
DNA Binding of the Cell Cycle Transcriptional Regulator GcrA Depends on N6-Adenosine Methylation in and Other
Several regulators are involved in the control of cell cycle progression in the bacterial model system Caulobacter crescentus, which divides asymmetrically into a vegetative G1-phase (swarmer) cell and a replicative S-phase (stalked) cell. Here we report a novel functional interaction between the enigmatic cell cycle regulator GcrA and the N6-adenosine methyltransferase CcrM, both highly conserved proteins among Alphaproteobacteria, that are activated early and at the end of S-phase, respectively. As no direct biochemical and regulatory relationship between GcrA and CcrM were known, we used a combination of ChIP (chromatin-immunoprecipitation), biochemical and biophysical experimentation, and genetics to show that GcrA is a dimeric DNA–binding protein that preferentially targets promoters harbouring CcrM methylation sites. After tracing CcrM-dependent N6-methyl-adenosine promoter marks at a genome-wide scale, we show that these marks recruit GcrA in vitro and in vivo. Moreover, we found that, in the presence of a methylated target, GcrA recruits the RNA polymerase to the promoter, consistent with its role in transcriptional activation. Since methylation-dependent DNA binding is also observed with GcrA orthologs from other Alphaproteobacteria, we conclude that GcrA is the founding member of a new and conserved class of transcriptional regulators that function as molecular effectors of a methylation-dependent (non-heritable) epigenetic switch that regulates gene expression during the cell cycle.
Published in the journal:
. PLoS Genet 9(5): e32767. doi:10.1371/journal.pgen.1003541
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1003541
Summary
Several regulators are involved in the control of cell cycle progression in the bacterial model system Caulobacter crescentus, which divides asymmetrically into a vegetative G1-phase (swarmer) cell and a replicative S-phase (stalked) cell. Here we report a novel functional interaction between the enigmatic cell cycle regulator GcrA and the N6-adenosine methyltransferase CcrM, both highly conserved proteins among Alphaproteobacteria, that are activated early and at the end of S-phase, respectively. As no direct biochemical and regulatory relationship between GcrA and CcrM were known, we used a combination of ChIP (chromatin-immunoprecipitation), biochemical and biophysical experimentation, and genetics to show that GcrA is a dimeric DNA–binding protein that preferentially targets promoters harbouring CcrM methylation sites. After tracing CcrM-dependent N6-methyl-adenosine promoter marks at a genome-wide scale, we show that these marks recruit GcrA in vitro and in vivo. Moreover, we found that, in the presence of a methylated target, GcrA recruits the RNA polymerase to the promoter, consistent with its role in transcriptional activation. Since methylation-dependent DNA binding is also observed with GcrA orthologs from other Alphaproteobacteria, we conclude that GcrA is the founding member of a new and conserved class of transcriptional regulators that function as molecular effectors of a methylation-dependent (non-heritable) epigenetic switch that regulates gene expression during the cell cycle.
Introduction
Epigenetic signals, such as methylation of DNA, play an important role in the regulation of gene expression in eukaryotes. Methylation of adenines in the N6 position (m6A) has been described in Bacteria, Archaea, Protists and Fungi. Though known for its protective role in bacterial restriction/modification systems [1], m6A also fulfills cellular functions in Gammaproteobacteria, including the initiation of DNA replication, transposition, mismatch repair, and virulence gene expression [2]–[4]. In the Alphaproteobacteria such as Caulobacter crescentus, Sinorhizobium meliloti, Brucella abortus and Agrobacterium tumefaciens, the solitary methyltranferase CcrM is required for efficient growth, presumably through gene expression control of critical cell cycle genes [5]–[8].
The cell cycle role of CcrM was originally described in C. crescentus [5], [7]. At each cell division, C. crescentus generates two different cells (stalked and swarmer) committed to specific stages of the cell cycle [9]. The stalked cell is able to replicate the DNA (S-phase) and it possesses an extension of the external envelope and membranes, called stalk. The swarmer cell is instead motile and non-replicative (G1-like) possessing a polar flagellum and several pili. Upon nutrient availability the swarmer cell differentiates in a stalked cell, resembling the eukaryotic G1→S transition. In this cyclical progression, a crucial role is played by CtrA, an essential transcriptional regulator that targets many cell cycle genes [10]. Its activity and abundance are precisely regulated in time and space through phosphorylation, proteolysis and transcription. In G1, CtrA∼P inhibits DNA replication by repression of the origin of replication [11] and only upon CtrA proteolysis or dephosphorylation, DnaA-mediated chromosome replication initiation occurs [12] committing cells to the S phase. The re-synthesis of CtrA requires transcription of ctrA that relies on the methylation-sensitive ctrAP1 promoter [13] whose activation depends on GcrA, an enigmatic factor that is encoded in the genomes of Alphaproeobacteria and several Caulophages [14], [15]. While in Caulobacter GcrA accumulates in early S phase and is confined to stalked cells [16] for the activation of ctrAP1, a second, auto-regulatory promoter, ctrAP2, reinforces ctrA transcription later in S-phase. Upon CtrA synthesis, an essential phosphorelay, composed of CckA and ChpT [17], phosphorylates CtrA that in turn activates transcription of the ccrM gene. After the introduction of m6A marks in the context of GAnTC sequences [5] CcrM is proteolyzed prior to cell division by the Lon protease [7]. How the m6A marks, introduced by CcrM, affect transcription is unclear, but the marks are transient as DNA replication converts the (full) methylation on both DNA strands to the hemi-methylated state, until strands are re-methylated in a distributive manner [18] once CcrM has accumulated at the end of S-phase. The time a given genomic locus spends in the hemi-methylated state is thus pre-determined by its physical proximity to the origin of replication [19], a feature that might be exploited to couple activation of certain promoters, such as ctrAP1, with replication progression [19]. GcrA and CcrM are implicated in the transcriptional regulation of ctrAP1, suggesting linked roles. While an underlying biochemical relationship is also hinted by the analysis of the gene occurrence pattern in the Alphaproteobacterial genomes [20], this link remains experimentally untested.
Here we use chromatin-immunoprecipitation, biochemical, genetic and biophysical methods to explore the basis of transcriptional activation by GcrA. We uncovered that GcrA binds preferentially m6A-marked DNA and that it associates with the RNA polymerase, presumably to facilitate transcription initiation at methylated promoters. To assess if this mechanism is specific for Caulobacter or instead evolutionarily conserved, we performed experiments with GcrA orthologs in other Alphaproteobacteria, observing essentially the same behaviour. We conclude that GcrA and CcrM define an important regulatory pair, which is evolutionarily conserved and has been appropriated for epigenetic control of cell cycle transcription in Alphaproteobacteria.
Results/Discussion
GcrA forms an elongated, partially unfolded dimer
Because GcrA is a conserved protein that lacks primary structural resemblances to known functional domains, we investigated the features of the primary structure of GcrA by bioinformatic prediction using SMART [21]; first, a non-significant homology (E-value equal to 734) to helix-turn-helix domains was detected (13–55 aa). Also GcrA has a high content of positively charged residues such as arginine and lysine mostly located in the central region (45–80 aa). Those features may support the ability of GcrA to bind DNA directly (see next sections) through the N-terminal part. Consistently, the N-terminal part is also the region of GcrA that is more conserved at the evolutionary level across orthologs of GcrA shown in the Figure S1. This conservation suggests an important functional role of the region, for example in the specific DNA binding and also putative interactions with other cellular factors.
With the goal of investigating the interactions of GcrA with DNA and its targets in vivo we purified an N-terminally hexa-histidine tagged variant of GcrA (His6-GcrA) from an E. coli overexpression strain by sequential affinity and gel filtration chromatography and characterized its biophysical properties (see Materials and Methods). SDS-PAGE (Figure S2) and dynamic light scattering (DLS) analysis (data not shown) indicated a highly pure (>95%) and monodisperse preparation of His6-GcrA. Prediction of unfolded regions using RONN suggest that regions 41–105 aa and 145–173 aa of GcrA are disordered, while the software SOPMA [22] suggested that GcrA is partially structured in the N-terminal region (Figure S3) with an organization in three predicted alpha helices suggesting a folded structure. To gain insight into the spatial organization, we conducted Small Angle X-ray Scattering (SAXS) analysis (Protocol S1 and Table S1) that allows the determination of shape, size and oligomerization status of macromolecules in solution (Figure S4A). SAXS estimates the molecular mass of His6-GcrA at 42 kDa, which corresponds to a dimeric organization. Also the dimensions of His6-GcrA, by using computed radius of gyration (Rg) and maximum dimension (Dmax) values, combined with the pair-distance distribution function, P(R), shape and the Kratky plot representation, are consistent with an elongated form and partially disordered conformation of His6-GcrA dimers in solution (See legend of Figure S4A for more technical details). Possibly the interaction of GcrA with other proteins and with DNA can induce a stabilization of the disordered regions.
Next, we performed limited proteolysis followed by MALDI-TOF mass spectrometry (MS) analysis in order to identify regions of His6-GcrA that were more resistant to proteolysis indicating its more compact (structured) nature. Two different proteases, Thermolysin and V8 (see Materials and Methods) were used and the most resistant fragments to proteolysis were analyzed by MS (Figure S4B). This analysis suggests that the N-terminal part of GcrA (from 1 to 115 ca.), although containing proteolytic sites for both proteases, was more stable than the C-terminal part, as also indicated by the prediction of alpha helical structures in the N-terminal portion of GcrA.
Genome-wide occupancy of GcrA at promoters in vivo
In light of these structural features suggesting that the N-terminal domain of GcrA binds DNA, we sought specific in vivo targets of GcrA that could be used to probe DNA-binding of GcrA in vitro. Previous non-quantitative chromatin-immunoprecipitation (ChIP) experiments using polyclonal antibodies to GcrA, provided support for the notion that GcrA affects the transcription of cell cycle genes by, directly or indirectly, associating with specific chromosomal sites [16]. To illuminate the basis for this selectivity and the underlying mechanism of transcriptional regulation by GcrA in Alphaproteobacteria, we subjected ChIP samples from NA1000 wild-type cells to deep sequencing (ChIP-Seq) [23] (Protocol S2). By mapping the reads onto the genome, we determined the binding profile of GcrA on genomic regions.
First, we used a peak finding strategy to identify regions bound by GcrA with high affinity (Protocol S2); this analysis allowed to identify 218 peaks that were subsequently associated to the closest genes (Table S2). Inspection of the top 50 targets (Figure 1A) revealed wide peaks (ca. 1 kbp wide, data not shown). We found that half of these GcrA-bound sequences were significantly associated with CcrM methylation sites (GAnTC). To explore this association more in details, we calculated the average number of methylation sites in 1 kbp windows centered on the peaks and found it to be close to 2 (1.78), in comparison with 0.58 sites per random 1 kbp genomic regions (Figure 1B). These results clearly indicate a significant enrichment of methylation sites in genomic regions bound in vivo by GcrA, raising the possibility that methylation enhances GcrA binding to its targets.

Next, in all promoter regions, defined from 300 bp upstream to 100 bp downstream a gene's start codon, we calculated the number of ChIp-Seq reads (see Protocol S2) (Table S3). We obtained (Z-score ≥2) 161 GcrA-bound promoter regions, 89 of which also contained a GAnTC methylation site (data not shown). This list contained many known GcrA-controlled targets such as mipZ, encoding a division regulator [24], podJ encoding a polarity factor [25], [26] and ctrA. We observed a remarkably small overlap with the genes previously identified as GcrA-dependent by DNA microarrays [16]. Only 5 genes passed the threshold (Z score ≥2), including those encoding CCNA_01542 (ice nucleation protein), CCNA_01556 (LacI family transcriptional regulator), CCNA_01766 (hypothetical protein), CCNA_02005 (inosine-uridine preferring nucleoside hydrolase), CCNA_02086 (sporulation domain containing protein), CCNA_02246 (mipZ), CCNA_02401 (encoding a transcriptional regulator) and CCNA_03325 (encoding a hypothetical protein). Since microarrays detect both direct and indirect mRNA abundance changes, it is likely that many genes whose expression was affected by GcrA depletion were, in fact, indirect targets of GcrA presumably under the control of other transcription factors, such as CtrA.
GcrA defines a new class of specific DNA–binding proteins
In order to test if GcrA binds DNA in vitro, we set up an electrophoretic mobility shift assay (EMSA) with His6-GcrA and regions identified as in vivo targets of GcrA by the ChIP-seq experiment described above. We selected 5 regions as EMSA probes, each with distinct features: 1) the preferred target (with the maximum number of reads in Table S2) corresponding to the N-terminal coding sequence of gene CCNA_00697 that has three GAnTC sites; 2) the intergenic sequence between CCNA_00278 and CCNA_00279 that is efficiently bound by GcrA in vivo, but has no predicted GAnTC methylation site; 3) the promoter of the gene mipZ, which was also discovered previously by microarray as a GcrA-dependent gene and has two juxtaposed GAnTC sites; 4), the P1 promoter of ctrA (ctrAP1) that has one GAnTC site (there is another GAnTC sequence after the transcription start site of the ctrAP2 promoter) and is thought to be activated by GcrA [16]; 5) a negative control, corresponding to the intergenic region between CCNA_01926 and CCNA_01927 which GcrA binds non-specifically in vivo based on ChIP-seq data. Probes were designed as 70-mer double stranded oligo-nucleotides, in which the central part corresponds to the genomic region with the highest number of ChIP-seq reads. The EMSA (Figure 2) showed that His6-GcrA shifted four out of five probes, indicating the formation of a His6-GcrA•DNA complex with sequences identified by ChIP-Seq, except for the intergenic sequence between CCNA_01926 and CCNA_01927 (i.e., the negative control). All positive probes gave rise to two distinct His6-GcrA•DNA complexes with similar migration properties, possibly reflecting different oligomeric states of His6-GcrA with different apparent affinities for the DNA (see below). In particular, probe CCNA_00697 was the most efficiently bound by His6-GcrA (Kd = 4±0.5 µM); probes ctrA (Kd = 6.5±0.5 µM) and mipZ (Kd = 8.5±0.5 µM) also showed DNA binding however the complex forms only at a higher concentration of His6-GcrA, mirroring, with the exception of the intergenic region between CCNA_00278 and CCNA_00279 (Kd>9 µM), the binding preference ChIP-seq in vivo. The EMSA results also demonstrate that His6-GcrA binds DNA in a specific fashion in vitro. Considering also that the conserved GcrA protein has no homology with known DNA binding proteins at the primary structure level, we conclude that GcrA defines a new class of alphaproteobacterial DNA binding proteins that directly interacts with target promoters to control transcription of many Caulobacter S-phase genes, including the gene encoding the master regulator CtrA. Although methylation sites are associated with GcrA-bound regions, GcrA apparently can also bind sequences that do not contain GAnTC methylation sites, based on the methylation-dependent binding experiments described below, we suggest that multiple DNA constrains exist in the GcrA specificity, perhaps involving m6A marks in different sequences contexts or a different type of methylation mark altogether. In this context, it is noteworthy that two putative cytosine methyltransferases are encoded in the C. crescentus genome [27].

DNA binding of GcrA is enhanced by CcrM-dependent methylation
To test if GAnTC methylation modulates the binding of His6-GcrA to some of its targets in vitro, we conducted EMSA competition experiments with the un-methylated region of PmipZ, CCNA_00697 and ctrAP1 as biotinylated probes and double stranded synthetic oligos harboring N6-adenosine methylation at GAnTC on either one or both strands as competitors. In these experiments, His6-GcrA was pre-incubated with the unlabeled competitor DNA, followed by the addition of the biotinylated probe. The more the unlabeled competitor DNA reduces the abundance of the shifted His6-GcrA•DNA complex, the higher the affinity of His6-GcrA is for the unlabeled competitor. As shown in Figure 3, we observed a clear preference of GcrA for the methylated competitors over the un-methylated one, with those carrying methylation on both strands (“full”-methylation) competing better than either one harbouring the methylation on one of the two strands (“hemi”-methylation). Remarkably, in the case of the CCNA_00697 and mipZ competitors, hemi-methylation on the “sense” strand is a better competitor than the hemi-methylated competitor with the modification on the other strand. For promoters of ctrA and mipZ, the calculated Kds provided quantitative confirmation of the results shown in Figure 3 (Figure S9B and S9C).

In order to assess if methylation alters the disposition of GcrA on its target DNA, we conducted DNase I protection assay using fully and hemi-(GAnTC) methylated fluorescently-labeled ctrAP1 promoter probes. As shown in Figure 4A, GcrA protects specific regions of the probe in a methylation-dependent manner, giving rise to a larger region of protection spanning the −35 to the −10 of the ctrAP1 promoter with the fully-methylated (i.e. on both strands) probe. By contrast, the protection of the hemi-methylated (on the plus strand) probe was confined to a region adjacent to the methylation site itself. Importantly, the un-methylated probe and the hemi-methylated probe carrying the modification on the minus strand did not show protection by His6-GcrA at any concentration. We conclude that methylation induces different oligomerization or conformational states in strand-specific manner.

Next, we complemented the DNase I protection experiments of the target, with protection experiments of His6-GcrA by limited proteolysis in the presence or absence of the various methylated ctrAP1 probes (Figure 4B). We found that efficiency of proteolysis is accelerated in the presence of methylated probes, suggesting that conformational rearrangements are induced by the methylated probe to expose protease hypersensitive sites, akin to the DNase I hypersensitive sites of the target DNA that become exposed only when GcrA associates with a methylated target, but not in the presence of the un-methylated site. We also ruled out the possibility that the oligos affected the DNase I activity by incubating another protein (His6-ChpT) [28], which was proteolyzed identically with or without the DNAs (data not shown).
CcrM-dependent methylation of ctrAP1 was previously proposed as an essential element of a transcriptional regulatory switch, culminating in the GcrA-dependent activation of ctrAP1 upon the conversion from full - to hemi-methylation [19]. Intriguingly, our results reveal that His6-GcrA binds hemi-methylated versus fully methylated ctrAP1 in strikingly different manner, with the latter covering a much larger area. This raises the possibility that cooperative interactions, induced by the transition from hemi - to full-methylation mediated by CcrM, can lead to a wider and stronger association of GcrA with the target DNA. As His6-GcrA wraps the entire region from −35 to +7 of fully methylated ctrAP1, it may interfere with RNA polymerase holo-enzyme (RNAP) at ctrAP1; a possibility that must be explored in future work. By contrast, on the hemi-methylated plus strand of ctrAP1, His6-GcrA occupies a 12 nt stretch overlapping the −35 region and adjacent GAnTC site and with lower affinity a 12 nt region from +5 onwards. GcrA could compete with RNAP for binding to ctrAP1 or alternatively tether it at the promoter, preventing promoter clearance, i.e. the switch from transcription initiation to the elongation phase. Furthermore the methylation strand-specificity opens the possibility of an “asymmetric” mechanism of gene regulation, in which only one of the two replicated loci is preferentially bound and transcriptionally regulated by GcrA before re-methylation by CcrM in the pre-divisional cell. Such, a regulatory bias could have far reaching consequences in all forms of spatiotemporal and/or of gene-dosage regulation for all living cells, as it has been suggested before for PapI-promoted Lrp binding to hemi-methylated sites in uropathogenic E. coli [29].
GcrA–dependent interactions with RNAP
To explore the models described above, we tested whether GcrA can directly or indirectly associate with RNAP. To this end, we passed a soluble C. crescentus cell lysate, in which DNA was fragmented by a mild DNase I treatment, over a nickel-NTA column that had been pre-loaded or not with His6-GcrA. Following extensive washes with buffer containing up to 1 M NaCl, we eluted His6-GcrA and associated proteins with buffer containing imidazole (see Materials and Methods). Blots harbouring these samples were then probed with antibodies to the β subunit of core RNAP, revealing that RNAP β in the eluate from the His6-GcrA pre-loaded column only (Figure 5A).

We extended these findings by showing that E. coli RNAP core enzyme can associate with the DNA•GcrA complex in an EMSA using ctrAP1 promoter. Increasing concentration of RNA polymerase clearly showed the formation of lower mobility complex whose formation was dependent on the presence of GcrA (Figure 5B). This interaction of RNAP with GcrA bound to its target was also observed with the mipZ promoter (Figure S5). Taken together, these results indicate that GcrA binds components of the RNA polymerase core complex and they provide a mechanistic explanation for how GcrA might affect gene transcription.
Transcriptional activation and promoter binding by GcrA in vivo requires methylation
The connection between methylation by CcrM and DNA-binding of GcrA, seen in vitro, together with the association of GcrA to RNAP, prompted us to explore if other GcrA target promoters are regulated in a methylation-dependent manner in vivo. To this end, we fused several promoters that have methylation sites and that emerged as in vivo targets of GcrA in the ChIP-seq experiments to the promoter-less lacZ reporter gene. We first confirmed the GcrA-dependence of these promoters by measuring lacZ-encoded β–galactosidase activities under GcrA-replete and deplete conditions using a ΔgcrA::Ω; xylX::Pxyl-gcrA strain [16] in which GcrA expression is induced in the presence of xylose and repressed in the presence of glucose (Figure 6A). After 5 h of depletion of GcrA in glucose, the β–galactosidase activities of the PmipZ-, PpodJ-, PflaY - and PpleC-lacZ reporters dropped by ca. 60% compared to the WT grown in glucose or the Pxyl-gcrA strain grown in xylose, and immunoblotting revealed a strong reduction in MipZ and PodJ abundance (Figure S6). It was previously also shown that activation of the ctrAP1 promoter requires GcrA [19]. By contrast, the PCCNA_00697-lacZ reporter only exhibited a 31% reduction in β–galactosidase activity under the same condition, possibly because as the preferred in vivo target of GcrA, residual GcrA that remains in the cell clings to the CCNA_00697 promoter (Figure 6B).

Next, we asked if a mutation of the GAnTC influences the promoter activity. To this end, all GAnTC sites in a promoter fragment of the lacZ reporter construct were mutated to GCnTC and the activity of the mutant promoters assayed by β–galactosidase measurements. The mutant promoters were crippled by 60–80% and immunoblotting showed the PodJ and MipZ failed to accumulate in cells lacking CcrM (Figure S6). Interestingly, the mutant PCCNA_00697-lacZ reporter showed a different response, retaining WT (100%) activity. Interestingly, the effect on PCCNA_00697-lacZ is mirrored for ctrAP1 whose activity was also unchanged by mutation of the GAnTC site to prevent CcrM-dependent methylation (Figure 6C) [19]. To test if this response was typical of GcrA-dependent promoters that are distal to the replication terminus, we analysed the methylation/GcrA dependency of another promoter, tipF (CCNA_00747), at a comparable location with respect to the origin of replication (). Unlike ctrAP1 and the region at CCNA_00697, tipF promoter activity requires GcrA and an intact GAnTC methylation site (see below).
To explore if methylation at GAnTC is required for GcrA to associate with its target sites in vivo, we compared the genome-wide promoter occupancy of GcrA in WT and ΔccrM cells by ChIP-seq (Figure 7A). Analysis of the two data sets (Table S3) unearthed a major role of GAnTC methylation in directing GcrA to target promoters, with 80 loci requiring CcrM to be efficiently bound by GcrA, including CCNA_00697, mipZ, podJ, flaY (encoding a putative flagellar regulator), pleC (encoding a developmental histidine kinase/phosphatase) and to a lesser extent ctrA. However for ctrAP1, detailed analysis of the ChIP-Seq traces (Figure S7) revealed that GcrA binding dropped in proximity to the GAnTC methylation sites. Immunoblotting confirmed that no apparent difference in the GcrA steady-state levels were discernible in WT and ΔccrM cells (Figure 7A).

To corroborate the ChIP-seq data, we performed ChIP analysis of WT and ΔccrM cells and measured the abundance of precipitated (GcrA-bound) mipZ and podJ promoters by quantitative real-time PCR (qChIP). As shown in Figure 7B, in the absence of CcrM, GcrA occupancy is reduced by 70% and 60%, respectively.
If CcrM-dependent GAnTC methylation is required for GcrA binding to its targets, then the corresponding promoters should not be methylated in ΔccrM cells. Because other methyltransferases might also contribute to adenosine methylation at the N6 position (m6A), we first determined the abundance of m6A across the genomes of WT and ΔccrM cells by ChIP-Seq analysis using an m6A-specific polyclonal antibody (Figure 7C). This analysis revealed that chromosomal loci, particularly towards the replication terminus, carry abundant CcrM-dependent m6A marks (Table S3). We validated this conclusion by qChIP experiments for two promoters proximal to the terminus, PmipZ and PpodJ, (Figure 7D) and a distal one, PtipF (Figure S10).
Methylation-dependent DNA binding of GcrA orthologs
To explore if GcrA-controlled functions are conserved across the Alphaproteobacteria, we introduced the GcrA ortholog [20] from Brucella melitensis biovar abortus 2308 (BAB1_0329) and Sinorhizobium meliloti Rm1021 (SMc02139) under the control of an xylose-inducible promoter on a low-copy plasmid [30] in C. crescentus, harbouring a temperature sensitive allele of gcrA with a Thr→Pro mutation at position 10 and evaluated their ability to support growth at the restrictive temperature [16]. As shown in Figure 8A, both B. abortus and S. meliloti gcrA orthologs are able to support viability of the strain gcrAts at the restrictive temperature (37°C) following induction with xylose. Orthologs of GcrA from S. meliloti and B. abortus, although able to complement GcrA functions, revealed morphological diversities in C. crescentus (Figure 8B), likely owing to differences in abundance, activity and/or target specificity of these GcrA versions. Regardless, the complementation of the temperature-sensitive phenotype indicates that the function and target site specificity of GcrA orthologs are similar. We confirmed this result by testing directly the ability of GcrA orthologs to bind the Caulobacter target promoters. Therefore B. abortus and S. meliloti GcrA with an N-terminal His6 tag were purified from E. coli overexpression strains (Figure S2). EMSA experiments using target sites of C. crescentus GcrA revealed that these GcrAs are able to bind DNA efficiently and with the same apparent specificity (Figure 9A). Surprisingly the B. abortus and S. meliloti GcrA orthologs are able to form multiple complexes with different migration properties, likely due to structural and/or charge differences.
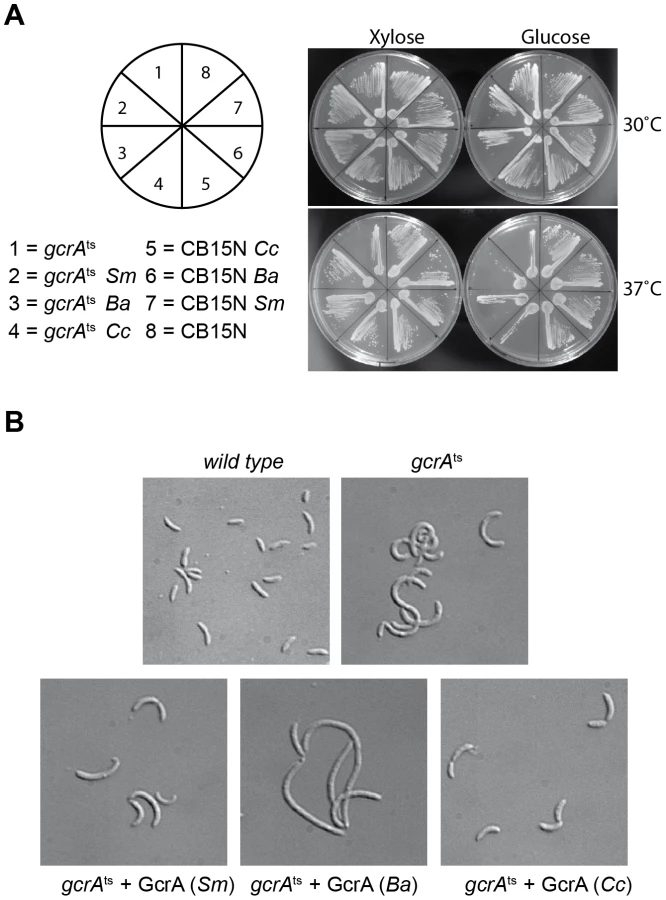

Finally we tested if binding of B. abortus and S. meliloti GcrAs is also stimulated by GAnTC methylation. EMSAs showed that methylation still affects GcrA binding, as fully methylated probes showed a stronger binding affinity in comparison with hemi-methylated and even more with non-methylated DNA. However the asymmetry in binding efficiency found for certain regions of DNA (Figure 9B) appeared different in other GcrAs with respect to the C. crescentus one.
Concluding remarks
Despite the pervasive effects of adenosine methylation on transcription in various bacterial lineages, our understanding of the underlying operating principles in these systems is still limited. With the identification and genetic/biochemical characterizations of the m6A-marked promoters and the transcriptional effector(s) recognizing them, we elucidated a crucial first step towards understanding the physiological underpinnings and the evolution of these epigenetic control systems in Alphaproteobacteria. Studies with the methylation-sensitive ctrAP1 promoter of C. crescentus as model led to the appealing model that replication of a given locus by DNA polymerase converts the promoter from the fully to the hemi-methylated state at a specific time in the cell-cycle that is dictated by the relative distance of the promoter from the origin of replication (ori). For ctrAP1, the hemi-methylated state was thought to be a prerequisite for GcrA-mediated activation, while full (re)-methylation by CcrM at the end of S-phase was viewed as the event leading to promoter silencing.
Our experiments not only establish GcrA as a methylation-dependent transcription factor binding ctrAP1 and other promoters in vivo and in vitro (Figure 10), but they may suggest an elegant explanation for the methylation-induced regulation of expression. While activation of the hemi-methylated plus strand of ctrAP1 correlates with localized binding of GcrA to 13 nt over the −35 region, in the fully methylated state more than 40 nt of ctrAP1, are covered. Once the DNA replication fork moves through the fully methylated ctrA locus in the ensuing cell cycle the binding state for hemi-methylated DNA is reinstated. Methylation seems to help recruiting GcrA to promoters but GcrA might interact with RNAP even in the absence of target DNA. Perhaps in the hemi-methylated state this binding allows the initiation of transcription and release of the polymerase, while in the fully methylated state GcrA could sequester RNAP, preventing its movement through the coding sequence. It is likely that the partially unstructured dimeric GcrA adopts compacter structure upon interacting with methylated target DNA or possibly RNAP, thus conferring these properties.

Contrary to the view that CcrM - and GcrA-dependent control of ctrAP1 in response to DNA replication applies to all GcrA target promoters, we note that many ori-distal promoters (such as PmipZ, PpodJ, and PpleC), but also terminus-distal promoters (such as PtipF) that fire in early S-phase, also carry CcrM-dependent m6A marks that are required for the recruitment of GcrA. Promoters near the terminus will be replicated late in S-phase and are, thus, thought to reside in the hemi-methylated state only during a short window before the synthesis of CcrM. This begs the question what purpose of m6A marks at these promoters may serve, since methylation change by replication should not be temporally correlated with promoter activation. As many promoters of cell division (e.g., mipZ), motility (e.g., flaY) and polarity genes (e.g., podJ) carry m6A marks that are recognized by GcrA (Figure 10), CcrM-dependent methylation might serve as a quality control function or coupling mechanism to prepare these promoters for activation in the ensuing cell division cycle once GcrA is expressed. In the gammaproteobacterium Vibrio cholerae, the origin-binding protein of chromosome II RctB is recruited to sites carrying m6A marks that have been introduced by the GATC-specific Dam methyltransferase [4]. Thus, while full methylation also has been adopted for regulatory purposes, different effectors and processes have been selected during evolution.
Materials and Methods
Strains and growth conditions
C. crescentus strains were grown in peptone-yeast extract (PYE, rich medium) at 30°C [31] or 37°C as necessary, tetracycline (1 µg/ml), kanamycin (25 µg/ml), spectinomycin/streptomycin (100-5 µg/ml) 0.1% glucose, or 0.1% xylose, as required. E. coli strains were grown at 20°C or 37°C in LB broth supplemented with ampicillin (100 µg/ml), as necessary. Plasmids were transformed into C. crescentus and E. coli BL21 (DE3) by electroporation. Plasmids and strains are listed in Table S5. To construct the ΔccrM mutant UG2212, the ΔccrM::Ω mutation was transduced by ϕCr30-mediated generalized transduction from LS2144 [6] into NA1000. One transductant was selected on spectinomycin/streptomycin-containing medium and subjected to whole genome Illumina sequencing by Fasteris SA (Geneva, Switzerland). The genome sequence of UG2212 failed to reveal point mutations or insertions/deletions compared to the parent.
Cloning
DNA fragments from C. crescentus, S. meliloti and B. abortus were amplified by PCR using cell lysates or genomic DNA for Brucella using Pfu-Turbo (Life Technologies, www.lifetechnologies.com/) following a protocol as recommended by the manufacturer. Primers are listed in the Figure S8. PCR products were then transferred in pENTR by Directional TOPO Cloning (Life technologies, www.lifetechnologies.com/), sequence verified and then transferred in pET derivatives His6-tagged destination vectors for E. coli BL21 expression, or pMR20 destination vector for xylose inducible expression in C. crescentus strains [30].
Transcriptional reporters were made by cloning PCR-amplified promoters fragments (Figure S8) into plac290 using EcoRI/XbaI [32].
Purification of GcrAs from C. crescentus, S. meliloti, and B. abortus
The full-length DNA fragment of GcrA was cloned into the pET15 derivative, pML375 vector [30], obtaining a recombinant protein with a His6-Thrombin cleavage site tag in N-terminus of the protein. Overexpression of GcrA was induced in E. coli BL21 (DE3) at OD (600 nm) 0.6–0.8 by 500 µM isopropyl-b-D-thiogalactoside (IPTG) O/N at 20°C. The cells were harvested by centrifugation and then resuspended in lysis buffer (PBS 1× pH 7.5, 0.2 M NaCl, 1 mM DTT, 0.1% Triton, supplemented with Complete Protease Inhibitor Cocktail (Roche, http://www.roche.com/) and DNase I (Euromedex, www.euromedex.com/) and lysed by Emulsiflex (Avestin, www.avestin.com/) at 10°C.
From the supernatant, GcrA was purified in two steps of purification, first, using Ni2+-nitrilotriacetate affinity resin (Ni-NTA) (Qiagen, www.qiagen.com/) equilibrated with lysis buffer and eluted by PBS 1× (pH 7.5), 0.5 M Imidazole, followed by Gel filtration step using HiLoad 16/60 Superdex 75 prep grade (GE Healthcare, www.gehealthcare.com/) equilibrated with running buffer (0.1 M Tris pH 8.5, 0.2 M NaCl, 5% Glycerol) optimized after DLS (See below the experimental procedure).
DLS measurements by the Zetasizer nano ZS (Malvern, www.malvern.com/) with an accuracy of 0.1°C were performed immediately after both the size exclusion step and the concentration step in order to find the best buffer composition. DLS was employed to estimate the thermo-stability of protein samples in different buffer solutions from to 15°C to 64°C, one degree steps. DLS was also used for the estimation of monodispersity of GcrA preparation.
Limited proteolysis
Purified His6-GcrA was digested with proteases Thermolysin (Sigma-Aldrich, www.sigmaaldrich.com/) and Endoproteinase GluC V8 (New England Biolabs, www.neb.com/) (25°C with 0.5 mg/ml GcrA in 20 mM TRIS pH 8, 150 mM NaCl for digestion with Thermolysin and 20 mM Tris (pH 7.6), 1 mM CaCl2 in case of digestion with V8). The protease/substrate ratio was 1∶100 (w/w) in each case. At different time intervals, aliquots of the proteolysis reactions were stopped with loading buffer. The protein samples were then analyzed by SDS-PAGE and the fragments analyzed by Trypsin digestion and mass spectrometry. Proteolysis control of His6-ChpT [28] in presence of differentially methylated DNAs was performed as described above.
Affinity chromatography for RNAP detection
Nickel columns loaded by His6-GcrA were also used for affinity chromatography showed in Figure 5B. A 1 ml HisPur-Ni-NTA Chromatography Cartridge (Qiagen, www.qiagen.com/), equilibrated with running buffer (0.1 M Tris pH 8.5, 0.15 M NaCl, 5% Glycerol) was loaded at 15°C with 23 mg of histidine-tagged C. crescentus GcrA (His6-GcrA) that was prepared as previously described, and washed with 15 volumes of running buffer. Meanwhile, 2 liters of C. crescentus cells (OD 600 nm of 0.6) were harvested by centrifugation (5000 rpm, 20 min, 4°C) and resuspended in 30 ml of lysis buffer (0.1 M Tris pH 8.5, 0.15 M NaCl, 1 mM DTT, 0.1% Triton, supplemented with Complete Protease Inhibitor Cocktail (Roche, www.roche.com/) and DNase I (Euromedex, www.euromedex.com/)) and lysed by Emulsiflex (Avestin, www.avestin.com/) at 10°C. The lysate was then centrifuged at 9500 rpm, 20 min, 4°C and the supernatant obtained was applied to the column. The column was eluted with running buffer NaCl gradient from 0.15 M and 1 M of NaCl. A last wash was done in presence of Imidazole (0,1 M Tris pH 8,5, 0,15 M NaCl, 5% Glycerol, 0.5 M Imidazole) in order to remove the His6-GcrA and proteins still bound at the column.
The negative control to this experiment was performed doing the same procedure with a 1 ml HisPur Ni-NTA Chromatography Cartridge without His6-GcrA.
The eluted samples were run in SDS-PAGE gel and transferred to nitrocellulose membrane. The membrane was blocked with PBS, 0.1% NP-40 and 3% dry milk for 1 hour at room temp. The membrane was incubated with anti-RNA polymerase B-subunit antibody (Thermo Scientific, www.pierce-antibodies.com/) against the β-subunit (1∶5000) at 4°C overnight. Each membrane was washed 5 times each for 10 min with PBS containing 0.1% NP-40, followed by incubation with the secondary antibody (1∶50,000) for 45 min. The membrane was developed following the procedure described under immunoblot section.
DNA binding in vitro assays
EMSAs were performed using the LightShift Chemiluminescent EMSA Kit (Thermo Scientific). Briefly, different versions of GcrA were incubated at room temperature in 10 mM Tris pH 7.5, 100 mM KCl, 0.5 mM DTT, 50 ng/µl poly(dI-dC), and 0.05% Nonidet P-40 binding buffer with 5 fmol of a biotin-labeled DNA fragment for 25 minutes.
After 25 min incubation at room temperature, samples were resolved by a 10% non-denaturing polyacrylamide gel prepared in TBE buffer (450 mM Tris, 450 mM boric acid and 0.01 mM EDTA). The samples were blotted onto a 0.45-µm Biodyne B nylon membrane (Thermo Scientific, www.piercenet.com/) at constant current of 300 mA for 45 min at 4°C, and then cross-linked to the membrane using a 312 nm UV Transilluminator (Uvitec, www.uvitec.com.) for 10 min. Membranes were processed as recommended in the Chemiluminescent Nucleic Acid Detection Module Kit (Thermo Scientific, www.piercenet.com/).
Competitive EMSAs were performed as described above, adding a preincubation step of 20 min at room temperature of GcrA and competitor DNAs before the usual 25 min GcrA/biotin-labeled DNA fragment incubation.
EMSA in presence of RNA polymerase core enzyme (Epicentre, www.epibio.com/) was performed by pre-incubating GcrA in presence of RNAP for 20 min at room temperature before the usual incubation with biotin-labeled DNA.
For detecting the binding region of GcrA, a 120 bp probe from ctrAP1 (Figure S8) was synthesized and labeled with Fam-6 (Eurogentec, www.eurogentec.com/). Single stranded probes containing m6A were also synthesized, which were later assembled into double stranded probes in different combinations. Five fmoles of probes were incubated at room temperature with increasing concentrations of purified GcrA as done with EMSA for 30 min. The samples were digested with approximately 7U of DNaseI (Euromedex, www.euromedex.com/) at room temp for 3 min. DNaseI was inactivated by adding 0.1 M EDTA followed by incubation at 75°C for 10 min. The digested fragments were eluted using the mini-elute columns (Qiagen, www.qiagen.com/). The samples were run in a 3130 Genetic Analyzer (Life Technologies) as described before [33], analyzed by GelQuest (SequentiX, www.sequentix.de/). Sequencing reactions were also performed using Thermo Sequenase Dye Primer Manual Cycle Sequencing Kit (Affymetrix, www.affymetrix.com/) using the probe region as a template and a sequencing primer labeled with FAM at the 5 primes.
β-Galactosidase assays
β-Galactosidase assays were performed at 30°C as described earlier [34], [35].
Immunoblots
PVDF (polyvinylidenfluoride) membranes (Merck-Millipore, www.merckmillipore.com) were blocked with PBS, 0.05% tween 20 and 5% dry milk for 1 h and then incubated for 1 h with the primary antibodies diluted in PBS, 0.05% tween 20, 5% dry milk. The membranes were washed 4 times for 5 min in PBS and incubated 1 h with the specific secondary antibody diluted in PBS, 0.05% tween 20 and 5% dry milk. The membranes were finally washed again 4 times for 5 min in PBS and revealed with Immobilon Western Blotting Chemoluminescence HRP substrate (Merck Millipore, www.merckmillipore.com/). The different antisera were used at the following dilutions: anti-CcrM (1∶10;000), anti-DnaA (1∶10;000), anti-GcrA (1∶10,000), anti-CtrA (1∶10,000). Anti-RNA polymerase beta antibodies (Abcam, www.abcam.com/) were used using the protocol described in “Affinity chromatography for RNAP detection”.
Quantitative Chromatin Immunoprecipitation (qChIP) assays
Mid-log phase cells were cross-linked in 10 mM sodium phosphate (pH 7.6) and 1% formaldehyde at room temperature for 10 min and on ice for 30 min thereafter, washed thrice in phosphate buffered saline (PBS) and lysed in a Ready-Lyse lysozyme solution (Epicentre, Madison, WI) according to the manufacturer's instructions. Lysates were sonicated (Sonifier Cell Disruptor B-30) (Branson Sonic Power. Co., www.bransonic.com/) on ice using 10 bursts of 20 sec at output level 4.5 to shear DNA fragments to an average length of 0.3–0.5 kbp and cleared by centrifugation at 14,000 rpm for 2 min at 4°C. Lysates were normalized by protein content, diluted to 1 mL using ChIP buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris-HCl [pH 8.1], 167 mM NaCl plus protease inhibitors (Roche, www.roche.com/) and pre-cleared with 80 µL of protein-A agarose (Roche, www.roche.com/) and 100 µg BSA. Ten % of the supernatant was removed and used as total chromatin input DNA.
Polyclonal antibodies to GcrA [16] and m6A (Synaptic Systems GmbH, www.sysy.com/) were added to the remains of the supernatant (1∶1,000 dilution), incubated overnight at 4°C with 80 µL of protein-A agarose beads pre-saturated with BSA, washed once with low salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl (pH 8.1), 150 mM NaCl), high salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl (pH 8.1), 500 mM NaCl) and LiCl buffer (0.25 M LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris-HCl (pH 8.1) and twice with TE buffer (10 mM Tris-HCl (pH 8.1) and 1 mM EDTA). The protein•DNA complexes were eluted in 500 µL freshly prepared elution buffer (1% SDS, 0.1 M NaHCO3), supplemented with NaCl to a final concentration of 300 mM and incubated overnight at 65°C to reverse the crosslinks. The samples were treated with 2 µg of Proteinase K for 2 h at 45°C in 40 mM EDTA and 40 mM Tris-HCl (pH 6.5). DNA was extracted using phenol∶chloroform∶isoamyl alcohol (25∶24∶1), ethanol-precipitated using 20 µg of glycogen as carrier and resuspended in 100 µl of water.
Real-time PCR
Real-time PCR was performed using a Step-One Real-Time PCR system (Applied Biosystems, www.appliedbiosystems.com/) using 5% of each ChIP sample (5 µL), 12.5 µL of SYBR green PCR master mix (Quanta Biosciences, www.quantabio.com/), 0.5 µL of primers (10 µM) and 6.5 µL of water per reaction. Standard curve generated from the cycle threshold (Ct) value of the serially diluted chromatin input was used to calculate the percentage input value of each sample. Average values are from triplicate measurements done per culture. The final data was generated from three independent cultures. The DNA regions analyzed by real-time PCR were from nucleotide −167 to +43 relative to the start codon of podJ, from −208 to +9 relative to the start codon of mipZ, from −185 to −16 relative to the start codon of ctrA.
Supporting Information
Zdroje
1. WionD, CasadesúsJ (2006) N6-methyl-adenine: an epigenetic signal for DNA-protein interactions. Nat Rev Microbiol 4 : 183–192 doi:10.1038/nrmicro1350.
2. MarinusMG, CasadesusJ (2009) Roles of DNA adenine methylation in host-pathogen interactions: mismatch repair, transcriptional regulation, and more. FEMS Microbiol Rev 33 : 488–503 doi:10.1111/j.1574-6976.2008.00159.x.
3. BrunetYR, BernardCS, GavioliM, LloubèsR, CascalesE (2011) An epigenetic switch involving overlapping fur and DNA methylation optimizes expression of a type VI secretion gene cluster. PLoS Genet 7: e1002205 doi:10.1371/journal.pgen.1002205.
4. DemarreG, ChattorajDK (2010) DNA adenine methylation is required to replicate both Vibrio cholerae chromosomes once per cell cycle. PLoS Genet 6: e1000939 doi:10.1371/journal.pgen.1000939.
5. ZweigerG, MarczynskiG, ShapiroL (1994) A Caulobacter DNA methyltransferase that functions only in the predivisional cell. J Mol Biol 235 : 472–485 doi:10.1006/jmbi.1994.1007.
6. StephensC, ReisenauerA, WrightR, ShapiroL (1996) A cell cycle-regulated bacterial DNA methyltransferase is essential for viability. Proc Natl Acad Sci USA 93 : 1210–1214.
7. WrightR, StephensC, ShapiroL (1997) The CcrM DNA methyltransferase is widespread in the alpha subdivision of proteobacteria, and its essential functions are conserved in Rhizobium meliloti and Caulobacter crescentus. J Bacteriol 179 : 5869–5877.
8. RobertsonGT, ReisenauerA, WrightR, JensenRB, JensenA, et al. (2000) The Brucella abortus CcrM DNA methyltransferase is essential for viability, and its overexpression attenuates intracellular replication in murine macrophages. J Bacteriol 182 : 3482–3489.
9. TsokosCG, LaubMT (2012) Polarity and cell fate asymmetry in Caulobacter crescentus. Curr Opin Microbiol 15 : 744–750 doi:10.1016/j.mib.2012.10.011.
10. QuonKC, MarczynskiGT, ShapiroL (1996) Cell cycle control by an essential bacterial two-component signal transduction protein. Cell 84 : 83–93.
11. QuonKC, YangB, DomianIJ, ShapiroL, MarczynskiGT (1998) Negative control of bacterial DNA replication by a cell cycle regulatory protein that binds at the chromosome origin. Proc Natl Acad Sci USA 95 : 120–125.
12. HottesAK, ShapiroL, McAdamsHH (2005) DnaA coordinates replication initiation and cell cycle transcription in Caulobacter crescentus. Mol Microbiol 58 : 1340–1353 doi:10.1111/j.1365-2958.2005.04912.x.
13. DomianIJ, ReisenauerA, ShapiroL (1999) Feedback control of a master bacterial cell-cycle regulator. Proc Natl Acad Sci USA 96 : 6648–6653.
14. GillJJ, BerryJD, RussellWK, LessorL, Escobar GarciaDA, et al. (2012) The Caulobacter crescentus phage phiCbK: genomics of a canonical phage. BMC Genomics 13 : 542 doi:10.1186/1471-2164-13-542.
15. PanisG, LambertC, ViollierPH (2012) Complete genome sequence of Caulobacter crescentus bacteriophage ϕCbK. J Virol 86 : 10234–10235 doi:10.1128/JVI.01579-12.
16. HoltzendorffJ, HungD, BrendeP, ReisenauerA, ViollierPH, et al. (2004) Oscillating global regulators control the genetic circuit driving a bacterial cell cycle. Science 304 : 983–987 doi:10.1126/science.1095191.
17. BiondiEG, ReisingerSJ, SkerkerJM, ArifM, PerchukBS, et al. (2006) Regulation of the bacterial cell cycle by an integrated genetic circuit. Nature 444 : 899–904 doi:10.1038/nature05321.
18. AlbuRF, JurkowskiTP, JeltschA (2012) The Caulobacter crescentus DNA-(adenine-N6)-methyltransferase CcrM methylates DNA in a distributive manner. Nucleic Acids Res 40 : 1708–1716 doi:10.1093/nar/gkr768.
19. ReisenauerA, ShapiroL (2002) DNA methylation affects the cell cycle transcription of the CtrA global regulator in Caulobacter. EMBO J 21 : 4969–4977.
20. BrilliM, FondiM, FaniR, MengoniA, FerriL, et al. (2010) The diversity and evolution of cell cycle regulation in alpha-proteobacteria: a comparative genomic analysis. BMC Syst Biol 4 : 52 doi:10.1186/1752-0509-4-52.
21. LetunicI, DoerksT, BorkP (2012) SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Res 40: D302–305 doi:10.1093/nar/gkr931.
22. GeourjonC, DeléageG (1995) SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci 11 : 681–684.
23. RobertsonG, HirstM, BainbridgeM, BilenkyM, ZhaoY, et al. (2007) Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods 4 : 651–657 doi:10.1038/nmeth1068.
24. ThanbichlerM, ShapiroL (2006) MipZ, a spatial regulator coordinating chromosome segregation with cell division in Caulobacter. Cell 126 : 147–162 doi:10.1016/j.cell.2006.05.038.
25. ViollierPH, SternheimN, ShapiroL (2002) Identification of a localization factor for the polar positioning of bacterial structural and regulatory proteins. Proc Natl Acad Sci USA 99 : 13831–13836 doi:10.1073/pnas.182411999.
26. HinzAJ, LarsonDE, SmithCS, BrunYV (2003) The Caulobacter crescentus polar organelle development protein PodJ is differentially localized and is required for polar targeting of the PleC development regulator. Mol Microbiol 47 : 929–941.
27. NiermanWC, FeldblyumTV, LaubMT, PaulsenIT, NelsonKE, et al. (2001) Complete genome sequence of Caulobacter crescentus. Proc Natl Acad Sci USA 98 : 4136–4141 doi:10.1073/pnas.061029298.
28. FioravantiA, ClantinB, DewitteF, LensZ, VergerA, et al. (2012) Structural insights into ChpT, an essential dimeric histidine phosphotransferase regulating the cell cycle in Caulobacter crescentus. Acta Crystallogr Sect F Struct Biol Cryst Commun 68 : 1025–1029 doi:10.1107/S1744309112033064.
29. HerndayA, BraatenB, LowD (2004) The intricate workings of a bacterial epigenetic switch. Adv Exp Med Biol 547 : 83–89.
30. SkerkerJM, PrasolMS, PerchukBS, BiondiEG, LaubMT (2005) Two-component signal transduction pathways regulating growth and cell cycle progression in a bacterium: a system-level analysis. PLoS Biol 3: e334 doi:10.1371/journal.pbio.0030334.
31. ElyB (1991) Genetics of Caulobacter crescentus. Meth Enzymol 204 : 372–384.
32. GoberJW, ShapiroL (1992) A developmentally regulated Caulobacter flagellar promoter is activated by 3′ enhancer and IHF binding elements. Mol Biol Cell 3 : 913–926.
33. YindeeyoungyeonW, SchellMA (2000) Footprinting with an automated capillary DNA sequencer. BioTechniques 29 : 1034–1036, 1038, 1040–1041.
34. HuitemaE, PritchardS, MattesonD, RadhakrishnanSK, ViollierPH (2006) Bacterial birth scar proteins mark future flagellum assembly site. Cell 124 : 1025–1037 doi:10.1016/j.cell.2006.01.019.
35. ViollierPH, ShapiroL (2003) A lytic transglycosylase homologue, PleA, is required for the assembly of pili and the flagellum at the Caulobacter crescentus cell pole. Mol Microbiol 49 : 331–345.
36. GoujonM, McWilliamH, LiW, ValentinF, SquizzatoS, et al. (2010) A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Research 38: W695–W699 doi:10.1093/nar/gkq313.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2013 Číslo 5
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Using Extended Genealogy to Estimate Components of Heritability for 23 Quantitative and Dichotomous Traits
- Encodes CDF Transporters That Excrete Zinc from Intestinal Cells of and Act in a Parallel Negative Feedback Circuit That Promotes Homeostasis
- HDAC7 Is a Repressor of Myeloid Genes Whose Downregulation Is Required for Transdifferentiation of Pre-B Cells into Macrophages
- Female Bias in and Regulation by the Histone Demethylase KDM6A
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy