
UTX and UTY Demonstrate Histone Demethylase-Independent Function in Mouse Embryonic Development
UTX (KDM6A) and UTY are homologous X and Y chromosome members of the Histone H3 Lysine 27 (H3K27) demethylase gene family. UTX can demethylate H3K27; however, in vitro assays suggest that human UTY has lost enzymatic activity due to sequence divergence. We produced mouse mutations in both Utx and Uty. Homozygous Utx mutant female embryos are mid-gestational lethal with defects in neural tube, yolk sac, and cardiac development. We demonstrate that mouse UTY is devoid of in vivo demethylase activity, so hemizygous XUtx− Y+ mutant male embryos should phenocopy homozygous XUtx− XUtx− females. However, XUtx− Y+ mutant male embryos develop to term; although runted, approximately 25% survive postnatally reaching adulthood. Hemizygous X+ YUty− mutant males are viable. In contrast, compound hemizygous XUtx− YUty− males phenocopy homozygous XUtx− XUtx− females. Therefore, despite divergence of UTX and UTY in catalyzing H3K27 demethylation, they maintain functional redundancy during embryonic development. Our data suggest that UTX and UTY are able to regulate gene activity through demethylase independent mechanisms. We conclude that UTX H3K27 demethylation is non-essential for embryonic viability.
Published in the journal:
. PLoS Genet 8(9): e32767. doi:10.1371/journal.pgen.1002964
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002964
Summary
UTX (KDM6A) and UTY are homologous X and Y chromosome members of the Histone H3 Lysine 27 (H3K27) demethylase gene family. UTX can demethylate H3K27; however, in vitro assays suggest that human UTY has lost enzymatic activity due to sequence divergence. We produced mouse mutations in both Utx and Uty. Homozygous Utx mutant female embryos are mid-gestational lethal with defects in neural tube, yolk sac, and cardiac development. We demonstrate that mouse UTY is devoid of in vivo demethylase activity, so hemizygous XUtx− Y+ mutant male embryos should phenocopy homozygous XUtx− XUtx− females. However, XUtx− Y+ mutant male embryos develop to term; although runted, approximately 25% survive postnatally reaching adulthood. Hemizygous X+ YUty− mutant males are viable. In contrast, compound hemizygous XUtx− YUty− males phenocopy homozygous XUtx− XUtx− females. Therefore, despite divergence of UTX and UTY in catalyzing H3K27 demethylation, they maintain functional redundancy during embryonic development. Our data suggest that UTX and UTY are able to regulate gene activity through demethylase independent mechanisms. We conclude that UTX H3K27 demethylation is non-essential for embryonic viability.
Introduction
Post-translational modifications of histones establish and maintain active or repressive chromatin states throughout cell lineages. Thus, the enzymes that catalyze these modifications often have crucial roles in establishing genomic transcriptional states in developmental decision-making. Histone methylation can stimulate gene activation or repression depending on which residues are targeted. Methylation of histone H3 on Lysine 4 (H3K4me) is an active chromatin modification, while methylation on histone H3 Lysine 27 (H3K27me) is associated with repression of gene activity [1].
The polycomb repressive complex 2 (PRC2) methylates H3K27 [2], [3], [4], [5]. Within this complex, enhancer of zeste homolog 2 (EZH2) catalyzes di and tri-methylation of H3K27. Embryonic ectoderm development (EED) and suppressor of zeste homolog 12 (SUZ12) are additional PRC2 core components indispensible for PRC2 activity [6], [7], [8]. EZH1 is a secondary, less efficient H3K27 methyl-transferase that shares some overlapping redundancy with EZH2 in ES cells and epidermal stem cells [9], [10], [11], [12]. The PRC1 complex is recruited through H3K27 trimethylation for additional histone modification and chromatin compaction [13]. In embryonic stem (ES) cells, PRC2 targets and represses genes essential for developmental events [14], [15], [16], [17]. The promoters of these PRC2 targets typically contain “bivalent” chromatin marks with both active H3K4 and repressive H3K27 methylation [18], [19], [20]. Loss of PRC2 activity de-represses these genes but does not alter ES cell pluripotency [14]. However, mouse mutations in any of the three PRC2 core components are early embryonic lethal with gastrulation defects [7], [21],[22].
H3K27 trimethylation is reversible as a family of histone demethylases catalyzes the removal of this epigenetic mark [23], [24], [25], [26]. JMJD3 (KDM6B) is an autosomal H3K27 demethylase upregulated during specific differentiation events [25], [27]. UTX (KDM6A) is a broadly expressed X-linked H3K27 demethylase that escapes X-inactivation [23], [24], [26], [28]. UTY is the Y chromosome homolog of UTX. Both UTX and JMJD3 demethylate H3K27 di and tri-methyl residues; however, UTY lacks this activity in vitro [26], [29]. Based on cell culture models, UTX and JMJD3 mediated H3K27 demethylation is vital in a wide array of functions including cell cycle regulation, M2 macrophage differentiation, neuronal stem cell specification, skin differentiation, and muscle differentiation [27], [30], [31], [32], [33], [34], [35]. In contrast, the biological function of UTY remains unknown.
Utx and Uty are genetically amenable to delineate H3K27me3 demethylation dependent versus demethylation independent function in mouse development. Comparative amino acid sequence analysis of UTX and UTY reveals 88% sequence similarity in humans (83% identity) and 82% sequence similarity in mouse. Across the annotated JmjC histone demethylase domain, the similarity is at 98% and 97% for human and mouse respectively. In the TPR (tetratricopeptide repeat) domain, the similarity is at 94%. So while UTY is reported to have lost H3K27 demethylase activity, it is remarkably well conserved with respect to UTX. Recent discoveries have revealed that JMJD3 functions in macrophage lipopolysaccharide response and lymphocyte Th1 response through H3K27 demethylase independent gene regulation [36], [37], suggesting that function of this family of proteins is not limited to histone demethylation. It has been hypothesized that X and Y chromosome homologs will escape X-inactivation in instances where the Y homolog has not lost functional activity and male to female dosage remains balanced [38]. Therefore, it is possible that UTX and UTY have functional overlap in H3K27 demethylase independent gene regulatory processes.
A recent publication by Lee et al. characterized heart defects in Utx homozygous embryos [39]. Cell culture experiments suggested that the phenotype resulted from H3K27 demethylase activity. Utx hemizygotes were reported to have a wide range of abnormalities, but it was not clear if any phenotypes overlap with the Utx homozygotes as no comparative data were illustrated. Given that Uty remained intact in these studies, it was not possible to conclude definitively whether Utx demethylase activity was essential for early embryonic development. Furthermore, it is not known whether mouse UTY is capable of H3K27 demethylation. The classification of UTY as having no demethylase activity is based on in vitro assays only. The possibility of in vivo demethylase activity due to other co-factors remains a possibility. Also, mouse UTY has considerable sequence divergence from human UTY. The two proteins are 75% identical overall, and 95% identical in the JmjC demethylase domain. Thus, it is possible that mouse UTY has retained demethylase activity.
In our study, we have generated mouse mutations in both Utx and Uty. Hemizygous Utx mutant male mice (XUtx− Y+) were runted at birth with only a small number surviving to adulthood. In contrast, Utx homozygous females (XUtx− XUtx−) had severe phenotypes mid-gestation, with developmental delay, neural tube closure, yolk sac, and heart defects. Unlike homozygotes, Utx hemizygotes lack mid-gestational cardiovascular defects and are recovered in Mendelian frequencies at E18.5. Furthermore, compound hemizygous male embryos (XUtx− YUty−) carrying mutations of both Utx and Uty phenocopy the Utx homozygotes. Thus, the disparity in hemizygous and homozygous Utx phenotypes is due to compensation by Uty in the hemizygous male embryos. We have utilized an in vivo H3K27 demethylation assay to demonstrate that mouse UTY is not capable of H3K27 demethylation. Additionally, cell culture data indicate UTX and UTY may function in gene activation as both proteins associate with the H3K4 methyl-transferase complex, the BRG1 chromatin remodeler, as well as heart transcription factors. Our results implicate a crucial H3K27 demethylase independent function for UTX and UTY in mouse embryonic development. This is the first ascribed function for UTY, and the first example of developmental redundancy for X and Y chromosome homologous genes. Notably, our data suggest the H3K27 demethylase activity of UTX is not essential for embryonic viability.
Results
Hemizygous Utx mutant male mice have reduced peri-natal viability
We developed mutant mouse lines to assess the contribution of UTX H3K27 demethylase function in mouse development. Two alleles for Utx were obtained from public resources. The BayGenomics gene trap line Kdm6aGt(RRA094)Byg is designated as XUtxGT1 (Figure 1A). RT-PCR and PCR genotyping verified the identity of this allele in both ES cells and mutant mice (Figures S1 and S2A–S2C). Additionally, we obtained the EUCOMM Kdm6a knockout line (project 26585, Kdm6atm1a(EUCOMM)Wtsi), designated as XUtxGT2fl, which inserts a gene trap in intron 2 along with a floxed 3rd exon (Figure 1A). Southern blotting and PCR genotyping verified the identity of this allele (Figures S1 and S2D–S2F). Notably, quantitative RT-PCR comparison of tail RNA from XUtxGT1 YUty+ versus XUtxGT2fl YUty+ mice demonstrated that Utx gene trap 1 is more effective than gene trap 2 (a 96% reduction compared to a 61% reduction in Figures S2C and S2F). Because XUtxGT2fl demonstrated incomplete trapping, the 3rd exon was deleted with Cre recombinase to establish XUtxGT2Δ (containing both the gene trap and deleted 3rd exon, Figure 1A). Deletion of the third Utx exon produces a frameshift and introduction of a translational stop codon when Utx is spliced from exon 2 to exon 4. XUtxGT1 and XUtxΔ are null alleles as UTX protein was eliminated in western blotting of these embryonic lysates (Figure 1B, 1C). Consistent with RT-PCR data, XUtxGT2fl exhibits a reduction but not absence of UTX protein (Figure 1D).

Heterozygous Utx female mice were crossed to wild type male mice to produce hemizygous Utx mutant males. At weaning, the hemizygous XUtxGT1 YUty+, XUtxGT2Δ YUty+, and XUtxGT2fl YUty+ mice all exhibited reductions of 68%, 83%, and 55% respectively from the expected genotype frequencies based on these crosses, yet expected genotype frequencies were observed at embryonic day E18.5 (Table 1). At E18.5, most of the hemizygous Utx males appeared phenotypically normal; however a small percentage of the fetuses exhibited exencephaly. At birth, the hemizygous Utx males were small and exhibited a failure to thrive phenotype. Those males that survived through this phenocritical phase reached adulthood and were fertile. Hemizygous Utx mutant males were runted compared to wild type littermates and remained smaller than controls throughout their lifespan (Figure 2A, 2B). Backcross of the Utx allele onto a C57BL/6J or 129/SvJ background affected postnatal viability, but hemizygous Utx male embryos were still readily obtained at E18.5 (Table S1).


Homozygous Utx females are mid-gestational embryonic lethal
Human UTY lacks demethylase activity based on in vitro assays, so we hypothesized that XUtx− XUtx− homozygous females will phenocopy XUtx− YUty+ hemizygous males in demethylase dependent function (UTX specific), but may demonstrate a more severe phenotype in demethylase independent roles. Homozygous XUtxGT1 XUtxGT1 and XUtxGT2Δ XUtxGT2Δ females were never observed at weaning or embryonic day E18.5 (Table 1), but were observed at expected genotype frequencies at E10.5. However, these embryos were dead and resorbed by E12.5 (Table 1). Notably, at E10.5 all homozygous XUtxGT1 XUtxGT1 and XUtxGT2Δ XUtxGT2Δ females were smaller in size and had open neural tubes in the midbrain region (Figure 3A-ii, iii, vi, vii). Variation in severity of the Utx homozygous phenotypes was observed in mutant embryos, ranging from medium sized with typical E10.5 features (Figure 3A-ii, vi) to much smaller embryos resembling the E9.5 timepoint (Figure 3A-iii, vii). The XUtxGT1 and XUtxGT2Δ alleles failed to complement, as trans-heterozygous XUtxGT1 XUtxGT2Δ female embryos resembled individual homozygous alleles (Figure 3A-viii). Hemizygous XUtxGT1 YUty+ male embryos appeared phenotypically normal at E10.5 (Figure 3A-iv). Homozygous XUtxGT2fl XUtxGT2fl females exhibited a slight reduction in phenotypic severity; about half of the mutant embryos had open neural tubes and some survival to E12.5 (Table 1). To distinguish between embryonic and extraembryonic contribution of UTX towards the homozygous phenotype, we crossed the Sox2Cre transgene into the Utxfl background. In this cross, paternally inherited Sox2Cre expression will drive Utx deletion specifically in embryonic tissue [40]. No XUtxfl XUtxfl, Sox2Cre female embryos were recovered at E18.5, whereas XUtxfl YUty+, Sox2Cre male embryos were recovered at expected frequencies (Table S2). At E10.5, XUtxfl XUtxfl, Sox2Cre embryos produced phenotypes largely identical to Utx homozygotes. In summary, Utx homozygous females demonstrate a significantly more severe embryonic phenotype in comparison to Utx hemizygous males.

Mid-gestational lethality is typically associated with defective cardiovascular development. Accordingly, we observed both heart and yolk sac vasculature/hematopoietic phenotypes in Utx homozygotes. Utx homozygous mutant hearts were small and underdeveloped, and more severe embryos exhibited peri-cardial edema (Figure 3A-ii, iii, vi, vii). The yolk sac vasculature of Utx homozygotes was pale with a reduction in the amount of vascular blood (Figure 3B-ii). In more severe examples, homozygous yolk sacs were completely pale with an unremodeled vascular plexus (Figure 3B-iii). Thus, abnormal cardiovascular function may be a source of lethality and developmental delay in Utx homozygous mutant embryos.
UTX and UTY have redundant function in embryonic development
The most likely explanation for the disparity between Utx hemizygotes and homozygotes is that UTY can compensate for the loss of UTX in embryonic development. We tested Utx and Uty expression in embryonic development to assess any overlap in expression patterns. Utx expression was initially gauged utilizing the B-galactosidase reporter in XUtx+ XUtxGT1 and XUtxGT1 XUtxGT1 whole mount E10.5 embryos. Utx was expressed at lower levels throughout the E10.5 embryo with a particular enrichment in the neural tube and otic placode (Figure S3A-ii, iii, iv). In situ hybridization for both Utx and Uty demonstrated similar expression patterns characterized by widespread low-level expression with particular enrichment in the neural tube (Figure S3B-ii, iii, v, vi). Our analysis of publicly available RNA-seq data sets [41], [42] revealed similar low-levels of expression for Utx and Uty.
To determine whether UTY can compensate for the loss of UTX, we obtained the Welcome Trust Sanger Institute gene trap line UtyGt(XS0378)Wtsi, designated as YUtyGT (Figure 4A). This line, inserted in intron 4, traps the Uty transcript in a similar position of the coding sequence as the Utx alleles (compare to Figure 1A). This gene trap line was verified by RT-PCR in ES cells and subsequent mice (Figures S1 and S2G), and it achieved a 99% reduction in Uty expression from XUtx+ YUtyGT mouse tail RNA (Figure 4B). Hemizygous Uty mutant males, XUtx+ YUtyGT, were viable and fertile (Table 1). However, no compound hemizygous XUtxGT1 YUtyGT and XUtxGT2Δ YUtyGT embryos were recovered at E18.5 (Table 1). At E10.5, expected genotype frequencies of XUtxGT1 YUtyGT and XUtxGT2Δ YUtyGT males were observed, but these embryos phenocopied the developmental delay, neural tube closure, cardiac, and yolk sac defects observed in Utx homozygous embryos (Figure 4C-iii, iv).

UTX and UTY redundancy is essential for progression of cardiac development
We performed a more detailed phenotypic assessment of Utx and Uty mutant hearts to scrutinize the extent of phenotypic overlap between XUtx− YUty+, XUtx− XUtx−, and XUtx− YUty− embryos. Analysis of cardiac development in similar sized E10.5 embryos (Figure 5A-i, ii, iii, iv) revealed that Utx homozygotes and Utx/Uty compound hemizygotes failed to complete heart looping (Figure 5A-vi, viii), whereas Utx heterozygotes and hemizygotes were phenotypically normal (Figure 5A-v, vii). Additionally, homozygotes and compound hemizygotes had smaller hearts with a lack of constriction between the left and right ventricles. Sectioning of E10.5 hearts confirmed that Utx homozygotes and Utx/Uty compound hemizygotes have small hearts with a reduction in ventricular myocardial trabeculation and little or no initiation of interventricular septum formation (Figure 5B-ii, iv). The outer ventricular wall of these embryos is much thinner, and the overall number of cardiomyocytes and myocardial structure is severely deficient (Figure 5C-ii, iv). In summary, while mid-gestational hearts appear normal in XUtx− YUty+ hemizygous males, XUtx− XUtx−homozygous females and XUtx− YUty− compound hemizygous males display identical deficiencies in cardiac development. Therefore, UTY compensates for the loss of UTX in hemizygous Utx mutant males, rescuing mid-gestational cardiac phenotypes.

Mouse and human UTY are incapable of H3K27 demethylation in vivo
UTX and UTY have redundant function in embryonic development, but it is not known whether mouse UTY is capable of H3K27 demethylation. Two independent publications demonstrated that human UTY has no catalytic activity in H3K27 demethylation in vitro [26], [29]. It is possible that human UTY (and not mouse UTY) has accumulated a specific polymorphism rendering it demethylase deficient. Additionally, in vitro assays remove UTY from its natural cellular context and may lack co-factors required to promote H3K27 demethylation. Therefore, we utilized an intracellular, in vivo demethylation assay, whereby HEK293T cells transiently over-expressing the UTX carboxy-terminus (encoding the JmjC and surrounding domains essential for proper structure and function) exhibit a reduction in H3K27me3 immunofluorescence levels [43]. In our assay, wild type and mutant constructs were expressed at similar levels (Figure S4A), and individual cells expressing similar, medium-high expression levels of each construct were selected for analysis (Figure 6). Expression of Flag-tagged human and mouse UTX demethylated H3K27me3 and H3K27me2, while a mutation known to disrupt activity (H1146A) was unable to demethylate H3K27 (Figure 6A and 6B, Figure S5A). Human UTX expression had no effect on other histone modifications we tested, such as H3K4me2 (Figure S5B). In contrast, neither human nor mouse UTY were capable of demethylating H3K27me3 and H3K27me2 (Figure 6A and Figure S5A). Cells expressing medium-to-high levels of UTY (N>100) never exhibited a reduction in H3K27me3 levels relative to nearby untransfected controls.

Our previous structural analysis of human UTX [43], combined with sequence alignments (Figure 6C and Figure S6), suggested several amino acid substitutions in human and mouse UTY sequences might make them catalytically inactive. We introduced these mutations into the human UTX C-terminal fragment (Y1135C, T1141I, SNR1025NKS, G1172D/G1191S, I1267P, I1267V, and H1329P), and examined their effects on the in vivo demethylation activity. Of all the mutations tested, only the Y1135C and T1143I mutations completely abolished the ability of UTX to demethylate H3K27 (Figure 6B, 6D). Complete loss of activity was similarly caused by mutations of the corresponding residues in JMJD3 (Y1377C and T1385I, Figure 6D). All qualitative data was also confirmed by immunofluorescence quantification (Figure S4B). Y1135 is conserved throughout all H3K27 demethylases (Figure S7), and in the crystal structure [43], it interacts with two of the three methyl groups of the H3K27me3 side chain, as well as N-oxalylglycine (NOG; an analog of the cofactor alpha-ketoglutarate) (Figure 6E). The smaller C947 side chain of mouse UTY would not effectively maintain either interaction. T1143 is conserved throughout H3K27, H3K9, and H3K36 demethylases (Figure S8), and also interacts with NOG (Figure 6E). Its replacement with bulky isoleucine not only removes the hydroxyl group for interaction with alpha-ketoglutarate, but also may sterically hinder its binding. These observations are consistent with the fact that no H3K27 demethylation activity has been detected for mouse UTY, and we therefore conclude that the catalytic domain of mouse UTY has crucial amino acid replacements that render the protein incapable of H3K27 demethylation. On the other hand, we failed to identify why human UTY is catalytically inactive. Notably, restoring the 2 crucial mouse UTY polymorphisms (M-UTY C947Y, I955T) failed to recover H3K27 demethylase activity (Figure 6B). These data suggest that unidentified structural elements in the UTY C-terminal region are also responsible for the lack of H3K27 demethylase activity.
UTX and UTY associate in common protein complexes and are capable of H3K27 demethylase independent gene regulation
Although human and mouse UTY have lost the ability to demethylate H3K27, they retain considerable sequence similarity with UTX, suggesting a conserved function. To gain more insight into the overlap in UTX and UTY activities, we performed a biochemical analysis of tagged constructs to determine if UTX and UTY can associate in common protein complexes. Co-transfection of Flag tagged UTX or UTY with HA-UTX followed by immunoprecipitation demonstrates that UTX can form a multimeric complex with itself and UTY (Figure 7A). UTX associates with a H3K4 methyl-transferase complex containing MLL3, MLL4, PTIP, ASH2L, RBBP5, PA-1, and WDR5 [23], [44]. To examine incorporation into this complex, we performed immunoprecipitations with Flag tagged UTX and UTY constructs. Both UTX and UTY were capable of associating with RBBP5 (Figure 7B). Thus, UTX and UTY are incorporated into common protein complexes.

To identify common gene targets of UTX and UTY mediated regulation we generated E10.5 mouse embryonic fibroblast (MEF) cell lines containing mutations in Utx and Uty (alleles XUtxGT2Δ and YUtyGT). The gene traps in these MEFs efficiently trapped Utx and Uty transcripts (Figure 7C). These MEFs did not demonstrate differences in levels of global H3K27me3 (Figure S9A). Genome-wide UTX promoter occupancy has been mapped in fibroblasts [30]. Therefore, we screened our mutant MEFs for misregulated genes affected by the loss of both Utx and Uty that had been documented as direct UTX targets. The FNBP1 promoter is bound by UTX [30]. We verified UTX and UTY binding to the Fnbp1 promoter by ChIP (Figure S9B and S9C). Fnbp1 expression was reduced to 68% of WT levels in XUtx− YUty+ MEFs, but was further compromised to 42% in XUtx− XUtx− lines and 48% in XUtx− YUty− MEFs in which all Utx and Uty activity was lost (Figure 7C). Analysis of E12.5 MEFs of a secondary allele (XUtxGT2fl) also demonstrated diminished Fnbp1 expression in both XUtx− XUtx− and XUtx− YUty− MEFs (Figure 7D). Therefore, Fnbp1 expression is positively regulated by both UTX and UTY.
To examine the role of UTX and UTY in Fnbp1 regulation, we performed H3K27me3 ChIP on E12.5 XUtx+ YUty+ or XUtx− XUtx− MEFs (Figure 7E). Quantitative PCR for an intergenic region served as a negative control, while HoxB1 served as a positive control for H3K27me3. Quantitative PCR demonstrated that the Fnbp1 promoter has relatively low levels of H3K27me3 with no additional accumulation in XUtx− XUtx− MEFs (Figure 7E). Alternatively, H3K4me3 significantly accumulated at the Fnbp1 promoter (Figure 7F). Notably, a loss of Fnbp1 H3K4me3 was observed in XUtx− XUtx− MEFs (Figure 7F). Therefore, UTX and UTY appear to function in Fnbp1 activation by regulating promoter H3K4 methylation rather than H3K27 demethylation.
UTX and UTY can both associate with heart transcription factors to regulate downstream target genes
It has been documented that UTX can associate with heart transcription factors and with the SWI/SNF chromatin remodeler, BRG1 [39]. It has been hypothesized that UTX association with these factors mediates H3K27 demethylase dependent and demethylase independent induction of the cardiomyocyte specification program. As UTX and UTY have redundant demethylase independent function in embryonic development, we examined whether UTY can also associate with these proteins. Co-transfection of Myc-UTY with Flag-BRG1 followed by immunoprecipitation demonstrated that UTY associates with BRG1 (Figure 8A). Myc-UTY also co-immunoprecipitated with Flag-NKX2–5, Flag-TBX5, and Flag-SRF (Figure 8B and Figure S10A). Thus, UTY can form the same protein complexes as UTX with respect to BRG1 and heart transcription factors.
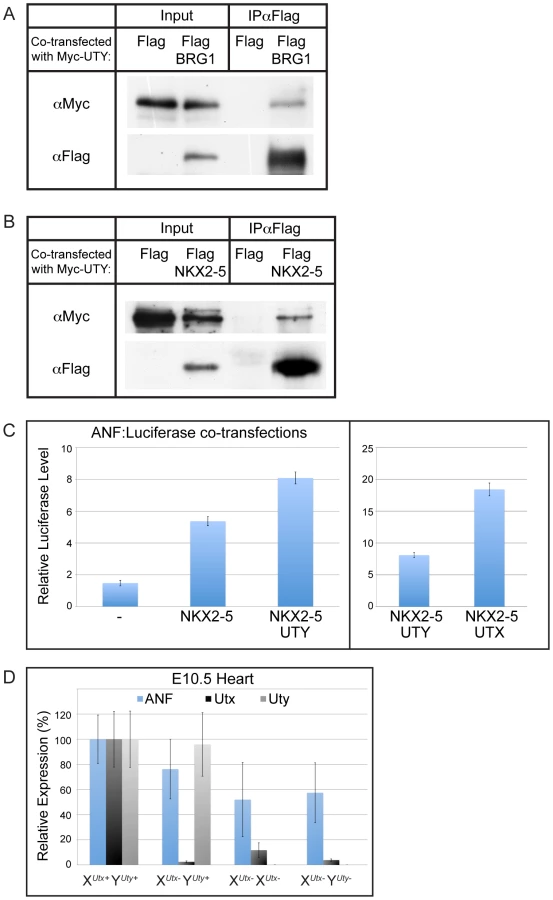
To examine function of UTY in directing activation of downstream heart transcription factor targets, we assessed the regulation of one previously characterized target, atrial natriuretic factor (ANF) [39]. Co-transfection of NKX2–5 with a ANF promoter-Luciferase reporter construct demonstrated a significant upregulation in expression off the ANF promoter (Figure 8C). The reporter expression was significantly enhanced when NKX2–5 was co-transfected with UTY (Figure 8C). The level of ANF reporter transcriptional enhancement was relatively weaker with UTY as compared to UTX, but this is most likely due to a reduction in the transfection efficiency of full-length UTY relative to UTX (as demonstrated in Figure 7A and 7B). UTY also significantly enhanced the ANF reporter response to TBX5 (Figure S10B). Finally, ANF expression was significantly affected in the hearts of only XUtx− XUtx− and XUtx− YUty− embryos (with 52% and 57% level of expression respective to XUtx+ YUty+ controls, Figure 8D). XUtx− YUty+ hemizygotes only had a moderate loss of ANF expression (76% expression level respective to XUtx+ YUty+) that was not statistically significant from wild type controls due to the variability in ANF expression. In summary, both UTX and UTY can associate with heart transcription factors to modulate expression of downstream targets.
Discussion
We have undertaken a rigorous genetic analysis contrasting UTX and UTY function in mouse embryonic development. In alignment with current literature, Utx homozygous females are lethal in mid-gestation with a block in cardiac development [39]. We now demonstrate that Utx hemizygous mutant males are viable at late embryonic timepoints in expected Mendelian frequencies. In fact, approximately 25% are capable of reaching adulthood. Our comprehensive phenotypic analysis of Utx hemizygous males illustrates that these embryos are phenotypically normal at mid-gestation and lack the cardiovascular dysfunction of Utx homozygous females. This stark phenotypic disparity suggests that UTY may compensate for the loss of UTX in the male embryo. Compound hemizygous Utx/Uty mutant male embryos phenocopy the cardiovascular and gross developmental delay of homozygous females, proving that UTX and UTY have redundant function in embryonic development. As we have demonstrated that mouse UTY lacks H3K27 demethylase activity in vivo, the overlap in embryonic UTX and UTY function is due to H3K27 demethylase independent activity. Given the widespread developmental delay and pleiotropy, it is difficult to assess the primary defect and tissue(s) responsible for UTX and UTY redundancy. The presence of functional UTY in Utx hemizygous males is not capable of preventing peri-natal runting and lethality, suggesting that UTX and UTY are not completely overlapping in activity. These later phenotypes could be due to H3K27 demethylase dependent activity of UTX. Furthermore, the lack of phenotype in Uty hemizygotes demonstrates the absence of any essential UTY specific function in mouse development.
The UTY Jumonji-C domain has maintained high conservation in the absence of catalytic H3K27 demethylase activity. JMJD3 mediated regulation of lymphocyte Th1 response requires an intact Jumonji-C domain, but is also not dependent on H3K27 demethylation [37]. Therefore, this domain may be an essential structural protein component, a protein binding domain, or a domain that may demethylate non-histone substrates. UTX and UTY can associate in a common protein complex and can both interact with RBBP5 of the H3K4 methyl-transferase complex. UTX, UTY, and JMJD3 all associate with H3K4 methyl-transferase complexes from multiple mouse and human cell types [23], [25], [44], [45]. The Fnbp1 promoter is bound by UTX, and gene expression is positively regulated by both UTX and UTY in MEFs. Based on our histone profiling at this locus, UTX and UTY affect the deposition of H3K4 methylation, not H3K27me3 demethylation. Therefore, the common UTX/UTY pathway in embryonic development may involve gene activation rather than removal of gene repression. JMJD3 has been linked more directly to transcriptional activation as the protein complexes with and facilitates factors involved in transcriptional elongation [46]. One cardiac target of UTX regulation, atrial natriuretic factor (ANF), was misregulated in ES cell differentiation [39]. Cell culture experiments suggest that ANF may be a target of both H3K27 demethylase dependent and demethylase independent regulation; however, this study could not distinguish UTX versus UTY function in ES cell differentiation. Both UTX and UTY affect the transcriptional response of an exogenous ANF reporter in the presence of heart specific transcription factors, suggesting that UTX and UTY can operate more directly by aiding in transcriptional activation of this gene rather than altering chromatin structure. Consistently, ANF expression was affected in XUtx− XUtx− and XUtx− YUty− embryonic hearts. UTX and UTY can both associate with the SWI/SNF chromatin remodeler BRG1, which has been hypothesized to mediate histone demethylase independent gene regulation, but the relevance and mechanism of this interaction is not known. Drosophila UTX associates with BRM (orthologous to BRG1) and CBP (a H3K27 acetyl-transferase), and the coupling of H3K27 demethylation with H3K27 acetylation may be essential for switching from a silent to active state [47].
Female cells are subject to gene silencing of one X-chromosome (X-inactivation) to balance gene dosage with males. Theory on establishing X-inactivation for X-Y chromosome homologs hypothesizes that the initial entry step is loss of function or expression of the Y homolog to create dosage imbalance [38]. This prediction also dictates that conservation of X-Y homolog function will maintain gene dosage between sexes, and the female X-homolog will not experience pressure to inactivate. Utx and Uty represent a unique paradox to this untested theory; UTY has lost demethylation activity yet Utx escapes X-inactivation. We now demonstrate that UTX and UTY have retained embryonic redundancy, verifying the presumed correlations between X-inactivation escape and functional dosage balance. Zfx, Sox3, and Amelx represent unbalanced X-chromosome genes; they have similar hemizygous and homozygous mutant phenotypes indicating that the Y chromosome homologs have lost redundant function [48], [49], [50], [51], [52], [53], [54]. Zfx and Sox3 are inactivated, while the Amelx inactivation status is unknown [55], [56]. Of all mouse X and Y chromosome homologs, only Utx, Kdm5c, and Eif2s3x are known to escape X-chromosome inactivation [28], [56], [57], [58]. Interestingly, both KDM5C (SMCX) and its Y chromosome homolog, KDM5D (SMCY) have retained catalytic activity in demethylation of H3K4 di and tri-methyl residues [59], [60], [61], [62]. In contrast to Utx X-chromosome escape driven by demethylation independent redundancy, Kdm5c may escape inactivation due to demethylation dependent redundancy. Our study is the first to demonstrate that an X-Y homologous pair that escapes X inactivation maintains functional conservation, and this escape may stem from an evolutionary benefit to maintain UTY demethylation independent function.
H3K27 demethylases are hypothesized to function in early developmental activation of “bivalent” PRC2 targets by coordinating H3K27 demethylation with H3K4 methylation. The H3K27 demethylation dependent phenotype (UTX specific) of Utx hemizygotes is not apparent until birth. The UTX H3K27 demethylase activity is dispensable for function in C. elegans [63]. Remarkably, the mammalian embryo, having numerous examples of H3K27me3 repression in early development, can survive to term without UTX histone demethylation. It is possible that there is further redundancy between UTX and JMJD3. JMJD3 mutant mice are not well characterized, but have been reported to be peri-natal lethal with distinct features in comparison to Utx hemizygotes [32]. Therefore, it is likely that JMJD3 has distinct targets in development. Overall, the earliest H3K27 demethylation dependent phenotypes for all members of this gene family do not manifest until late embryonic development. This timepoint is much later than the converse early embryonic phenotypes from mutations in the H3K27 methyl-transferase complex [7], . Thus, there appears to be a lack of interplay between H3K27 methylation and demethylation in gene regulation, and the early embryonic removal of H3K27me3 from PRC2 mediated processes (such as ES cell differentiation, reactivation of the inactive X-chromosome, or establishing autosomal imprinting) may involve other mechanisms such as histone turnover or chromatin remodeling. H3K27 demethylases may certainly have crucial roles in the specification of progenitor cell populations of organ systems essential in peri-natal or postnatal viability, and genetic model systems will best assess the functional impact that H3K27 demethylation plays in these processes.
Methods
Cell culture and constructs
HEK293T were maintained in DMEM supplemented with Glutamine, Pen-Strep, and 10%FBS. Flag-Human UTX (Plasmid #17438) and UTY (Plasmid #17439) were obtained through Addgene [29]. The N-terminus of H-UTX and H-UTY were deleted with QuikChange Lightning (Agilent) as directed producing H-UTX C-terminus 880–1401 (Genbank: NP_066963.2) and H-UTY C-terminus 827–1343 (Genbank: NP_009056.3, an N-terminal His tag was also incorporated into both constructs). Site directed mutagenesis was performed via QuikChange Lightning (Agilent) as directed to produce point mutations. The mouse UTX C-terminus (880–1401) deviated from Human UTX at 2 residues, R1073K and S1263N (According to the Sanger Vega server, the primary Utx transcript Kdm6a-001 encodes for Genbank: CAM27157, we also detected this transcript in E14 ES cell RT-PCR). These changes were created in the H-UTX C-terminus to generate the M-UTX construct. The Flag-tagged mouse UTY C-terminus (692–1212, Genbank: NP_033510.2) was subcloned by RT-PCR of E14 ES cell RNA and introduced into the same vector as the other UTX constructs (PCS2+MT backbone). HA tagged H-UTX was obtained through Addgene [24]. Flag tagged mouse JMJD3 was generously provided by Burgold et al. [27]. Flag tagged BRG1 was obtained through Addgene (Plasmid #19143). Flag tagged NKX2–5 (Plasmid #32969), TBX5 (Plasmid #32968), and SRF (Plasmid #32971) were obtained through Addgene and recombined into DEST26 (Invitrogen). Flag tagged NKX2–5 was also generously provided by Benoit Bruneau [64].
Transfections, immunofluorescence, Western blotting, antibodies
Transfection of HEK293T was accomplished with Lipofectamine 2000 as directed (Invitrogen). Lipid complexes were removed 24 hours post-transfection, and analysis was performed after 48 hours total. Fixation, extraction, and immunofluorescence were performed as described [65]. Immunofluorescence antibodies include anti-Flag (Sigma F3165, 1∶500), anti-H3K27me3 (Millipore 07-449, 1∶500), anti-H3K27me2 (Millipore 07-452, 1∶500), and anti-H3K4me2 (Millipore 07-030, 1∶500). Cells were imaged with Zeiss axiovision software. Image stacks were deconvolved and z-projected. Quantification of H3K27me3 immunofluorescence was performed on deconvolved z-projected stacks (with no pixel saturation in images) using ImageJ software (NIH). The average mean H3K27me3 signal was calculated for untransfected and transfected cells in a given image. For each image, the relative % H3K27me3 was determined, and the average relative % H3K27me3 was calculated for >15 images per construct. For western blotting, nuclear lysates were prepared according to Invitrogen's nuclear extraction protocol. Immunoprecipitations were carried out with 50 µl Flag beads (Sigma A2220) in buffer A as described, using 500 µg (UTY-UTX, RBBP5, BRG1, TBX5, SRF associations) or 1 mg (UTY-NKX2–5 association) of lysate [44]. Immunoprecipitation reactions were boiled off beads and run with 10% input on an 8% SDS-PAGE gel. Histone extractions were prepared as described [66]. Western blotting was performed as described [67] with anti-Flag (Cell Signaling 2368, 1∶4000), anti-RBBP5 (Bethyl Labs A300-109A, 1∶5000), anti-HA (Roche 11867423001, 1∶10000), anti-Myc (Abcam ab9132, 1∶5000), anti-H3K27me3 (Millipore 07-449, 1∶2000), anti-H3 (Millipore 06-755, 1∶5000), and anti-UTX [24].
MEF generation, RT–PCR, and ChIP
E10.5 and E12.5 MEFs were generated by removal of the head and interior organs of respective embryos. The remaining body was passed through a 20G needle 6× and plated in DMEM supplemented with Glutamine, Pen-Strep, and 15%FBS. After 3 passages, RNA was isolated with Trizol, and cleaned with an RNeasy kit (Quiagen). RNA from 3 distinct WT XUtx+ YUty+ MEF lines was compared to 3 XUtxGT2Δ XUtxGT2Δ lines on an Illumina bead array (University of Tennessee Health Science Center). All genes significantly decreased in XUtxGT2Δ XUtxGT2Δ MEFs were cross-referenced to the list of UTX bound promoters in human fibroblasts [30]. These genes were analyzed by qRT-PCR (Bio-Rad SsoFast EvaGreen, CFX96 real time system) in all mutant MEF combinations to identify UTY regulated genes. ChIP was performed on these MEFs according to Rahl et al. [68]. MEFs (5×106 cells) were sonicated by a Branson Sonifier at 15% duty cycle (0.7 s on 0.3 s off). ChIP was performed with anti-H3K27me3 (Millipore 07-449, 10 µl), anti-H3K4me3 (Abcam ab1012, 5 µl), anti-Myc (Abcam ab9132, 10 µl), anti-UTX (Santa Cruz H-300, 50 µl), or Rabbit IgG (Sigma, I5006) and qPCR was performed as described above.
Luciferase assay
We received the ANF promoter-Luciferase reporter construct from Benoit Bruneau [69]. This construct was co-transfected in the presence of NKX2–5 or TBX5 with or without UTX or UTY. Luciferase activity was measured using the Promega Dual Luciferase Reporter Assay System on the Promega Glomax Multi Detection System. All readings were normalized to a Renilla Luciferase control that was co-transfected with all samples.
Mouse strains
Kdm6aGt(RRA094)Byg (XUtxGT1), Kdm6atm1a(EUCOMM)Wtsi (XUtxGT2fl), and UtyGt(XS0378)Wtsi (YUtyGT) ES cells were obtained from BayGenomics (through MMRRC), EUCOMM, and SIGTR (through MMRRC) respectively. All ES cells were injected into C57BL/6J host blastocysts for chimera generation. Chimeras were crossed to CD1 to assess germline transmission, and were maintained on either a mixed CD1 background or were backcrossed to 129/SvJ or C57BL/6J. Sox2Cre and RosaFlp transgenes were obtained from The Jackson Laboratory [40]. The VasaCre transgene was developed by Gallardo et al. [70].
Mouse crosses
All mouse experimental procedures were approved by the University of North Carolina Institutional Animal Care and Use Committee. Utx homozygous data was generated either by crosses between Utx hemizygous males and Utx heterozygous females, or by crosses between XUtxGT2fl YUty+ VasaCre males and XUtx+ XUtxGT2Δ heterozygous females. XUtxGT2fl YUty+ VasaCre males were utilized because of an initial difficulty in generating XUtxGT2Δ YUty+ males and due to the efficient and specific activity of VasaCre in the male germline [70]. Utx hemizygous phenotypic data was developed from the previously mentioned homozygous crosses or through crosses between a WT male and heterozygous Utx female. Compound hemizygous Utx/Uty embryos were generated by crossing heterozygous Utx females with hemizygous Uty males. Embryos were PCR genotyped from yolk sac samples for Utx and were sexed by a PCR genotyping scheme to distinguish Utx from Uty. All primer sequences are available upon request.
Histology, in situ hybridization, and LacZ staining
Histology samples, in situ hybridization, and LacZ staining were performed as described [71]. In situ hybridization probes were generated to be identical to previous literature [72].
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. MartinC, ZhangY (2005) The diverse functions of histone lysine methylation. Nat Rev Mol Cell Biol 6 : 838–849.
2. CaoR, WangL, WangH, XiaL, Erdjument-BromageH, et al. (2002) Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298 : 1039–1043.
3. MullerJ, HartCM, FrancisNJ, VargasML, SenguptaA, et al. (2002) Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111 : 197–208.
4. CzerminB, MelfiR, McCabeD, SeitzV, ImhofA, et al. (2002) Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell 111 : 185–196.
5. KuzmichevA, NishiokaK, Erdjument-BromageH, TempstP, ReinbergD (2002) Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev 16 : 2893–2905.
6. MontgomeryND, YeeD, ChenA, KalantryS, ChamberlainSJ, et al. (2005) The murine polycomb group protein Eed is required for global histone H3 lysine-27 methylation. Curr Biol 15 : 942–947.
7. PasiniD, BrackenAP, JensenMR, Lazzerini DenchiE, HelinK (2004) Suz12 is essential for mouse development and for EZH2 histone methyltransferase activity. EMBO J 23 : 4061–4071.
8. CaoR, ZhangY (2004) SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell 15 : 57–67.
9. ShenX, LiuY, HsuYJ, FujiwaraY, KimJ, et al. (2008) EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell 32 : 491–502.
10. MargueronR, LiG, SarmaK, BlaisA, ZavadilJ, et al. (2008) Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell 32 : 503–518.
11. EzhkovaE, PasolliHA, ParkerJS, StokesN, SuIH, et al. (2009) Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 136 : 1122–1135.
12. EzhkovaE, LienWH, StokesN, PasolliHA, SilvaJM, et al. (2011) EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev 25 : 485–498.
13. MargueronR, ReinbergD (2011) The Polycomb complex PRC2 and its mark in life. Nature 469 : 343–349.
14. ChamberlainSJ, YeeD, MagnusonT (2008) Polycomb repressive complex 2 is dispensable for maintenance of embryonic stem cell pluripotency. Stem Cells 26 : 1496–1505.
15. LeeTI, JennerRG, BoyerLA, GuentherMG, LevineSS, et al. (2006) Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 125 : 301–313.
16. BoyerLA, PlathK, ZeitlingerJ, BrambrinkT, MedeirosLA, et al. (2006) Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441 : 349–353.
17. BrackenAP, DietrichN, PasiniD, HansenKH, HelinK (2006) Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev 20 : 1123–1136.
18. AzuaraV, PerryP, SauerS, SpivakovM, JorgensenHF, et al. (2006) Chromatin signatures of pluripotent cell lines. Nat Cell Biol 8 : 532–538.
19. BernsteinBE, MikkelsenTS, XieX, KamalM, HuebertDJ, et al. (2006) A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125 : 315–326.
20. KuM, KocheRP, RheinbayE, MendenhallEM, EndohM, et al. (2008) Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet 4: e1000242 doi:10.1371/journal.pgen.1000242.
21. FaustC, SchumacherA, HoldenerB, MagnusonT (1995) The eed mutation disrupts anterior mesoderm production in mice. Development 121 : 273–285.
22. O'CarrollD, ErhardtS, PaganiM, BartonSC, SuraniMA, et al. (2001) The polycomb-group gene Ezh2 is required for early mouse development. Mol Cell Biol 21 : 4330–4336.
23. LeeMG, VillaR, TrojerP, NormanJ, YanKP, et al. (2007) Demethylation of H3K27 regulates polycomb recruitment and H2A ubiquitination. Science 318 : 447–450.
24. AggerK, CloosPA, ChristensenJ, PasiniD, RoseS, et al. (2007) UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 449 : 731–734.
25. De SantaF, TotaroMG, ProsperiniE, NotarbartoloS, TestaG, et al. (2007) The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell 130 : 1083–1094.
26. LanF, BaylissPE, RinnJL, WhetstineJR, WangJK, et al. (2007) A histone H3 lysine 27 demethylase regulates animal posterior development. Nature 449 : 689–694.
27. BurgoldT, SpreaficoF, De SantaF, TotaroMG, ProsperiniE, et al. (2008) The histone H3 lysine 27-specific demethylase Jmjd3 is required for neural commitment. PLoS ONE 3: e3034 doi:10.1371/journal.pone.0003034.
28. GreenfieldA, CarrelL, PennisiD, PhilippeC, QuaderiN, et al. (1998) The UTX gene escapes X inactivation in mice and humans. Hum Mol Genet 7 : 737–742.
29. HongS, ChoYW, YuLR, YuH, VeenstraTD, et al. (2007) Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci U S A 104 : 18439–18444.
30. WangJK, TsaiMC, PoulinG, AdlerAS, ChenS, et al. (2010) The histone demethylase UTX enables RB-dependent cell fate control. Genes Dev 24 : 327–332.
31. SeenundunS, RampalliS, LiuQC, AzizA, PaliiC, et al. (2010) UTX mediates demethylation of H3K27me3 at muscle-specific genes during myogenesis. EMBO J 29 : 1401–1411.
32. SatohT, TakeuchiO, VandenbonA, YasudaK, TanakaY, et al. (2010) The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat Immunol 11 : 936–944.
33. SenGL, WebsterDE, BarraganDI, ChangHY, KhavariPA (2008) Control of differentiation in a self-renewing mammalian tissue by the histone demethylase JMJD3. Genes Dev 22 : 1865–1870.
34. BarradasM, AndertonE, AcostaJC, LiS, BanitoA, et al. (2009) Histone demethylase JMJD3 contributes to epigenetic control of INK4a/ARF by oncogenic RAS. Genes Dev 23 : 1177–1182.
35. AggerK, CloosPA, RudkjaerL, WilliamsK, AndersenG, et al. (2009) The H3K27me3 demethylase JMJD3 contributes to the activation of the INK4A-ARF locus in response to oncogene - and stress-induced senescence. Genes Dev 23 : 1171–1176.
36. De SantaF, NarangV, YapZH, TusiBK, BurgoldT, et al. (2009) Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J 28 : 3341–3352.
37. MillerSA, MohnSE, WeinmannAS (2010) Jmjd3 and UTX play a demethylase-independent role in chromatin remodeling to regulate T-box family member-dependent gene expression. Mol Cell 40 : 594–605.
38. JegalianK, PageDC (1998) A proposed path by which genes common to mammalian X and Y chromosomes evolve to become X inactivated. Nature 394 : 776–780.
39. LeeS, LeeJW, LeeSK (2012) UTX, a Histone H3-Lysine 27 Demethylase, Acts as a Critical Switch to Activate the Cardiac Developmental Program. Dev Cell
40. HayashiS, LewisP, PevnyL, McMahonAP (2002) Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech Dev 119 Suppl 1: S97–S101.
41. CloonanN, ForrestAR, KolleG, GardinerBB, FaulknerGJ, et al. (2008) Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods 5 : 613–619.
42. MortazaviA, WilliamsBA, McCueK, SchaefferL, WoldB (2008) Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5 : 621–628.
43. SengokuT, YokoyamaS (2011) Structural basis for histone H3 Lys 27 demethylation by UTX/KDM6A. Genes Dev 25 : 2266–2277.
44. ChoYW, HongT, HongS, GuoH, YuH, et al. (2007) PTIP associates with MLL3 - and MLL4-containing histone H3 lysine 4 methyltransferase complex. J Biol Chem 282 : 20395–20406.
45. LeeJ, SahaPK, YangQH, LeeS, ParkJY, et al. (2008) Targeted inactivation of MLL3 histone H3-Lys-4 methyltransferase activity in the mouse reveals vital roles for MLL3 in adipogenesis. Proc Natl Acad Sci U S A 105 : 19229–19234.
46. ChenS, MaJ, WuF, XiongLJ, MaH, et al. (2012) The histone H3 Lys 27 demethylase JMJD3 regulates gene expression by impacting transcriptional elongation. Genes Dev 26 : 1364–1375.
47. TieF, BanerjeeR, ConradPA, ScacheriPC, HartePJ (2012) Histone Demethylase UTX and Chromatin Remodeler BRM Bind Directly to CBP and Modulate Acetylation of Histone H3 Lysine 27. Mol Cell Biol 32 : 2323–2334.
48. LuohSW, BainPA, PolakiewiczRD, GoodheartML, GardnerH, et al. (1997) Zfx mutation results in small animal size and reduced germ cell number in male and female mice. Development 124 : 2275–2284.
49. Galan-CaridadJM, HarelS, ArenzanaTL, HouZE, DoetschFK, et al. (2007) Zfx controls the self-renewal of embryonic and hematopoietic stem cells. Cell 129 : 345–357.
50. ArenzanaTL, Smith-RaskaMR, ReizisB (2009) Transcription factor Zfx controls BCR-induced proliferation and survival of B lymphocytes. Blood 113 : 5857–5867.
51. WeissJ, MeeksJJ, HurleyL, RaverotG, FrassettoA, et al. (2003) Sox3 is required for gonadal function, but not sex determination, in males and females. Mol Cell Biol 23 : 8084–8091.
52. RizzotiK, Lovell-BadgeR (2007) SOX3 activity during pharyngeal segmentation is required for craniofacial morphogenesis. Development 134 : 3437–3448.
53. RizzotiK, BrunelliS, CarmignacD, ThomasPQ, RobinsonIC, et al. (2004) SOX3 is required during the formation of the hypothalamo-pituitary axis. Nat Genet 36 : 247–255.
54. BarronMJ, BrookesSJ, KirkhamJ, ShoreRC, HuntC, et al. (2010) A mutation in the mouse Amelx tri-tyrosyl domain results in impaired secretion of amelogenin and phenocopies human X-linked amelogenesis imperfecta. Hum Mol Genet 19 : 1230–1247.
55. AdlerDA, BresslerSL, ChapmanVM, PageDC, DistecheCM (1991) Inactivation of the Zfx gene on the mouse X chromosome. Proc Natl Acad Sci U S A 88 : 4592–4595.
56. YangF, BabakT, ShendureJ, DistecheCM Global survey of escape from X inactivation by RNA-sequencing in mouse. Genome Res 20 : 614–622.
57. EhrmannIE, EllisPS, MazeyratS, DuthieS, BrockdorffN, et al. (1998) Characterization of genes encoding translation initiation factor eIF-2gamma in mouse and human: sex chromosome localization, escape from X-inactivation and evolution. Hum Mol Genet 7 : 1725–1737.
58. AgulnikAI, MitchellMJ, MatteiMG, BorsaniG, AvnerPA, et al. (1994) A novel X gene with a widely transcribed Y-linked homologue escapes X-inactivation in mouse and human. Hum Mol Genet 3 : 879–884.
59. ChristensenJ, AggerK, CloosPA, PasiniD, RoseS, et al. (2007) RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell 128 : 1063–1076.
60. IwaseS, LanF, BaylissP, de la Torre-UbietaL, HuarteM, et al. (2007) The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell 128 : 1077–1088.
61. LeeMG, NormanJ, ShilatifardA, ShiekhattarR (2007) Physical and functional association of a trimethyl H3K4 demethylase and Ring6a/MBLR, a polycomb-like protein. Cell 128 : 877–887.
62. EissenbergJC, LeeMG, SchneiderJ, IlvarsonnA, ShiekhattarR, et al. (2007) The trithorax-group gene in Drosophila little imaginal discs encodes a trimethylated histone H3 Lys4 demethylase. Nat Struct Mol Biol 14 : 344–346.
63. VandammeJ, LettierG, SidoliS, Di SchiaviE, Norregaard JensenO, et al. The C. elegans H3K27 demethylase UTX-1 is essential for normal development, independent of its enzymatic activity. PLoS Genet 8: e1002647 doi:10.1371/journal.pgen.1002647.
64. GargV, KathiriyaIS, BarnesR, SchlutermanMK, KingIN, et al. (2003) GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 424 : 443–447.
65. TuckerKE, BercianoMT, JacobsEY, LePageDF, ShpargelKB, et al. (2001) Residual Cajal bodies in coilin knockout mice fail to recruit Sm snRNPs and SMN, the spinal muscular atrophy gene product. J Cell Biol 154 : 293–307.
66. ShechterD, DormannHL, AllisCD, HakeSB (2007) Extraction, purification and analysis of histones. Nat Protoc 2 : 1445–1457.
67. HebertMD, SzymczykPW, ShpargelKB, MateraAG (2001) Coilin forms the bridge between Cajal bodies and SMN, the spinal muscular atrophy protein. Genes Dev 15 : 2720–2729.
68. RahlPB, LinCY, SeilaAC, FlynnRA, McCuineS, et al. c-Myc regulates transcriptional pause release. Cell 141 : 432–445.
69. ArgentinS, ArdatiA, TremblayS, LihrmannI, RobitailleL, et al. (1994) Developmental stage-specific regulation of atrial natriuretic factor gene transcription in cardiac cells. Mol Cell Biol 14 : 777–790.
70. GallardoT, ShirleyL, JohnGB, CastrillonDH (2007) Generation of a germ cell-specific mouse transgenic Cre line, Vasa-Cre. Genesis 45 : 413–417.
71. ChandlerRL, ChandlerKJ, McFarlandKA, MortlockDP (2007) Bmp2 transcription in osteoblast progenitors is regulated by a distant 3′ enhancer located 156.3 kilobases from the promoter. Mol Cell Biol 27 : 2934–2951.
72. XuJ, DengX, WatkinsR, DistecheCM (2008) Sex-specific differences in expression of histone demethylases Utx and Uty in mouse brain and neurons. J Neurosci 28 : 4521–4527.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Enrichment of HP1a on Drosophila Chromosome 4 Genes Creates an Alternate Chromatin Structure Critical for Regulation in this Heterochromatic Domain
- Normal DNA Methylation Dynamics in DICER1-Deficient Mouse Embryonic Stem Cells
- The NDR Kinase Scaffold HYM1/MO25 Is Essential for MAK2 MAP Kinase Signaling in
- Functional Variants in and Involved in Activation of the NF-κB Pathway Are Associated with Rheumatoid Arthritis in Japanese
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy