amount of RNA–seq studies, our automated TSS annotation will not only facilitate transcriptome annotation for a wider range of pro - and eukaryotes but can also be adapted for the analysis among different growth or stress conditions. Our comparative dRNA–seq analysis revealed conservation of most TSS, but also single-nucleotide-polymorphisms (SNP) in promoter regions, which lead to strain-specific transcriptional output. Furthermore, we identified strain-specific sRNA repertoires that could contribute to differential gene regulation among strains. In addition, we identified a novel minimal CRISPR-system in Campylobacter of the type-II CRISPR subtype, which relies on the host factor RNase III and a trans-encoded sRNA for maturation of crRNAs. This minimal system of Campylobacter, which seems active in only some strains, employs a unique maturation pathway, since the crRNAs are transcribed from individual promoters in the upstream repeats and thereby minimize the requirements for the maturation machinery. Overall, our study provides new insights into strain-specific transcriptome organization and sRNAs, and reveals genes that could modulate phenotypic variation among strains despite high conservation at the DNA level.
Published in the journal:
. PLoS Genet 9(5): e32767. doi:10.1371/journal.pgen.1003495
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003495
Summary
Campylobacter jejuni is currently the leading cause of bacterial gastroenteritis in humans. Comparison of multiple Campylobacter strains revealed a high genetic and phenotypic diversity. However, little is known about differences in transcriptome organization, gene expression, and small RNA (sRNA) repertoires. Here we present the first comparative primary transcriptome analysis based on the differential RNA–seq (dRNA–seq) of four C. jejuni isolates. Our approach includes a novel, generic method for the automated annotation of transcriptional start sites (TSS), which allowed us to provide genome-wide promoter maps in the analyzed strains. These global TSS maps are refined through the integration of a SuperGenome approach that allows for a comparative TSS annotation by mapping RNA–seq data of multiple strains into a common coordinate system derived from a whole-genome alignment. Considering the steadily increasing amount of RNA–seq studies, our automated TSS annotation will not only facilitate transcriptome annotation for a wider range of pro - and eukaryotes but can also be adapted for the analysis among different growth or stress conditions. Our comparative dRNA–seq analysis revealed conservation of most TSS, but also single-nucleotide-polymorphisms (SNP) in promoter regions, which lead to strain-specific transcriptional output. Furthermore, we identified strain-specific sRNA repertoires that could contribute to differential gene regulation among strains. In addition, we identified a novel minimal CRISPR-system in Campylobacter of the type-II CRISPR subtype, which relies on the host factor RNase III and a trans-encoded sRNA for maturation of crRNAs. This minimal system of Campylobacter, which seems active in only some strains, employs a unique maturation pathway, since the crRNAs are transcribed from individual promoters in the upstream repeats and thereby minimize the requirements for the maturation machinery. Overall, our study provides new insights into strain-specific transcriptome organization and sRNAs, and reveals genes that could modulate phenotypic variation among strains despite high conservation at the DNA level.
Introduction
Deep RNA–sequencing (RNA–seq) has been revolutionizing transcriptome analyses of both pro - and eukaryotes [1], [2]. Several recent RNA–seq studies have revealed an unexpectedly complex transcriptional output from bacterial genomes and have been successfully used for the global identification of small RNA (sRNA) genes as well transcriptional start sites (TSS) [2], [3]. For example, our recently developed differential RNA–seq (dRNA–seq) approach, which is selective for the analysis of primary transcriptomes, allowed us to provide a genome-wide map of TSS in Helicobacter pylori [4]. In addition, a steadily growing number of studies report cis-encoded antisense RNAs as a widespread layer of gene-expression control in bacteria [5], [6]. Despite this rapid accumulation of transcriptome data, the bioinformatics-based data mining is still lagging behind. Thus, in most cases transcriptome features such as TSS and novel non-coding RNAs still have to be manually annotated, which is laborious and time-consuming. The problem is compounded for comparative transcriptomics of several species within a genus. Therefore, most RNA–seq studies have been limited to single bacterial strains so far (reviewed in [2], [3]). However, a comparative approach would not only allow for refining the transcriptome annotation of the individual species by integrating the information from multiple strains but can also reveal differences in transcriptome organization or gene expression among strains for which different phenotypes cannot be explained by the genome sequences alone.
Epsilonproteobacteria, including several important human pathogens such as H. pylori and Campylobacter jejuni, show a significant strain-to-strain variability on the phenotypic and genomic level and, thus, represent good model organisms for such a comparative approach. For example, multilocus sequence typing (MLST) analysis revealed that C. jejuni, the most prevalent food-borne bacterial pathogen in the industrialized world to date [7], [8], is genetically diverse, with a weakly clonal population structure and high rates of intraspecies recombination [9], [10]. It has been suggested that this extensive genetic diversity could lead to different clinical outcomes and facilitate adaptation to and persistence in the host [11], [12], [13]. Since comparison among multiple species or strains has mainly been examined at the genomic level, the differences in transcriptome structure that have an impact on phenotypic flexibility remain currently unknown [14], [15].
C. jejuni is a commensal of chicken but leads to gastroenteritis in humans, where it has also been associated with the development of secondary autoimmune disorders such as the Guillain-Barré or Miller-Fisher syndromes [16]. Except for a cytolethal distending toxin (CDT) [17] and homologs of a type-IV secretion system on the pVir plasmid of strain 81–176 [18], C. jejuni lacks most classical virulence factors of other gastrointestinal pathogens. Therefore, it has been suggested that mainly the motility and metabolic capabilities of Campylobacter are required for virulence and colonization of the host [16], [19].
Besides the lack of classical virulence determinants, little is known about the transcriptome structure of Campylobacter. The 1.6 megabase A/T-rich (∼31% G+C content) genome of C. jejuni encodes only three sigma factors and a few transcriptional regulators [20], indicating additional layers of gene regulation. Bacterial sRNAs are an emerging class of post-transcriptional gene expression regulators which have been implicated in bacterial stress response and virulence regulation [21]. As do 50% of all bacteria, Campylobacter lacks a homolog of the RNA chaperone Hfq, which is a key player in sRNA–mediated regulation in enterobacteria [22], [23]. Despite the prediction of five sRNA candidates in C. jejuni NCTC11168 using conventional, strand-insensitive RNA–seq combined with predictions of conserved RNA structures [24], knowledge about post-transcriptional regulation in this pathogen is still very limited. Furthermore, a global TSS map including sRNA and antisense RNA promoters is still missing. However, such sRNA regulators could also contribute to phenotypic diversity by mediating strain-specific gene regulation.
To gain insight into the sRNA repertoires as well as differences in primary transcriptomes among multiple strains of one bacterial species, we applied the dRNA–seq approach in a comparative manner to four C. jejuni isolates. In addition, we introduce a novel automated, comparative TSS annotation method for a fast and accurate generation of genome-wide TSS maps. Using this method we identified conserved and strain-specific TSS, some of which carry SNPs in promoter regions. Moreover, we detected 15 conserved and 24 strain-specific sRNA candidates, highlighting differential sRNA expression among strains. Furthermore, expression and conservation analysis reveals the presence of a minimal CRISPR system, an RNA–based immune system, in C. jejuni. Overall, defining the Campylobacter transcriptome structure including genome-wide promoter maps provides new insights into gene regulation and transcriptome evolution not only in this species but also in other pathogens.
Results
dRNA–seq analysis of multiple Campylobacter jejuni strains
The determination of exact transcript boundaries and identification of novel transcripts facilitates genome annotation and also the discovery of regulatory elements which control gene expression. Our recently developed dRNA–seq approach allows for an efficient global TSS annotation by differential sequencing of two cDNA libraries which discriminate primary and processed 5′ ends: one library (−) is generated from untreated total RNA, whereas the second library (+) is generated after treatment with terminator exonuclease (TEX) which specifically degrades processed RNAs with a 5′-mono-phosphate [4].
To identify conserved and strain-specific transcriptome features, we have applied the dRNA–seq approach to three human and one chicken isolate of C. jejuni (see Table 1 and Figure S1 in Text S1). Strain NCTC11168 [20], originally isolated from a case of human enteritis in the UK, was the first Campylobacter strain for which the genome was sequenced but displays only poor motility and virulence. C. jejuni 81–176 [25], isolated from a diarrheal outbreak in the U.S., is highly pathogenic and carries two large plasmids, pVir [18] and pTet [26]. Strain 81116 [27], a human isolate from a waterborne outbreak in the UK in 1983, is a genetically stable and lab-adapted strain which is still infective for chicken [28]. The virulence potential of the chicken isolate RM1221 [14] is unknown.

Upon construction of two sets of dRNA–seq libraries from biological replicates of mid-log growth RNA samples for each of the four strains, we sequenced between 2.3 to 5.5 Mio cDNA reads per library which were subsequently mapped to the individual genome sequences (Table S1). Sequencing of dRNA–Seq libraries leads to a characteristic enrichment of cDNA reads at TSS in the TEX-treated sample [4]. These enrichment patterns allowed us to determine TSS in the four strains, and in many cases we observed an enrichment of cDNA reads at a given TSS in all four strains (for an example see Figure S2 in Text S1, rpsL). Moreover, our comparative dRNA–seq data showed that homologous genes might share the same TSS, even if the promoter regions are not conserved. Thus, based on sequence information alone it can be unclear whether a gene is expressed in the individual strains, whereas the comparative dRNA–seq data can provide this information. However, for several homologous genes, a clear TSS enrichment pattern (more than twofold in TEX+ vs. TEX−) was observed in only some of the strains despite highly conserved promoter regions (Figure S2 in Text S1, Cj1380). Nevertheless, a sharp flank in the cDNA distribution for all four strains indicates that transcription starts at exactly the same position. Without the comparative information from the other strains such cases could not unambiguously be defined as a TSS. Overall, a combination of sequence conservation and comparative TSS enrichment pattern analyses can be used to refine global TSS maps.
Automated and comparative TSS analysis using a SuperGenome approach
Manual annotation of TSS is labor - and time-consuming for a single strain and impractical for the analysis of multiple strains or larger genomes. To facilitate TSS comparisons, we mapped the four dRNA–seq data sets to a common coordinate system, the so-called SuperGenome [29], derived from a multiple whole genome alignment of the four strains (Figure 1A). Overall, 65% of the 2,115,275 SuperGenome positions were identical among the four strains (Table S2). The SuperGenome allows for a parallel visualization of the individual TEX+ and TEX - cDNA distributions of the four strains in a genome browser and thereby for a direct comparison of TSS enrichment patterns. The comparative dRNA–seq data show that the majority of TSS are enriched and detected in multiple strains (for an example locus see Figure 1B, black arrows) but that there are also differences among strains. For example, we observed a TSS within kpsM, which encodes for one of the capsule export genes, in only two of the strains and a TSS upstream of homologs of Cj1456c only in strain 81116 (Figure 1B, red arrow and blue arrows, respectively), indicating that there is strain-specific transcriptional output.

To automatically annotate TSS in a comparative manner, a two-step algorithm was employed: 1) TSS were detected independently for each strain based on dRNA–seq enrichment patterns and 2) TSS were mapped to the SuperGenome to allow for comparison and assignment of TSS among strains. Subsequently, all TSS of the individual strains were automatically classified as primary TSS (pTSS; main promoter of a gene) or secondary TSS (sTSS; alternative promoter upstream of pTSS of a gene), internal TSS (iTSS; promoter within gene), antisense TSS (asTSS; promoter antisense to a gene +/−100 nts) or orphan TSS (no association with annotation) according to their location relative to annotated genes (Figure S3A in Text S1 and Materials and Methods section). Our comparative approach enabled the annotation of a total of 3,377 TSS positions in the SuperGenome. 1,035 of the TSS were detected (but not necessarily enriched) in all four strains (Figure 2A, Table 2, and Table S3). Between 1,905 and 2,167 TSS were detected in the individual strains, including ∼300 strain-specific TSS (Table S4). The overall higher number of 2,167 TSS and 450 strain-specific TSS in RM1221 are mainly derived from TSS within its prophage elements (CMLP1, CJIE2-4) to which only cDNA reads from RM1221 were mapped in the SuperGenome (Table 1 and Figure 1A). On the two large plasmids, pVir (∼35 kB) and pTet (∼45 kB), of C. jejuni 81–176 we detected 70 and 58 TSS, respectively (Table S5). Approximately 60% of the conserved TSS are classified as pTSS or sTSS and in most cases drive transcription of mRNA genes. In contrast, only 21–31% of the strain-specific TSS are pTSS or sTSS and the majority of the strain-specific TSS (47–54%) are classified as antisense TSS (Table 2).


Comparative analysis of regulatory elements
Using MEME [30] we analyzed the promoter regions (−50 to +1 of all TSS) upstream of the total number of 8,019 TSS in all four strains for sequence motifs of at least 45 nt. This revealed a periodic A/T-rich pattern instead of a clear −35 box followed by an extended −10 box (TGxTATAAT) for ∼89% of the promoter regions in the four strains as a consensus motif for the housekeeping sigma factor, σ70 (Figure 2B). This motif fits with a previously predicted consensus sequence for a smaller number of Campylobacter promoters and for σ70 in H. pylori based on dRNA–seq, indicating that transcription predominantly initiates at an extended −10 box in Epsilonproteobacteria [4], [31]. Moreover, we identified motifs which resemble the consensus binding sites for the alternative sigma factors, σ28 (FliA) and σ54 (RpoN), for 141/8,019 (1.8%) and 36/8,019 (0.4%) TSS, respectively (Figure 2B). Separate analyses of the four strains indicated that there is no strong variation in their general promoter patterns among strains (Figure S4 in Text S1). Since some genes that are known to be regulated by the alternative sigma factors were missed in our MEME searches, we defined a consensus motif based on nine and eight TSS from strain NCTC11168 with a previously described FliA - and RpoN-dependent promoter, respectively (Table S6). Pattern searches with this consensus motif revealed additional 138 and 26 TSS fitting the FliA and RpoN motifs, respectively (Table S7).
Approximately 35% of all TSS in each strain are pTSS and around 10% are classified as sTSS (Table 2 and Figure S3 in Text S1). The majority of the 3,241 5′UTRs defined by pTSS and sTSS of mRNA genes have a length between 20 and 50 nt (Figure S5A in Text S1). MEME searches detected an “AAGGA”-motif with an upstream A/T-rich sequence as a consensus for the ribosome binding site in 2,990 out of the 3,158 5′UTRs with a length ≥8 nt.
Pairwise comparison of the 5′UTR lengths of genes with at least one ortholog in one of the other strains showed that the majority of the conserved genes have the same 5′UTR length but revealed also several length variations among strains (Figure S5B in Text S1). Despite a correction - where possible - for differences in start codon annotations (Tables S8 and S9), many of the 5′UTR length differences are due to different 5′ ends of the CDS of the respective genes (Table S10). In some cases, different 5′UTR lengths result from insertion of new genes upstream of the start codon and the acquisition of new promoters in certain strains (Figure 2C). For example, in three of the strains, a gene encoding for a restriction modification (RM) enzyme (CJE1195, C8J_0992, cjeI) is inserted upstream of npdA, encoding a NAD-dependent deacetylase, which is transcribed from a pTSS that was probably acquired together with cjeI. In contrast, strain 81–176 lacks the RM enzyme as well as the downstream promoter and npdA is co-transcribed as a polycistronic mRNA from a TSS upstream of murC.
Besides insertions of new upstream genes with a novel promoter, different promoters can lead to 5′UTR length variation among strains. For example, different TSS sets were detected in the four strains for homologs of a putative transporter, Cj0339 (Figure S6A in Text S1). Moreover, it has previously been reported that the asnA gene of strain 81–176 has acquired a sec-dependent secretion signal to the otherwise cytoplasmic asparaginase found in NCTC11168 and facilitates asparagine utilization in this strain [19]. Our dRNA–seq data indicate that the different asnA forms are transcribed from strain-specific promoters with ∼100-fold higher cDNA read counts for the secreted asparaginase compared to the cytoplasmic form (Figure S6B in Text S1).
SNP–dependent strain-specific promoter usage
Comparison of promoter motifs for conserved and strain-specific TSS revealed no difference in the general promoter patterns (Figure S7 in Text S1). However, our comparative TSS detection allowed us to identify regions with strain-specific promoter usage, e.g., regions for which we detected a TSS in only some of the strains although the region is present in the SuperGenome in all strains (Table S3, compare columns “mapCount” and “detCount”). We observed that in many cases single nucleotide polymorphisms (SNP) lead to disruption of promoters in a subset of strains (Figure 3). For example, the pTSS of pnk, encoding an inorganic polyphosphate/ATP-NAD kinase, is enriched and conserved in all four strains (Figure 3A). In contrast an iTSS within pnk is only detected in RM1221 and NCTC11168. In strains 81–176 and 81116 a C to T exchange in the “CGATTT” σ28 consensus seems to be sufficient to abolish transcription initiation from this TSS. Additional examples for mutations within conserved promoter elements are shown in Figure S8 in Text S1. Moreover, we noted that mutations within the periodic A/T-rich pattern upstream of the −10 box could also affect transcription (Figure 3B and Figure S9 in Text S1). For example, in strain NCTC11168, the Cj0004c and Cj0005c genes encode a monoheme cytochrome c and molybdopterin oxidoreductase, respectively, which allows C. jejuni to use sulphite as a respiratory electron donor [32]. This bi-cistronic operon along with the σ70 −10 box is conserved in all four strains (Figure 3B). However, mutations in the A/T rich upstream pattern of its pTSS in strain 81116 coincide with a loss of transcription either due to disturbing transcription initiation or binding of some regulatory factor. In line with this, we observed cytochrome c reduction as a measure for sulphite oxidation in only three of the strains (Figure 3C). This indicates, that although genes are conserved among strains and show high conservation in promoter regions, they may not necessarily be transcribed in all of them.

The non-coding RNA repertoire of C. jejuni
In addition to mRNA TSS, we identified many candidates for non-coding RNAs by our comparative dRNA–seq. For example, ∼45% of the TSS for each strain were classified as antisense TSS (asTSS) and 445 of the asTSS were detected in all four strains, indicating a large fraction of antisense transcription (Table 2, Figures S3 and S10 in Text S1). Furthermore, we detected several candidates for trans-encoded sRNAs in the chromosomes of the four strains and on the pVir and pTet plasmids (Figure 4, Figure S11 in Text S1 and Tables S11 and S12). Northern blot profiling under different growth phases confirmed expression of most of these sRNA candidates. Some sRNAs, such as CJnc60 or CJnc140, are highly conserved and show similar expression patterns in all strains. In contrast, some of the other conserved sRNAs, such as CJnc180 and CJnc190, which are encoded antisense to each other, show differential expression patterns among strains. Moreover, our Northern blots confirmed strain-specific sRNAs, such as CJnc30, which is only present in NCTC11168, or CJnc20, which is missing in RM1221. Most of our candidate sRNAs are transcribed from their own TSS, but we also found examples of sRNA candidates generated by processing, e.g., from 3′ ends of mRNAs (Figure S11 in Text S1 and Table S11). Furthermore, the majority of our sRNA candidates accumulated in exponential phase or during stationary phase growth.
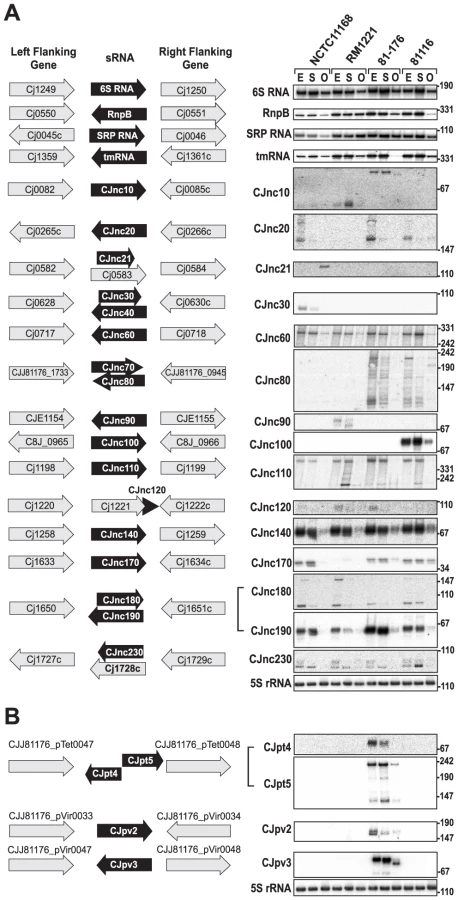
Conservation analysis of our sRNA candidates showed that the majority of them are restricted to Campylobacter jejuni (Figure 5), indicating that they either have a specific regulatory function in this species or that their sequence conservation is not high enough to detect homologs by BLAST searches. Even the housekeeping RNAs (SRP RNA, tmRNA, RNase P RNA, and 6S RNA) are not conserved at the sequence level outside Campylobacter species. Several of the C. jejuni sRNA candidates, e.g., CJnc170 and CJnc190, are highly conserved in diverse strains, and could have a more general regulatory role within C. jejuni. In contrast, some sRNA candidates such as CJnc30 and CJnc80 or the plasmid-encoded sRNAs are found in only some of the strains, indicating strain-specific sRNA repertoires which might contribute to strain-specific regulation of gene expression.

CRISPR loci in Campylobacter
One of the regions with highest numbers of cDNA reads in NCTC11168 and 811116 corresponded to the CRISPR (clustered regularly interspaced short palindromic repeats) locus. CRISPR loci are transcribed as precursors that are processed into mature crRNAs that together with Cas proteins silence invading foreign nucleic acids such as plasmids or phages [33], [34], [35]. Three of the four C. jejuni strains harbor a so-called type-II CRISPR/Cas system (Figure 6 and Figure S12 in Text S1), which requires a trans-encoded sRNA, TracrRNA, and the host factor RNase III for crRNA maturation [36], [37].

Our dRNA–seq analysis shows that the crRNAs and TracrRNA are actively transcribed and share a stretch of perfect complementarity in C. jejuni which allows for RNase III-dependent processing of the crRNAs. In line with this, we observe an accumulation of processed ∼38-nt spacer-repeat units and processed ∼62 nt TracrRNA (Figure 6B). Only weak CRISPR expression was detected in strain RM1221. Conservation analysis showed that the cas9 gene, which is required for the stability of crRNAs and cleavage of target DNA [36], [38], carries a stop mutation in strain RM1221 leading to a truncated protein (Figure 6C). In strain 81–176 the CRISPR region is replaced by two genes with very low G/C content compared to the flanking genomic regions.
The RNase III/TracrRNA–dependent processing leads to a cleavage event within the repeat region of the crRNAs [36]. Surprisingly, the mature spacer-repeat units are enriched in our TEX-treated libraries, indicative of primary transcripts starting at position five within each spacer. Moreover, the 3′ end of each repeat ends with “GGTAAAAT” resembling an extended −10 box. Thus, C. jejuni apparently employs promoter sequences within each repeat to initiate transcription of the associated spacer unit, so that only one processing event mediated by RNase III and TracrRNA in the repeat sequence is required to generate the 3′ end of the mature crRNA. In line with this model, we observed accumulation of longer transcripts upon deletion of RNase III (Figure S13 in Text S1). Primer extension analysis confirmed that these longer transcripts start at the proposed TSS within each spacer and that most of the TracrRNA species are transcribed from these upstream promoters. Thus, in contrast to other type-II systems, where the crRNAs are transcribed together with a leader sequence from one upstream promoter, each repeat in the C. jejuni CRISPR carries its own promoter (Figure 6D).
Discussion
Here we present the first comparative primary transcriptome analysis of four different C. jejuni strains including a novel automated TSS annotation method, which revealed strain-specific promoter usage and sRNA repertoires. The observed strain-specific transcriptional output reveals candidate genes which could contribute to phenotypic variation among strains or facilitate adaptation to different hosts or niches.
Previously, the generation of global TSS maps from RNA–seq data has mainly been performed on a manual or semi-automated basis [4], [39], [40], [41], [42], [43], [44], [45]. These strategies are typically very laborious and time-consuming with limited reproducibility and require even more effort for the comparison of transcriptomes from multiple conditions or strains. Therefore, we have developed a model-based TSS prediction method based on criteria used for manual TSS annotations. It provides detailed values (such as number of read starts, enrichment factor etc.) as well as classifications (pTSS, sTSS, asTSS, iTSS) for each TSS candidate which allows the user to review the underlying thresholds for TSS detection. Benchmarking of our TSS prediction method using the manually annotated TSS from H. pylori [4] showed that our approach achieved a sensitivity of 82% and a precision rate of 75%. Since our comparative approach allows for the integration of data sets from different strains or conditions, its performance can generally be increased by the inclusion and comparison of multiple data sets and replicates. Moreover, the TSS annotation will be further improved by adjusting the parameters to additional training sets based on experimental validation. Recently, Schmidtke et al. presented a fully automated approach for TSS annotation of a single genome based on read count data and a sophisticated statistical model, which calculates p-values for TSS candidates and thereby provides a confidence estimation for observing a TSS [46]. However, the specific properties of the TSS candidates, such as expression height and dRNA–seq enrichment factor, cannot be directly inferred from the resulting p-values. In future, a combination of both approaches would be promising especially for very weak TSS candidates. Furthermore, our automated TSS method also allows for a comparative TSS annotation among dRNA–seq libraries from multiple strains by the integration of the SuperGenome approach. Considering the rapidly increasing number of genome sequences as well as RNA–seq studies, our approach will facilitate a systematic TSS annotation among different strains or growth conditions and can also be adapted to the analyses of eukaryotic transcriptomes.
Comparative genomics of multiple Campylobacter strains or species provided the core genome of the genus and also revealed differences in genome content and structure which could support adaptation to different hosts [14], [15], [25], [47], [48]. Furthermore, comparison of genetically closely related C. jejuni strains isolated from different hosts indicated that Campylobacter uses high phenotypic flexibility and genetic microdiversity to reversibly adapt to changing environments [49]. In addition, variation in contingency loci contributes to rapid adaptation to novel hosts [50]. This genetic flexibility among strains might be supported by differences in transcription among strains and post-transcriptional regulation which we observed in our comparative dRNA–seq data. For this first comparative analysis we have selected widely used laboratory strains with available genome sequence. However, our comparative approach can be easily adapted to the analysis of multiple isolates from different hosts or isolation sources. For example, the use of our single-nucleotide resolution approach could help to understand the transcriptomic and phenotypic differences that were previously observed during the analysis of the genome-sequenced and original isolate of C. jejuni NCTC11168 [51].
Our comparative dRNA–seq approach allowed us to annotate between 1,905 and 2,167 TSS in the four C. jejuni strains. We observed that the majority of TSS are conserved among multiple strains, but we also found many examples where SNPs in promoter regions apparently resulted in strain-specific promoter usage. Thus, although some promoters are highly conserved and show almost perfect overlap to promoter consensus motifs - which would be indicative for active transcription - the respective genes are not necessarily expressed at the same level among strains. Therefore, comparative transcriptomics facilitates the identification of differences in the functional output from genomes which cannot be directly inferred from closely related DNA sequences.
SNPs can be adaptive and, thereby, lead to niche expansion [52]. In addition, point mutations in ORFs or rRNA genes can mediate antibiotics resistances in bacteria including Campylobacter [53]. In bacterial pathogens, a variety of pathoadaptive mutations have been described which can affect cell binding, host tissue tropism or virulence regulation [54], [55], [56], [57]. Furthermore, point mutations in ORFs could lead to truncated or non-functional proteins or changes in protein activity [58], [59], [60] and have also been shown to discriminate mRNA targets of bacterial sRNAs [61]. Even single regulatory genes or promoter inversions can be sufficient to alter bacterial host specificity [62], [63] and promoter SNPs have been associated with overproduction of virulence genes [64], [65]. Based on our global TSS maps, we identified several examples where SNPs disrupt conserved positions in promoter motifs recognized by the three sigma factors. Furthermore, we found several examples for disruptive SNPs in the A/T-rich upstream regions, indicating that this region is also required for transcription initiation in Epsilonproteobacteria. These promoters with SNPs might be good hints to genes which contribute to adaptation to different environments or hosts. For example, in strain 81116 we observed mutations in the promoter for monoheme cytochrome c and a molybdopterin oxidoreductase, which allow C. jejuni to use sulphite as a respiratory electron donor [32]. This indicates that strain 81116 has lost the ability to transcribe this operon and perhaps even lost the ability to respire sulphite (Figure 3). C. jejuni is unable to metabolize glucose since it lacks the enzyme phosphofructokinase. Instead, it has a complex branched respiratory chain and can utilize a variety of electron donors like formate, lactate, or sulphite. Sulphite respiration might also help C. jejuni to survive in sulphite-rich niches and foods, and might also be used for detoxification. Furthermore, a strain lacking the molybdopterin oxidoreductase has been shown to have a reduced ability to infect Caco-2 cells [66].
Since the SNPs in promoter regions could also interfere with binding of a transcriptional regulator and thereby affect transcription, such promoters with strain-specific expression patterns represent good candidates to fish for novel DNA-binding proteins. For example, a SNP in the Fur-binding site of the sodB promoter of certain Helicobacter pylori strains has been shown to affect direct binding of apo-Fur [67]. The global map of TSS and promoter SNPs may give hints as to how C. jejuni and other microbes with compact genomes and few transcription factors could adapt gene expression according to different environmental conditions.
Our comparative approach allowed us to annotate TSS by combining the dRNA–seq data from multiple strains. This approach improves the annotation accuracy of the individual strains by integrating the information from multiple transcriptomes and also reveals differences among strains. Previously, bacterial RNA–seq studies mainly focused on the analyses of single strains (reviewed in [2]) or were not strand-specific [68]. Two recent comparative transcriptome analyses using strand-specific cDNA sequencing mainly focused on the divergence of sRNA expression as well as long antisense RNAs [45], [69]. Apart from these studies, expression profiling of sRNAs in multiple strains has been reported only for a limited number of bacteria and was mainly based on Northern blot analysis [70], [71]. A global RNA–seq based comparison of regulatory elements including sRNAs was recently carried out between the two closely related species, Escherichia coli and Klebsiella pneumoniae, and revealed that the majority of orthologous operons were transcribed from different promoters [72].
Our global transcriptome maps revealed several candidates for conserved and strain-specific sRNAs. Small regulatory RNAs have been implicated as key regulators in metabolic pathways and during pathogenesis [73], [74]. The newly identified sRNAs could contribute to virulence gene regulation and host adaptation by modulating metabolic pathways which are important for host colonization in C. jejuni [19]. A study based on conventional, non strand-specific RNA–seq predicted five sRNAs in C. jejuni [24]. In our strand-specific dRNA–seq data, we observed expression for four of them (Figure S11 in Text S1). Moreover, a dRNA–seq study of C. jejuni NCTC11168 compared the transcriptome organization between C. jejuni and H. pylori [4] and identified around 20 trans-encoded sRNA candidates in C. jejuni, most of which are also detected in our study (I. Porcelli and A. van Vliet, personal communication). The majority of our sRNA candidates are expressed as independent transcripts. However, we also detected examples of processed sRNA species, which can be generated from the 3′ end of mRNAs. Recently, such 3′-end derived transcripts have been shown to stably associate with the RNA chaperone Hfq in Salmonella and to act as regulatory RNAs on trans-encoded mRNAs [75]. Since Campylobacter lacks Hfq, it will be interesting to see whether its sRNAs require a different RNA chaperone for their activity and stability or whether they act independently of an auxiliary protein. Moreover, future studies will be required to uncover the target genes and physiological roles of sRNAs in Campylobacter.
The most abundant sRNA in strain NCTC11168 corresponds to TracrRNA, which is required together with RNase III for maturation of CRISPR RNAs [36]. Due to the high variability of the spacer sequences, CRISPR loci have been used for strain genotyping including Campylobacter species [76]. Surprisingly, we found that the crRNAs in C. jejuni are transcribed from individual promoters within each repeat unit (Figure 6). In other prokaryotes, the crRNAs are transcribed from a leader sequence and several processing steps are required to generate the mature crRNAs. Therefore, the CRISPR locus of Campylobacter with its individual crRNA promoters and only three cas proteins represents a “minimal” system of the type-II subtype (Figure 6D) which requires only one processing event by RNase III within the repeats to generate the mature crRNAs. A similar CRISPR organization was also identified in Neisseria menigitidis (N. Heidrich and J. Vogel, personal communication). Furthermore, in strain RM1221, the crRNAs and TracrRNA were only weakly expressed probably due to a stop-mutation in cas9, while strain 81–176 completely lacks the CRISPR locus. Interestingly, these two strains carry prophages or plasmids, indicating that these horizontally acquired genetic elements could be mutually exclusive with an active CRISPR system. Conservation analysis in additional strains showed that strains with plasmids or integrated elements very often carry degenerated CRISPR loci (Figure S14 in Text S1). Moreover, it has recently been shown that ganglioside-like LOS structures of GBS-associated C. jejuni strains can confer efficient bacteriophage resistance and that the presence of sialyltransferases correlates significantly with an apparently non-functional CRISPR system [77]. Further studies of the influence of the CRISPR system on pathogenicity of Campylobacter will be required. Moreover, since components of type-II CRISPR systems have recently been adapted for genome editing in humans [78], [79], the minimal type-II systems might be useful for further improvements of such genome editing tools.
Overall our high-resolution transcriptome map revealed regulatory elements and their conservation in multiple Campylobacter jejuni strains on a genome-wide scale. The comparison of multiple strains improves annotation of transcriptome features such as TSS maps and reveals strain-specific TSS usage as well as sRNA repertoires. These strain-specific transcription patterns will provide new insights into genes which could promote phenotypic differences despite high conservation at the genome level. Our novel automated TSS annotation can easily be applied to a wider range of strains or conditions and may also be used for to the annotation of eukaryotic transcriptomes.
Materials and Methods
Bacterial strains and oligonucleotides
Campylobacter jejuni strains and DNA oligonucleotides used for cloning, as hybridization probes or for primer extension are listed in Tables S13 and S14, respectively.
Campylobacter jejuni growth
Bacteria were grown on Müller-Hinton agar plates supplemented with 10 µg/ml vancomycin at 37°C under microaerobic conditions (10% CO2, 5% O2). For liquid cultures, a starter culture was inoculated with bacteria grown on plates to a final OD600 of 0.04 in 20 ml of Brucella broth (BB) medium including 10 µg/ml vancomycin and incubated overnight at 37°C under microaerobic atmosphere and an agitation of 140 rpm. The next day, 50 ml BB including 10 µg/ml vancomycin were inoculated to a final OD600 of 0.04 using the starter culture and incubated as described above. When the cultures reached mid-exponential (6.5 hrs; OD600 between ∼0.3–0.4), stationary (13 hrs; OD600 between ∼0.5–1) and overnight phase (29 hrs; OD600 between ∼0.5–1), culture volumes of cells corresponding to a total amount of 2, 4 and 8 OD600, respectively, were mixed with 0.2 volumes of stop-mix (95% EtOH and 5% phenol, V/V), frozen in liquid N2 and stored at −80°C until RNA extraction. The four strains varied in the final OD600 values that they reached at these growth phases, e.g., strain 81–176 showed highest OD600 values of up to ∼1.0 in stationary and overnight phase, whereas strain 81116 reached only OD600 values of ∼0.5. However, despite these differences in OD600 the harvested samples corresponded to similar growth phases along the growth curves of the four strains.
RNA extraction and Northern blot
Frozen cell pellets were thawed on ice and resuspended in lysis solution containing 600 µl of 0.5 mg/ml lysozyme in TE buffer (pH 8.0) and 60 µl 10% SDS. Bacterial cells were lysed by incubating the samples for 1–2 minutes at 65°C. Afterwards, total RNA was extracted using the hot-phenol method described previously [80].
For Northern Blot analysis, 10 to 15 µg RNA was loaded per sample. After separation on 6% polyacrylamide (PAA) gels containing 7 M urea, RNA was transferred to Hybond-XL membranes, which were hybridized with γ32P-ATP end-labeled oligodeoxyribonucleotide probes indicated in Table S14. Candidate sRNAs with more than 50 reads (relative score) in untreated dRNA–seq libraries were probed on Northern blots.
Construction of cDNA libraries for dRNA–seq and Illumina sequencing
For each of the four selected C. jejuni strains, NCTC1116, 81–176, 81116, and RM1221, dRNA–seq libraries were constructed from biological duplicates of RNA samples harvested at mid-log growth in BB.
Residual genomic DNA was removed from the total RNA isolated by DNase I treatment. For depletion of processed transcripts, equal amounts of Campylobacter RNA were incubated with Terminator™ 5′-phosphate-dependent exonuclease (TEX) (Epicentre #TER51020) as previously described [4].
Libraries for Solexa sequencing (HiSeq) of cDNA were constructed by vertis Biotechnology AG, Germany (http://www.vertis-biotech.com/), as described previously for eukaryotic microRNAs [81] but omitting the RNA size-fractionation step prior to cDNA synthesis. For details see Supplementary Methods in Text S1.
The resulting cDNA libraries were sequenced using a HiSeq 2000 machine (Illumina) in single-read mode. The raw, de-multiplexed reads as well as coverage files (see Supplementary Methods in Text S1) have been deposited in the National Center for Biotechnology Information's Gene Expression Omnibus [82] under the accession GSE38883.
For a detailed description of the read mapping, expression graph construction and normalization of expression graphs see Supplementary Methods in Text S1. In total, we sequenced between 2.3 to 5.5 Mio cDNA reads for each of the cDNA libraries which were subsequently mapped to the individual genome sequences (Table S1).
Transcriptional start site (TSS) annotation and SuperGenome approach
A whole genome alignment of the four C. jejuni strains was computed with Mauve [83]. Based on this global alignment a common genomic coordinate system, the SuperGenome was defined into which all positional information can be projected that relates to the single genomes [29]. This resulted in a consensus sequence with the coordinates of the complete alignment and a mapping of each position of each single genome to a position in the alignment. Next, all genome-specific data (expression height graphs derived from mapped read data, genomic annotations, and sequences) were mapped to the common coordinate system.
Our automated TSS prediction approach, which uses this SuperGenome mapping for comparative analyses, consists of several steps: The initial detection of TSS in the single strains is based on the localization of positions, where a significant number of reads start. Thus, for each position i in the RNA–seq graph corresponding to the TEX+ library the algorithm calculates e(i)-e(i-1), where e(i) is the expression height at position i (Figure S15 in Text S1). In addition, the factor of height change is calculated, i.e. e(i)/e(i-1). To evaluate if the reads starting at this position are originating from primary transcripts, the enrichment factor is calculated as eTEX+(i)/eTEX−(i). For all positions where these values exceed the threshold (see Supplementary Material) a TSS candidate is annotated.
The TSS prediction procedure is applied to both replicates of each strain. TSS candidates, which are not detected in both replicates with a maximal positional difference of one nucleotide, are discarded. Afterwards, TSS candidates that are in close vicinity are grouped into a cluster and only the TSS candidate with the highest expression is kept. In the next step, the TSS candidates of each strain are mapped to the SuperGenome to assign each TSS to the corresponding TSS in the other strains. The final TSS annotations are then characterized on the SuperGenome level with respect to their occurrence in the different strains and in which strains they appear to be enriched. In the context of the individual strains the TSS are further classified according to their location relative to annotated genes. For this we used a similar classification scheme as previously described [4]. Thus, for each TSS it is decided if it is the primary or secondary TSS of a gene, if it is an internal TSS, an antisense TSS or if it cannot be assigned to one of these classes (orphan). A TSS is classified as primary or secondary if it is located ≤300 bp upstream of a gene. The TSS with the strongest expression considering all strains is classified as primary. All other TSS that are assigned to the same gene are classified as secondary. Internal TSS are located within an annotated gene on the sense strand and antisense TSS are located inside a gene or within ≤100 bp on the antisense strand. These assignments are indicated by a 1 in the respective column of Tables S4, S5, S6, S7, S8, S9. Orphan TSS, which are not in the vicinity of an annotated gene, are indicated by “0” in all four columns.
To validate our automated TSS detection we applied it to the previously generated dRNA–seq data of Helicobacter pylori grown under five different conditions [4]. In this study, we had manually annotated the TSS based on enrichment patterns in the TEX+ compared to TEX - libraries. We used these hand-curated TSS positions as benchmark and compared it to the results of the automated detection. We allowed a difference of up to one nucleotide when comparing an automatically detected TSS to a manually annotated TSS. With this threshold, the automated approach achieves a sensitivity of 82% and a precision rate of 75%. The parameters used for the TSS annotation in C. jejuni were selected according to this benchmarking with the manual TSS set of H. pylori (see also Supplementary Methods in Text S1).
Promoter and RBS motifs detection and data visualization
To detect potential promoter motifs, sequence regions corresponding to 50 nt upstream of the TSS positions and the TSS position itself were scanned by MEME version 4.8.1 [84] with width parameters (fixed width of 45 nt as well as flexible widths). For the detection of ribosome binding site (RBS) motifs, the 5′UTR sequences of mRNAs were inspected by MEME. The 5′UTR length distributions were visualized using R and the ggplot2 package.
Cytochrome c oxidoreductase activity assay
The assay was adapted from Kappler et al [85]. The activity of the oxidoreductase enzyme was measured as the increase in cytochrome c absorbance at 550 nm when converted from the oxidized to the reduced form. Briefly, 2 µl cells (corresponding to 0.01 OD600 nm in 10 mM Tris/HCl pH 8) were added to a freshly prepared mix containing the following: 45 µl 10 mM Tris/HCl pH 8 (with or without 2 mM sodium sulphite), 5 µl horse heart cytochrome c (10 mg/ml; #C2506, Sigma-Aldrich). Absorbance was measured after 15 minutes at 550 nm using an absorption coefficient of 20 mM−1cm−1 (spectrophotometer ND-1000; Peqlab). In the control sample, 2 µl of 10 mM Tris/HCl pH 8 was added instead of cells.
Conservation analysis of sRNAs
To study the conservation of sRNAs in Epsilonproteobacteria, homologous sequences were searched with blastn (part of the BLAST+ package version 2.2.26 [86]; the word-size parameter was set to 10 nt). The number of identical nucleotides of the best hits of each sRNA candidate was divided by the total number of nucleotides of the query sRNA and multiplied by 100 to calculate the percentage value of conserved nucleotides. For visualization, conservation values ≥40% identity were translated into a gray scale and values below 40% were depicted as white boxes.
Supporting Information
Zdroje
1. WangZ, GersteinM, SnyderM (2009) RNA–Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10 : 57–63.
2. CroucherNJ, ThomsonNR (2010) Studying bacterial transcriptomes using RNA–seq. Curr Opin Microbiol 13 : 619–624.
3. van VlietAH (2010) Next generation sequencing of microbial transcriptomes: challenges and opportunities. FEMS Microbiol Lett 302 : 1–7.
4. SharmaCM, HoffmannS, DarfeuilleF, ReignierJ, FindeissS, et al. (2010) The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464 : 250–255.
5. ThomasonMK, StorzG (2010) Bacterial antisense RNAs: how many are there, and what are they doing? Annu Rev Genet 44 : 167–188.
6. GeorgJ, HessWR (2011) cis-antisense RNA, another level of gene regulation in bacteria. Microbiol Mol Biol Rev 75 : 286–300.
7. DastiJI, TareenAM, LugertR, ZautnerAE, GrossU (2010) Campylobacter jejuni: a brief overview on pathogenicity-associated factors and disease-mediating mechanisms. Int J Med Microbiol 300 : 205–211.
8. YoungKT, DavisLM, DiritaVJ (2007) Campylobacter jejuni: molecular biology and pathogenesis. Nat Rev Microbiol 5 : 665–679.
9. SuerbaumS, LohrengelM, SonnevendA, RubergF, KistM (2001) Allelic diversity and recombination in Campylobacter jejuni. J Bacteriol 183 : 2553–2559.
10. DingleKE, CollesFM, WareingDR, UreR, FoxAJ, et al. (2001) Multilocus sequence typing system for Campylobacter jejuni. J Clin Microbiol 39 : 14–23.
11. LinzB, SchusterSC (2007) Genomic diversity in Helicobacter and related organisms. Res Microbiol 158 : 737–744.
12. SuerbaumS, JosenhansC (2007) Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat Rev Microbiol 5 : 441–452.
13. WassenaarTM, NewellDG (2000) Genotyping of Campylobacter spp. Appl Environ Microbiol 66 : 1–9.
14. FoutsDE, MongodinEF, MandrellRE, MillerWG, RaskoDA, et al. (2005) Major structural differences and novel potential virulence mechanisms from the genomes of multiple campylobacter species. PLoS Biol 3: e15 doi:10.1371/journal.pbio.0030015.
15. ParkerCT, QuinonesB, MillerWG, HornST, MandrellRE (2006) Comparative genomic analysis of Campylobacter jejuni strains reveals diversity due to genomic elements similar to those present in C. jejuni strain RM1221. J Clin Microbiol 44 : 4125–4135.
16. WassenaarTM, BlaserMJ (1999) Pathophysiology of Campylobacter jejuni infections of humans. Microbes Infect 1 : 1023–1033.
17. Lara-TejeroM, GalanJE (2000) A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 290 : 354–357.
18. BaconDJ, AlmRA, HuL, HickeyTE, EwingCP, et al. (2002) DNA sequence and mutational analyses of the pVir plasmid of Campylobacter jejuni 81–176. Infect Immun 70 : 6242–6250.
19. HofreuterD, NovikV, GalanJE (2008) Metabolic diversity in Campylobacter jejuni enhances specific tissue colonization. Cell Host Microbe 4 : 425–433.
20. ParkhillJ, WrenBW, MungallK, KetleyJM, ChurcherC, et al. (2000) The genome sequence of the food-borne pathogen Campylobacter jejuni reveals hypervariable sequences. Nature 403 : 665–668.
21. StorzG, VogelJ, WassarmanKM (2011) Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 43 : 880–891.
22. VogelJ, LuisiBF (2011) Hfq and its constellation of RNA. Nat Rev Microbiol 9 : 578–589.
23. ChaoY, VogelJ (2010) The role of Hfq in bacterial pathogens. Curr Opin Microbiol 13 : 24–33.
24. ChaudhuriRR, YuL, KanjiA, PerkinsTT, GardnerPP, et al. (2011) Quantitative RNA–seq analysis of the transcriptome of Campylobacter jejuni. Microbiology 157 : 2922–2932.
25. HofreuterD, TsaiJ, WatsonRO, NovikV, AltmanB, et al. (2006) Unique features of a highly pathogenic Campylobacter jejuni strain. Infect Immun 74 : 4694–4707.
26. BatchelorRA, PearsonBM, FriisLM, GuerryP, WellsJM (2004) Nucleotide sequences and comparison of two large conjugative plasmids from different Campylobacter species. Microbiology 150 : 3507–3517.
27. PearsonBM, GaskinDJ, SegersRP, WellsJM, NuijtenPJ, et al. (2007) The complete genome sequence of Campylobacter jejuni strain 81116 (NCTC11828). J Bacteriol 189 : 8402–8403.
28. ManningG, DuimB, WassenaarT, WagenaarJA, RidleyA, et al. (2001) Evidence for a genetically stable strain of Campylobacter jejuni. Appl Environ Microbiol 67 : 1185–1189.
29. HerbigA, JagerG, BattkeF, NieseltK (2012) GenomeRing: alignment visualization based on SuperGenome coordinates. Bioinformatics 28: i7–15.
30. BaileyTL, ElkanC (1994) Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2 : 28–36.
31. PetersenL, LarsenTS, UsseryDW, OnSL, KroghA (2003) RpoD promoters in Campylobacter jejuni exhibit a strong periodic signal instead of a -35 box. J Mol Biol 326 : 1361–1372.
32. MyersJD, KellyDJ (2005) A sulphite respiration system in the chemoheterotrophic human pathogen Campylobacter jejuni. Microbiology 151 : 233–242.
33. BrounsSJJ, JoreMM, LundgrenM, WestraER, SlijkhuisRJH, et al. (2008) Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321 : 960–964.
34. HaleCR, ZhaoP, OlsonS, DuffMO, GraveleyBR, et al. (2009) RNA–Guided RNA Cleavage by a CRISPR RNA–Cas Protein Complex. Cell 139 : 945–956.
35. MarraffiniLA, SontheimerEJ (2008) CRISPR Interference Limits Horizontal Gene Transfer in Staphylococci by Targeting DNA. Science 322 : 1843–1845.
36. DeltchevaE, ChylinskiK, SharmaCM, GonzalesK, ChaoY, et al. (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471 : 602–607.
37. MakarovaKS, HaftDH, BarrangouR, BrounsSJ, CharpentierE, et al. (2011) Evolution and classification of the CRISPR-Cas systems. Nat Rev Microbiol 9 : 467–477.
38. JinekM, ChylinskiK, FonfaraI, HauerM, DoudnaJA, et al. (2012) A Programmable Dual-RNA–Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science
39. MitschkeJ, GeorgJ, ScholzI, SharmaCM, DienstD, et al. (2011) An experimentally anchored map of transcriptional start sites in the model cyanobacterium Synechocystis sp. PCC6803. Proc Natl Acad Sci U S A 108 : 2124–2129.
40. AlbrechtM, SharmaCM, ReinhardtR, VogelJ, RudelT (2010) Deep sequencing-based discovery of the Chlamydia trachomatis transcriptome. Nucleic Acids Res 38 : 868–877.
41. JagerD, SharmaCM, ThomsenJ, EhlersC, VogelJ, et al. (2009) Deep sequencing analysis of the Methanosarcina mazei Go1 transcriptome in response to nitrogen availability. Proc Natl Acad Sci U S A 106 : 21878–21882.
42. FiliatraultMJ, StodghillPV, MyersCR, BronsteinPA, ButcherBG, et al. (2012) Genome-wide identification of transcriptional start sites in the plant pathogen Pseudomonas syringae pv. tomato str. DC3000. PLoS ONE 6: e29335 doi:10.1371/journal.pone.0029335.
43. SchluterJP, ReinkensmeierJ, DaschkeyS, Evguenieva-HackenbergE, JanssenS, et al. (2010) A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genomics 11 : 245.
44. WurtzelO, SapraR, ChenF, ZhuY, SimmonsBA, et al. (2010) A single-base resolution map of an archaeal transcriptome. Genome Res 20 : 133–141.
45. WurtzelO, SestoN, MellinJR, KarunkerI, EdelheitS, et al. (2012) Comparative transcriptomics of pathogenic and non-pathogenic Listeria species. Mol Syst Biol 8 : 583.
46. SchmidtkeC, FindeissS, SharmaCM, KuhfussJ, HoffmannS, et al. (2012) Genome-wide transcriptome analysis of the plant pathogen Xanthomonas identifies sRNAs with putative virulence functions. Nucleic Acids Res
47. PolyF, ReadTD, ChenYH, MonteiroMA, SerichantalergsO, et al. (2008) Characterization of two Campylobacter jejuni strains for use in volunteer experimental-infection studies. Infect Immun 76 : 5655–5667.
48. FriisC, WassenaarTM, JavedMA, SnipenL, LagesenK, et al. (2010) Genomic characterization of Campylobacter jejuni strain M1. PLoS ONE 5: e12253 doi:10.1371/journal.pone.0012253.
49. GrippE, HlahlaD, DidelotX, KopsF, MaurischatS, et al. (2011) Closely related Campylobacter jejuni strains from different sources reveal a generalist rather than a specialist lifestyle. BMC Genomics 12 : 584.
50. JeromeJP, BellJA, Plovanich-JonesAE, BarrickJE, BrownCT, et al. (2011) Standing genetic variation in contingency loci drives the rapid adaptation of Campylobacter jejuni to a novel host. PLoS ONE 6: e16399 doi:10.1371/journal.pone.0016399.
51. GaynorEC, CawthrawS, ManningG, MacKichanJK, FalkowS, et al. (2004) The genome-sequenced variant of Campylobacter jejuni NCTC 11168 and the original clonal clinical isolate differ markedly in colonization, gene expression, and virulence-associated phenotypes. J Bacteriol 186 : 503–517.
52. PerutzMF (1983) Species adaptation in a protein molecule. Mol Biol Evol 1 : 1–28.
53. AlfredsonDA, KorolikV (2007) Antibiotic resistance and resistance mechanisms in Campylobacter jejuni and Campylobacter coli. FEMS Microbiol Lett 277 : 123–132.
54. Wilmes-RiesenbergMR, FosterJW, CurtissR3rd (1997) An altered rpoS allele contributes to the avirulence of Salmonella typhimurium LT2. Infect Immun 65 : 203–210.
55. HorstmannN, SahasrabhojaneP, SuberB, KumaraswamiM, OlsenRJ, et al. (2011) Distinct single amino acid replacements in the control of virulence regulator protein differentially impact streptococcal pathogenesis. PLoS Pathog 7: e1002311 doi:10.1371/journal.ppat.1002311.
56. Van SteltenA, SimpsonJM, WardTJ, NightingaleKK (2010) Revelation by single-nucleotide polymorphism genotyping that mutations leading to a premature stop codon in inlA are common among Listeria monocytogenes isolates from ready-to-eat foods but not human listeriosis cases. Appl Environ Microbiol 76 : 2783–2790.
57. WeissmanSJ, MoseleySL, DykhuizenDE, SokurenkoEV (2003) Enterobacterial adhesins and the case for studying SNPs in bacteria. Trends Microbiol 11 : 115–117.
58. ChoiSS, ChiversPT, BergDE (2011) Point mutations in Helicobacter pylori's fur regulatory gene that alter resistance to metronidazole, a prodrug activated by chemical reduction. PLoS ONE 6: e18236 doi:10.1371/journal.pone.0018236.
59. SeshuJ, BoylanJA, HydeJA, SwingleKL, GherardiniFC, et al. (2004) A conservative amino acid change alters the function of BosR, the redox regulator of Borrelia burgdorferi. Mol Microbiol 54 : 1352–1363.
60. VegaY, DickneiteC, RipioMT, BockmannR, Gonzalez-ZornB, et al. (1998) Functional similarities between the Listeria monocytogenes virulence regulator PrfA and cyclic AMP receptor protein: the PrfA* (Gly145Ser) mutation increases binding affinity for target DNA. J Bacteriol 180 : 6655–6660.
61. PapenfortK, PodkaminskiD, HintonJC, VogelJ (2012) The ancestral SgrS RNA discriminates horizontally acquired Salmonella mRNAs through a single G-U wobble pair. Proc Natl Acad Sci U S A 109: E757–764.
62. MandelMJ, WollenbergMS, StabbEV, VisickKL, RubyEG (2009) A single regulatory gene is sufficient to alter bacterial host range. Nature 458 : 215–218.
63. SomvanshiVS, SloupRE, CrawfordJM, MartinAR, HeidtAJ, et al. (2012) A single promoter inversion switches Photorhabdus between pathogenic and mutualistic states. Science 337 : 88–93.
64. LiangX, HallJW, YangJ, YanM, DollK, et al. (2011) Identification of single nucleotide polymorphisms associated with hyperproduction of alpha-toxin in Staphylococcus aureus. PLoS ONE 6: e18428 doi:10.1371/journal.pone.0018428.
65. OkudaJ, NishibuchiM (1998) Manifestation of the Kanagawa phenomenon, the virulence-associated phenotype, of Vibrio parahaemolyticus depends on a particular single base change in the promoter of the thermostable direct haemolysin gene. Mol Microbiol 30 : 499–511.
66. TareenAM, DastiJI, ZautnerAE, GrossU, LugertR (2011) Sulphite : cytochrome c oxidoreductase deficiency in Campylobacter jejuni reduces motility, host cell adherence and invasion. Microbiology 157 : 1776–1785.
67. CarpenterBM, GanczH, Gonzalez-NievesRP, WestAL, WhitmireJM, et al. (2009) A single nucleotide change affects fur-dependent regulation of sodB in H. pylori. PLoS ONE 4: e5369 doi:10.1371/journal.pone.0005369.
68. Yoder-HimesDR, ChainPS, ZhuY, WurtzelO, RubinEM, et al. (2009) Mapping the Burkholderia cenocepacia niche response via high-throughput sequencing. Proc Natl Acad Sci U S A 106 : 3976–3981.
69. BernickDL, DennisPP, LuiLM, LoweTM (2012) Diversity of Antisense and Other Non-Coding RNAs in Archaea Revealed by Comparative Small RNA Sequencing in Four Pyrobaculum Species. Front Microbiol 3 : 231.
70. DiChiaraJM, Contreras-MartinezLM, LivnyJ, SmithD, McDonoughKA, et al. (2010) Multiple small RNAs identified in Mycobacterium bovis BCG are also expressed in Mycobacterium tuberculosis and Mycobacterium smegmatis. Nucleic Acids Res 38 : 4067–4078.
71. PichonC, FeldenB (2005) Small RNA genes expressed from Staphylococcus aureus genomic and pathogenicity islands with specific expression among pathogenic strains. Proc Natl Acad Sci U S A 102 : 14249–14254.
72. KimD, HongJS, QiuY, NagarajanH, SeoJH, et al. (2012) Comparative Analysis of Regulatory Elements between Escherichia coli and Klebsiella pneumoniae by Genome-Wide Transcription Start Site Profiling. PLoS Genet 8: e1002867 doi:10.1371/journal.pgen.1002867.
73. PapenfortK, VogelJ (2010) Regulatory RNA in bacterial pathogens. Cell Host Microbe 8 : 116–127.
74. BeiselCL, StorzG (2010) Base pairing small RNAs and their roles in global regulatory networks. FEMS Microbiol Rev 34 : 866–882.
75. ChaoY, PapenfortK, ReinhardtR, SharmaCM, VogelJ (2012) An atlas of Hfq-bound transcripts reveals 3′ UTRs as a genomic reservoir of regulatory small RNAs. EMBO J 31 : 4005–4019.
76. SchoulsLM, ReulenS, DuimB, WagenaarJA, WillemsRJ, et al. (2003) Comparative genotyping of Campylobacter jejuni by amplified fragment length polymorphism, multilocus sequence typing, and short repeat sequencing: strain diversity, host range, and recombination. J Clin Microbiol 41 : 15–26.
77. LouwenR, Horst-KreftD, de BoerAG, van der GraafL, de KnegtG, et al. (2012) A novel link between Campylobacter jejuni bacteriophage defence, virulence and Guillain-Barre syndrome. Eur J Clin Microbiol Infect Dis
78. MaliP, YangL, EsveltKM, AachJ, GuellM, et al. (2013) RNA–Guided Human Genome Engineering via Cas9. Science
79. CongL, RanFA, CoxD, LinS, BarrettoR, et al. (2013) Multiplex Genome Engineering Using CRISPR/Cas Systems. Science
80. BlombergP, WagnerEG, NordstromK (1990) Control of replication of plasmid R1: the duplex between the antisense RNA, CopA, and its target, CopT, is processed specifically in vivo and in vitro by RNase III. EMBO J 9 : 2331–2340.
81. BerezikovE, ThuemmlerF, van LaakeLW, KondovaI, BontropR, et al. (2006) Diversity of microRNAs in human and chimpanzee brain. Nat Genet 38 : 1375–1377.
82. EdgarR, DomrachevM, LashAE (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30 : 207–210.
83. DarlingAC, MauB, BlattnerFR, PernaNT (2004) Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res 14 : 1394–1403.
84. BaileyTL, BodenM, BuskeFA, FrithM, GrantCE, et al. (2009) MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–208.
85. KapplerU, BennettB, RethmeierJ, SchwarzG, DeutzmannR, et al. (2000) Sulfite:Cytochrome c oxidoreductase from Thiobacillus novellus. Purification, characterization, and molecular biology of a heterodimeric member of the sulfite oxidase family. J Biol Chem 275 : 13202–13212.
86. CamachoC, CoulourisG, AvagyanV, MaN, PapadopoulosJ, et al. (2009) BLAST+: architecture and applications. BMC Bioinformatics 10 : 421.
87. MakarovaKS, WolfYI, KooninEV (2013) Comparative genomics of defense systems in archaea and bacteria. Nucleic Acids Res doi: 10.1093/nar/gkt157.
88. Barrero-TobonAM, HendrixsonDR (2012) Identification and analysis of flagellar coexpressed determinants (Feds) of Campylobacter jejuni involved in colonization. Mol Microbiol 84 : 352–369.
89. WostenMM, WagenaarJA, van PuttenJP (2004) The FlgS/FlgR two-component signal transduction system regulates the fla regulon in Campylobacter jejuni. J Biol Chem 279 : 16214–16222.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Using Extended Genealogy to Estimate Components of Heritability for 23 Quantitative and Dichotomous Traits
- Encodes CDF Transporters That Excrete Zinc from Intestinal Cells of and Act in a Parallel Negative Feedback Circuit That Promotes Homeostasis
- HDAC7 Is a Repressor of Myeloid Genes Whose Downregulation Is Required for Transdifferentiation of Pre-B Cells into Macrophages
- Female Bias in and Regulation by the Histone Demethylase KDM6A
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy