
Genetic Modifiers of Chromatin Acetylation Antagonize the Reprogramming of Epi-Polymorphisms
Natural populations are known to differ not only in DNA but also in their chromatin-associated epigenetic marks. When such inter-individual epigenomic differences (or “epi-polymorphisms”) are observed, their stability is usually not known: they may or may not be reprogrammed over time or upon environmental changes. In addition, their origin may be purely epigenetic, or they may result from regulatory variation encoded in the DNA. Studying epi-polymorphisms requires, therefore, an assessment of their nature and stability. Here we estimate the stability of yeast epi-polymorphisms of chromatin acetylation, and we provide a genome-by-epigenome map of their genetic control. A transient epi-drug treatment was able to reprogram acetylation variation at more than one thousand nucleosomes, whereas a similar amount of variation persisted, distinguishing “labile” from “persistent” epi-polymorphisms. Hundreds of genetic loci underlied acetylation variation at 2,418 nucleosomes either locally (in cis) or distantly (in trans), and this genetic control overlapped only partially with the genetic control of gene expression. Trans-acting regulators were not necessarily associated with genes coding for chromatin modifying enzymes. Strikingly, “labile” and “persistent” epi-polymorphisms were associated with poor and strong genetic control, respectively, showing that genetic modifiers contribute to persistence. These results estimate the amount of natural epigenomic variation that can be lost after transient environmental exposures, and they reveal the complex genetic architecture of the DNA–encoded determinism of chromatin epi-polymorphisms. Our observations provide a basis for the development of population epigenetics.
Published in the journal:
. PLoS Genet 8(9): e32767. doi:10.1371/journal.pgen.1002958
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002958
Summary
Natural populations are known to differ not only in DNA but also in their chromatin-associated epigenetic marks. When such inter-individual epigenomic differences (or “epi-polymorphisms”) are observed, their stability is usually not known: they may or may not be reprogrammed over time or upon environmental changes. In addition, their origin may be purely epigenetic, or they may result from regulatory variation encoded in the DNA. Studying epi-polymorphisms requires, therefore, an assessment of their nature and stability. Here we estimate the stability of yeast epi-polymorphisms of chromatin acetylation, and we provide a genome-by-epigenome map of their genetic control. A transient epi-drug treatment was able to reprogram acetylation variation at more than one thousand nucleosomes, whereas a similar amount of variation persisted, distinguishing “labile” from “persistent” epi-polymorphisms. Hundreds of genetic loci underlied acetylation variation at 2,418 nucleosomes either locally (in cis) or distantly (in trans), and this genetic control overlapped only partially with the genetic control of gene expression. Trans-acting regulators were not necessarily associated with genes coding for chromatin modifying enzymes. Strikingly, “labile” and “persistent” epi-polymorphisms were associated with poor and strong genetic control, respectively, showing that genetic modifiers contribute to persistence. These results estimate the amount of natural epigenomic variation that can be lost after transient environmental exposures, and they reveal the complex genetic architecture of the DNA–encoded determinism of chromatin epi-polymorphisms. Our observations provide a basis for the development of population epigenetics.
Introduction
Recent studies have shown that individuals largely differ in their epigenomic chromatin signatures. This finding makes tracking epigenetic marks in natural populations attractive, including investigating their possible contribution to the variation of common physiological traits. So far, epigenomic intra-species diversity has been primarily studied at the level of the methylome (DNA methylation profile). Natural accessions of A. thaliana were found to differ in their methylation level at about 10% of all CCGG sites [1] and this variability was largely concentrated within genic regions [2]. In humans, numerous inter-individual differences of DNA methylation were also reported [3]–[6] and, importantly, the methylomes of monozygotic twins were shown to diverge during their lifetime [7]. Measuring this diversity at a genome-wide scale extended what had been observed earlier at individual loci in mice, where the level of transgene methylation was shown to strongly vary between laboratory strains [8], [9]. However, natural epigenomic variability is not restrained to DNA methylation. DNase-seq profiles of cell-lines from human families revealed ∼10,000 sites that were polymorphic in their chromatin signature [10] and it is likely that a significant fraction of them is not associated with DNA methylation differences but with other regulatory hallmarks. Natural variability was also reported at the level of high-order chromatin structure, when distinct A. thaliana accessions were compared for their level of genome compaction in response to light [11]. Finally, histone acetylation profiles also varies, as we previously described in a comparison of two unrelated wild strains of S. cerevisiae [12].
Unlike DNA variants that are irreversible and therefore tractable, epigenotypes are thought to be largely labile (i.e. able to change their state) on time scales ranging from seconds to multiple generations [13]. When the spontaneous epimutation rate of DNA methylation was estimated in 30-generations mutation accumulation lines of A. thaliana, it was found to be several orders of magnitude higher than the rate of DNA variation [14], [15]. Moreover, chromatin signatures not only change spontaneously but also in response to environmental conditions [16]. Various environmental factors have the potential to exert this effect. Temperature, for example, can induce dramatic epigenetic changes in plants. In the normal life cycle of many species, experiencing winter cold is essential for flowering later in spring: the FLC locus, whose expression prevents flowering, becomes silenced by a well-described mechanism after several weeks of vernalization (for a review, see [17]). In addition, extreme and stressful temperatures may be experienced, in which case the chromatin state of A. thaliana repetitive sequences can change to alleviate their silencing [18]–[20]. The response to subtle temperature variations was also shown to depend on the proper incorporation of histone variant H2A.Z [21]. In addition, specific extracellular signals such as hormones in animals can also trigger chromatin reprogramming at target loci, and the pathways involved provide many routes by which chromatin can sense environmental conditions. To a broader extent, diet represents a set of factors able to induce epigenome modifications [22]. Feeding animals with altered amounts of methyl donors can induce methylome reprogramming [23]. Such treatments have illustrated how environmental conditions may stably print epigenotypes across generations. In mice for example, reprogramming was observed in adult offsprings of males that had been on specific diets [24], [25].
In the particular case of chromatin acetylation, direct coupling between epigenetic signatures and energy metabolism (obviously related to diet) is known to happen at least at three levels. First, sirtuins are known to deacetylate histones and a number of other proteins in a NAD+-dependent manner [26], [27]. Secondly, the level of Acetyl-CoA, which donates the acetyl group transferred to histones, can vary according to glucose availability and efficient metabolism [28]. And thirdly, carbonyl compounds can inactivate class I Histone Deacetylases (HDAC) by alkylation of two cysteine residues [29]. And beyond dietary effects, some environments contain natural HDAC inhibitors such as Trichostatin-A (TSA) produced by Streptomyces platensis, or butyrate, a natural product of the intestinal flora [30]. Thus, individuals may harbor personalized epigenomes because they have experienced a specific history of past environmental exposures or stochastic transitions (Figure 1A).

Alternatively, epi-polymorphisms can be influenced by DNA variations that modify chromatin regulations, either in cis (i.e. locally) or in trans (i.e. distantly) [31]. Well-known examples of cis-modifiers are transposon insertions [32], [33], whose regional effects on chromatin states have been the basis for extremely informative genetic screens in yeast (as reviewed in [32]). In humans, several heritable disorders are caused by trinucleotide repeat expansions that perturb chromatin states locally [34]. One striking example is the non-coding repeat region of the FMR1 gene, where moderate expansions mediate hyper-acetylation of the locus and increased mRNA levels, resulting in Fragile X Tremor Ataxia Syndrome [35], whereas larger expansions induce chromatin silencing, decreased gene expression, and Fragile X Mental Retardation Syndrome [36]. The very few known trans-acting genetic modifiers of chromatin states are sequence changes within chromatin modifying enzymes [6], [11], but other DNA polymorphisms may also act in trans by affecting the activity of upstream regulators of chromatin modifying machineries. The numerous examples of DNA-encoded chromatin differences suggest that individuals may harbor distinct epigenotypes simply as a result of their different genetic content (Figure 1B).
We previously identified thousands of yeast nucleosomes carrying differential levels of H3K14 acetylation between two wild S. cerevisiae strains (BY and RM) [12]. Following this previous study, we define here Single Nucleosome Epi-Polymorphisms (SNEPs) as the intra-species variations of the level of an epigenetic mark carried on a nucleosome. The polymorphic mark may be any histone post-translational modification or the incorporation of a histone variant. A SNEP for one such mark then corresponds to the preferential presence of the mark at one nucleosomal position in some individuals or strains as compared to others. Consequently, SNEPs of various epigenetic marks may be carried on the same nucleosome. By tracking H3K14ac SNEPs, we describe here both an experimental reprogramming experiment and the genetic architecture of H3K14 acetylation variation. The results show that some epi-polymorphisms are reprogrammed after a transient perturbation of chromatin states whereas others persist, and this persistence can, at least partly, be explained by genetic determinants encoded in the DNA.
Results
The present study focuses on one epigenetic mark, the acetylation of histone H3 at Lysine 14 (H3K14ac). For simplicity, the terms ‘SNEP’ and ‘epi-polymorphism’ are used here interchangeably to refer to H3K14ac epi-polymorphisms.
A Transient Epi-Drug Treatment Reprograms a Subset of Epi-Polymorphisms
We previously described 5,442 SNEPs corresponding to acetylation variation between two S. cerevisiae strains (BY and RM). Here we assessed the stability of these epi-polymorphisms by transiently exposing the two strains to an extremely perturbing environment (Figure 1C). We sought to distinguish three types of SNEPs: Persistent SNEPs, corresponding to initial inter-strain differences that remained significant after the perturbation; Labile SNEPs, corresponding to original inter-strain differences that significantly changed after the perturbation; and Induced SNEPs, corresponding to inter-strain differences that appeared after the perturbation.
BY and RM cells were treated with high concentrations of TSA for 4–5 generations. As expected, this treatment caused a bulk increase of H3K14 acetylation in both strains (Figure S1). Cells were then washed and let grown for over 20 generations in standard medium lacking TSA. After this recovery period, the global level of H3K14 acetylation had returned to normal in both strains and we examined again inter-strain differences at single-nucleosome resolution, using chromatin immunoprecipitation and hybridization on whole genome tiling arrays, as previously described [12]. The protocol was applied on biological triplicates for each strain. Inter-strain acetylation ratios before treatment and after recovery were highly correlated (Figure 2A, Spearman r = 0.7).

We performed two complementary statistical analyses on the data. First, we specifically searched for induced and persistent SNEPs. To this end, we applied our previously described SNEP detection algorithm (NucleoMiner) to the newly generated dataset (see Methods). At a False Discovery Rate (FDR) of 0.0001, we detected 2,379 SNEPs after recovery. Interestingly, 898 of them were new ones: for these nucleosomes, the level of K14 acetylation was not significantly different between the strains before treatment. They were unlikely false negatives, because detection power was higher in the initial search than after recovery from TSA (12 versus 6 microarrays used). Rather, these induced SNEPs illustrate that epi-polymorphisms may indeed result from new environmental exposures. Interestingly, 524 of the 898 induced SNEPs were ‘isolated’, i.e. their two flanking nucleosomes were not SNEPs after treatment and recovery. Of these, 436 were initially in a context where neither of the flanking nucleosome was a SNEP. This specificity illustrates that SNEPs can be induced at precise nucleosomes and not necessarily on consecutive ones.
Of the 5,442 SNEPs originally detected in normal conditions [12], 1,481 were also significant post-recovery. All of them except one had the same directionality (i.e. same strain showing increased acetylation) before treatment and after recovery and these were therefore called ‘persistent’ (Figure 2A, 2B).
The remaining 3,961 initial SNEPs could be ‘labile’, but many of them may simply be false negatives that escaped detection post recovery. We therefore applied a different test to reliably search for cases of lability: we tested for all nucleosomes if the inter-strain acetylation ratio had changed (see Methods). This was the case for 4,484 nucleosomes (FDR = 0.001). Among these, 1,076 belonged to the list of nucleosomes containing initial SNEPs and we therefore qualified these SNEPs as ‘labile’ (Figure 2A, 2B). These labile SNEPs did not represent cases of high experimental noise, as they were not necessarily those with low initial statistical significance (Figure S2). In conclusion, three different types of acetylation epi-polymorphisms (induced, persistent and labile) could be detected in large proportions.
We previously reported that for ∼50% of SNEPs, no acetylation variation was detectable on their flanking nucleosomes [12] (see Figure 2B for an example). Here we observed that these ‘isolated’ epi-polymorphisms globally had reduced persistence (Figure 2C, Wilcoxon P<10−15) and contained more labile SNEPs than expected (51% versus 35% among non-isolated, P<10−15, χ2 test). This suggests that epi-polymorphisms carried on specific single nucleosomes are less stabilized than those established on consecutive nucleosomes.
The mechanism(s) by which labile SNEPs are established and lost remain unknown. However, when confronting our data to a published map of histone turnover rates [37], we observed that labile SNEPs corresponded to nucleosomes of faster histone replacement, as compared to persistent SNEPs (P = 0.003, see Text S1). This suggests that the increased dynamics of molecular replacement at these positions contributes to SNEP lability. In addition, we also observed an increased persistence among nucleosomes located within protein-coding genes or located within regions of conserved DNA sequence (Figure S3).
The reprogramming experiment presented here was designed to test the stability of SNEPs and not the effect of treatment in each strain. Assessing precisely the amount of reprogramming within each strain would require a dataset where all samples prior and post treatment are processed in parallel, by the same experimenter, using common batches of reagents. This was not the case here, and confounding experimental factors would likely bias any statistical inference of reprogramming within each strain. However, we made interesting observations when inspecting the fold change of acetylation between the levels before treatment and the levels after recovery. First, the mean fold change across all nucleosomes was similar between the two strains (Figure 2D). This is consistent with the similar levels of bulk acetylation seen on whole protein extracts (Figure S1). Secondly, the fold change in the BY strain presented elevated variability between nucleosomes, as compared to the RM strain (high variance in Figure 2D). This higher variability does not correspond to higher experimental error in the BY samples, as the between-replicates variance was similar between the two strains (Table S1). This suggests that more nucleosomes were reprogrammed in the BY strain than in the RM strain. To specifically look at this possibility, we plotted the distribution of fold changes in the 1,076 labile SNEPs, where reprogramming occurred. This highlighted a strong asymmetry in BY, with a majority of labile SNEPs having gained acetylation in this strain (Figure 2E). There are at least three possible interpretations of this. First, TSA may have imposed a stronger chromatin hyperacetylation in BY than in RM. Secondly, the BY strain may have recovered badly from treatment, with a chromatin remaining at an artificially high acetylation level despite the long recovery time. Alternatively, the BY strain may initially have had many nucleosomes with low levels of acetylation, which were reset to ‘normal’ levels by exposure to TSA. In the first two cases, the observed gain of acetylation is not expected to target specific nucleosomes. In contrast, in the latter case, the nucleosomes that were reprogrammed should correspond to those initially identified as poorly acetylated in BY. In other words, the presence of a SNEP before treatment should predict the treatment effect. To see if such a prediction could be made, we considered three classes of nucleosomes on the basis of observations made before treament only: those initially SNEPs as BY hypo-acetylated, those initially SNEPs as BY hyper-acetylated, and those not initially SNEPs. We then compared the extent of fold change between these three categories of nucleosomes. The classification was not predictive of the fold change in the RM strain (Figure 2F), but it was highly predictive of the effect in the BY strain (Figure 2G). This observation suggests that the BY strain possessed many nucleosomes that were initially hypoacetylated and predisposed to resetting at a higher level.
Genetic Dissection of H3K14ac Epigenomic Variation
We then investigated the genetic control of epi-polymorphisms. Using the maps of nucleosome positions previously generated for BY and RM [12], we associated every nucleosome with the nucleotide region that overlapped its position in both strains (see Methods). We then measured the level of acetylation of each of these regions in 60 meiotic segregants from the BYxRM cross [38]. This was done by culturing each segregant in standard laboratory conditions, and by performing single-nucleosome resolution chromatin immunoprecipitation as above. We defined one quantitative trait of acetylation per nucleosome, which reflected the abundance of the DNA region associated with the nucleosome in the immunoprecipitated material (see Methods). Using these trait values, we searched the genome for Quantitative Trait Loci of acetylation (aceQTL). A first scan was performed at a genome x epigenome scale. To do so, we selected 36,558 nucleosomes with H3K14ac heritability higher than 0.2, and for each of these we searched the entire genetic map for linkage. Calculations were done using a convenient platform called eQTNMiner, which was originally designed for the Bayesian Inference of nucleotide-resolution eQTLs [39]. eQTNMiner reports linkage evidence as a Bayes Factor (BF), which quantifies the relative support of the data in favor of the alternative hypothesis (there is a QTL) against the null hypothesis (there is no QTL). We recorded linkages at various Bayes Factor thresholds, and computed empirical significance of each threshold by a permutation test (Table S2, see Methods). At BF = 1000 (corresponding to FDR = 0.034), we found significant linkages for a total of 2,418 nucleosomes (Figure 3A). Of these, 77 were linked to 2 aceQTLs and all others to a single one.

We then applied a second scan to specifically search for cis-modifiers. For each nucleosome, linkage was searched across DNA polymorphisms located within 5 Kb. We chose this distance as a compromise between the small physical size of a nucleosome (147nt) and the usual large regions scanned for cis-eQTLs (10–50 Kb). At BF = 50 (corresponding to FDR = 0.0007), we found cis-linkages involving the control of 4,173 nucleosomes. There were 17% of SNEPs (908/5,442) for which an aceQTL could be found in at least one of the two scans. Given the rather small size of the segregating population examined, we can assume that some genetic linkages were missed. This fraction is therefore a lower-bound estimate of the ‘genetically encoded’ class of SNEPs. All further analysis was done on results obtained from the first scan only, as they reflect both cis and trans regulators, with effects strong enough to pass a stringent genome-by-epigenome significance level.
The number of nucleosomes controlled by each trans-acting aceQTLs varied greatly (Figure 3B). Seventeen loci, called ‘master-aceQTLs’ hereafter, were found to control more nucleosomes than expected by chance (Table 1, see Methods). One of them contained the locus controlling the cell mating type (MAT), which encodes different transcriptional co-factors in BY(MATα) and RM(MATa). Of the 148 nucleosomes controlled by this locus, 83 had a marked acetylation difference between BYα and RMa that could be detected without replicated experiments. To directly test if MAT accounted for the associated acetylation variation, we performed two additional ChIP-chip experiments on strains having reversed mating types (BYa and RMα) and we tested for acetylation variation between isogenic strains differing only at MAT. The expected difference was observed for 69 of the 83 nucleosomes tested (p<0.01, see Text S1), which validated MAT as the responsible polymorphism underlying this master-aceQTL. As examples, epigenomic profiles at the KAR4 locus are shown in Figure 3C, where the control of three SNEPs by MAT is apparent.

Only a small fraction (16%) of the nucleosomes controlled in cis were proximal to elements known to affect nearby chromatin (Ty transposons, rDNA, telomeres, HML and HMR loci). This suggests that many other causes for acetylation variability exist. Intuitively, trans-regulation could result from sequence variants targeting chromatin modifying enzymes. To examine this possibility, we analyzed relevant Gene Ontologies (GO). We saw that eight of the seventeen master-aceQTLs did not contain any gene annotated to participate in chromatin regulation (Table 1). Among all 141 trans-acting aceQTLs, 63 contained a gene with relevant annotation, which corresponded to the number expected by chance only (Text S1). Thus, trans-modifiers of acetylation are not necessarily restricted to chromatin modifying enzymes but may include upstream molecular players. This conclusion is analogous to the previous observation that trans-acting modifiers of gene expression (eQTLs) do not necessarily correspond to transcription factors [38].
In some cases, the genetic control of chromatin acetylation had a complex basis, such as digenic regulations by antagonistic aceQTLs (Figure 3D). This illustrates the quantitative nature of acetylation variation and reveals that subtle epigenomic variations can segregate as complex traits in natural populations.
Partial Overlap between aceQTLs and eQTLs
Acetylation of H3K14 is generally a mark of active transcription [40]. However, we previously described that higher acetylation of BY/RM SNEPs did not necessarily imply an increased expression of the overlapping gene [12]. This suggests that some SNEPs do not participate in transcriptional activation while others do. Thus, one would expect that only a fraction of the genetic regulations of acetylation are concordant with the genetic regulation of gene expression. We therefore examined the overlap between aceQTL and eQTL results, taking advantage of a transcriptomic dataset previously generated on the same strains and culture conditions [38]. This was done in two steps. First, inspection of master-aceQTLs showed that several of them (including MAT) corresponded to loci previously identified as master eQTLs [38] (Table 1). For example, the GPA1-S469I polymorphism targets a G-protein α subunit and underlies expression variation of many pheromone-responsive genes [38]. This polymorphism lies at an aceQTL affecting 24 nucleosomes that reside within or near these target genes. GPA1-S469I is therefore a likely regulator of both expression and acetylation at these loci. For similar reasons, a transposon insertion altering the HAP1 transcription factor is likely a regulator of both expression and acetylation of target genes (Table 1). However, the overall overlap between aceQTLs and eQTLs was only partial. For example, the AMN1 locus on chromosome II was previously linked to the expression level of 18 transcripts and was not detected as a master QTL of chromatin acetylation here. Conversely, 10 master-aceQTLs were located at positions not previously associated with major transcriptional variation [38] (Table 1).
In a second step, we systematically compared aceQTL and eQTL linkages without restricting the analysis to master-aceQTLs. To do so, we reduced aceQTLs of the first scan to 2,530 non-redundant linkages (i.e. pairs of one nucleosome and one genetic marker). For every linkage between a genetic marker m and a nucleosome ν, we examined if a significant eQTL could be found between m and a gene located within 10 Kb of ν. For 31% of aceQTLs, no such concordance could be found. Note that statistical power was much higher to detect eQTLs than aceQTLs because many more segregants were used. It is therefore unlikely that these cases corresponded to false negatives. Overall, the strength of linkage was poorly correlated between aceQTLs and eQTLs (Figure 3E–3G, Figure S4). We then used the same criteria as above to re-examine master-aceQTLs and classify them based on the fraction of their linkages that matched eQTLs (Table 1). Of the 17 master-aceQTLs, nine clearly corresponded to eQTLs, four had partial concordance and four did not affect the expression level of genes proximal to the target nucleosomes. Notably, expression of KAR4 was not affected by MAT alleles (Figure 3G). Our genetic dissection therefore unravelled the coexistence of two types of H3K14 acetylation epi-polymorphisms, one type associated with transcriptional variation and one disconnected from it. Although it is difficult to precisely estimate their relative proportions, the results argue that in at least 30% of cases, genetic polymorphisms modulate chromatin acetylation without altering gene transcription levels.
Antagonism between SNEPs Reprogramming and Genetic Control
When epi-polymorphisms result from DNA-encoded regulatory variation, they should persist (e.g. be maintained or return to their initial state) across extreme environmental perturbations because their causative variants do. We sought to test this principle by comparing the results obtained on SNEP persistence through temporary TSA exposure with the genetic properties of aceQTL control. We first examined the success rate of aceQTL mapping when searching for regulators of ‘isolated’ or ‘non-isolated’ SNEPs. More aceQTLs were found for SNEPs carried on consecutive nucleosomes (Figure 3H). Note that this enrichment does not imply that aceQTL targets are necessarily clustered: for 60% of nucleosomes controlled by an aceQTL, none of the flanking nucleosome was in linkage with the same aceQTL locus. However, finding more aceQTL for clustered SNEPs was concordant with the increased persistence of these SNEPs (Figure 2C). We therefore directly examined if the presence of genetic regulators correlated with the level of environmental persistence. Accordingly, aceQTLs were found 4 times more often for persistent SNEPs than for labile SNEPs (Figure 4A). Consistently, a Receiver Operating Curve applied to all SNEPs showed that persistence was strongly associated with successful aceQTL mapping (Figure 4B). In addition, if high environmental persistence is explained by strong genetic control, then it should correlate with high genetic linkage score. We therefore represented the strength of genetic linkage as a function of environmental persistence, which confirmed the expected trend (Figure 4C).

Finally, genetic linkage results alone may sometimes not reflect the strength of genetic determinism. For example, if numerous QTLs with small individual contribution altogether control the acetylation value of a nucleosome, then none of them may be found despite a complete overall genetic determinism. Similarly, a complete control by epistatic or antagonistic genetic loci may not be detected. However, even in such complex genetic cases, the overall determinism can still be estimated by the genetic heritability of the acetylation trait in the segregating population. We therefore examined heritability itself, and found that increasing heritability values were unambiguously associated with gradual shifts towards higher persistence (Figure 4D). Thus, genetic determinism was indeed correlated with elevated environmental persistence, regardless of the complexity of the underlying control.
Discussion
This study reveals that H3K14ac epi-polymorphisms are not equally sensitive to environmental reprogramming. Some of them can be lost after temporary perturbations while others persist. This persistence clearly correlates with the presence of genetic determinants that encode epi-polymorphisms in the DNA. This genetic control is complex and resembles architectures previously described for eQTLs, with both cis and trans regulators and the presence of master regulators affecting numerous targets.
Importantly, our results further highlight the quantitative nature of the variation of acetylation levels. At any given time, a nucleosome of a given cell is or is not acetylated. Thus, acetylation is sometimes considered a discrete variable. However, the average acetylation level of this nucleosome across a population of cells is quantitative, because it depends on the number of cells that carry the acetylation mark, which corresponds to an equilibrium state of the population resulting from many biochemical reactions. The fact that this level varies as a complex trait shows that aceQTLs change the proportion of cells that are acetylated at their target nucleosome. In other words, aceQTLs are genotypes that modify the probability that a given nucleosome is acetylated in a given cell at a given time. How this happens will probably remain unknown until new technologies are developped to interrogate single nucleosome states in single cells. Also, the quantitative variability studied here is different from several epimutations described in plants where strong silencing of large chromosomal domains is established by a combination of many molecular and structural changes.
Whether the genetic control of chromatin variability also controls the level of nearby gene expression appears to be context specific. In humans, Gibbs et al. did not find any consistent overlap between expression Quantitative Trait Loci (eQTL) modifying the transcriptome of brain tissues and Quantitative Trait Loci modifying the methylome of these tissues (methQTL) [5]. In contrast, Bell et al. reported a clear consistency between eQTL and methQTL in HapMap lymphoblastoid cell lines [6]. Here we observed that about 70% of aceQTLs linkages overlap with eQTLs. The remaining fraction of aceQTLs could correspond to regulations of non-coding transcripts, which were not interrogated by our study. Alternatively, given our previous association between SNEPs and transcriptional plasticity [12], it is possible that some aceQTLs modify the chromatin in a way that manifests only upon transcriptional stimulation. In other words, genetic modifiers could increase K14 acetylation of a locus, which would then become more responsive to transcriptional activation or repression upon specific conditions. In such cases, aceQTLs could participate in gene x environment interactions by creating epi-polymorphisms that personalize the way the genome responds to the environment.
We proved that the MAT locus affects chromatin acetylation of many target loci. This locus determines the cell's mating type by dictating specific transcriptional programs. The MATα allele encodes two regulatory proteins: α1, which activates α-specific genes, and α2, which represses expression of a-specific genes. The MATa allele encodes the a1 protein only, which heterodimerizes with α2 in diploid a/α cells to form a repressor of haploid-specific genes. How specific transcriptional programs are established in a and α cells has been the focus of many studies, revealing the interplay with chromatin acetylation regulation at specific target promoters. In α cells, the cooperative binding of α2 and Mcm1 recruits the Tup1-Ssn6 repressor, which is known to interact with several histone deacetylases [41]–[43]. In a cells, α-specific genes are occupied by Sum1 which is known to recruit the NAD+-dependent histone deacetylase Hst1 to repress transcription [44]. In our study, some loci controlled by MAT displayed an epigenomic profile totally predictable given the known transcriptional control. This was the case for the BAR1 gene for example, which encodes a secreted protease specifically expressed in a cells. The chromatin signature of the entire locus was affected by the mating type. MATa strains displayed occupancy and acetylation intensities typical of highly expressed genes [40], with a marked nucleosome-free region near the transcription start site, and a high and low level of H3K14 acetylation in the first and second half of the coding region, respectively (Figure S5). However, other loci controlled by MAT displayed unexpected patterns of chromatin variation. One such example was the KAR4 locus, which encodes two forms of a transcription factor essential for nuclear fusion during mating. The long form is expressed in mitotically growing cells, and the short form is induced in response to pheromone from a transcriptional site about 30 nucleotides downstream the first ATG [45]. Our study revealed marked differences between MATa and MATα growing cells in the 3′ part of the gene, which were not accompanied by differential transcriptional levels (Figure 3C and 3G). How a cells maintain elevated H3K14 acetylation on two nucleosomes at the end of the KAR4 coding region remains to be identified. It is possible that a and α cells do not use the same strategy to maintain the locus transcriptionally active and responsive to pheromone. Comparing Ste12, Tup1, or Sum1 occupancy between a and α cells might reveal some differences in this region. Alternatively, DNA replication initiated downstream KAR4, at the ARS304 site, could have an effect if its timing differs between a and α cells [46]. Another particular case of mating-type specific chromatin organization was the promoter of the SAG1 gene, which encodes the α-agglutinin specifically expressed in α cells. The repressed state of a cells corresponded to nucleosome occupancy downstream the TSS, and to hypoacetylation of H3K14 specifically at the -1 nucleosome (Figure S6). These three examples illustrate that the mechanism by which MAT alleles affect chromatin signatures at target genes is not simple: it can affect an entire locus (BAR1), or a specific set of nucleosomes in the promoter (SAG1) or the 3′ region (KAR4).
More generally, the fact that aceQTLs were not preferentially found at sites coding for chromatin modifying enzymes may seem counterintuitive: one could expect that DNA polymorphisms affect chromatin states by modifying the sequences of enzymes directly involved in chromatin regulation. However, protein complexes that regulate chromatin are themselves highly regulated, and any DNA polymorphism affecting these upstream regulators has the potential to induce chromatin modification indirectly. In fact, this is what happens with MAT alleles: they do not code for chromatin remodelling enzymes but they determine distinct recruitments of chromatin modifiers at specific sites. This observation is very similar to results from eQTL mapping, from which we know that genetic modifiers of gene expression do not necessarily reside in direct transcriptional regulators [38]. For example, the AMN1, GPA1, IRA2 and MKT1 yeast genes were all validated as eQTL players but they do not encode direct regulators of transcription [38], [47]. These polymorphisms affect gene expression by perturbing regulatory networks upstream of transcriptional machineries. The results presented here suggest that aceQTLs likely follow a similar rule: causative polymorphisms may reside not only within chromatin modifying complexes but also in their upstream regulators.
We show that a transient environmental change imposed by TSA treatment can reprogram a subset of H3K14ac epi-polymorphisms: numerous new SNEPs were induced, and numerous initial SNEPs were lost. An important consideration is that TSA imposed a perturbation but did not necessarily saturate the acetylation of H3K14 on all nucleosomes. In normal conditions, H3K14 acetylation levels result from a balance between the activity of histone acetyltransferases (HATs) and deacetylases (HDACs). In S. cerevisiae, at least three HATs are known to acetylate Lysine 14 of Histone H3: Gcn5p [48], [49], Sas3p [50], and Hpa2p [51], and deacetylation of Lysine 14 can be attributed to HDACs of all three classes: Hos3p and Rpd3p of class I [52], [53], Hda1p of class II [41] and Sir2p of class III [54]. TSA is known to induce a bulk hyperacetylation by inhibiting the activity of a subset of these HDACs: while Rpd3p and Hda1p are sensitive, Hos3p and Sir2p remain active. Thus, the perturbation applied in our experiment did not necessarily saturate K14 acetylation on the entire chromatin. In addition to the direct effect of TSA on HDACs that deacetylate H3K14, the treatment may have perturbed this lysine residue indirectly. The very slow growth in presence of TSA (not shown) suggests that cells profoundly reshaped molecular profiles during treatment, with possible consequences on the regulations of HATs and HDACs.
The reprogramming observed preferentially corresponded to a gain of acetylation in the BY strain, with a majority of labile SNEPs corresponding to hypo-acetylated nucleosomes in the BY strain that returned to levels comparable to those of the RM strain. An intuitive interpretation of this asymmetry would be that TSA was more efficient to induce hyperacetylation in BY than in RM. The strains are probably not equally sensitive to TSA, given the two previously mapped QTLs of growth fitness in the presence of TSA that segregate in the BYxRM cross [55]. However, the possibility that BY suffered a more pronounced hyperacetylation does not explain why only a subset of nucleosomes were preferentially reprogrammed. Alternatively, the strains may differ in their recovering efficiency. Although after 20 generations all HDAC complexes are young enough to consider they never bound the chemical inhibitor, it is still possible that the chromatin of the BY strain did not fully return to equilibrium. Then again, why would an incomplete recovery target preferentially a subset of nucleosomes? Our observation that the nucleosomes affected are largely those initially hypoacetylated suggests a third and complementary interpretation: the BY strain may have accumulated hypoacetylation ‘epimutations’ that were cured by the treatment. BY is a strain that has been maintained in laboratories for decades and is known to possess many deleterious mutations that would likely be counter-selected in the wild. Our results raise the possibility that it has also drifted at the epigenetic level, and it will be very exciting to test this hypothesis in future experiments.
More generally, it will be essential to question the origin of the ‘labile’ SNEPs: those which gained but also those which lost acetylation in BY, and the few where the change happened in RM. Theoretically, the differences in these epigenotypes may have occurred any time between the initial divergence of the strains and the last hours before the stocks were frozen in our laboratory. In other words, our study identified their lability but not their origin and age. A related question is how stable are labile and newly induced SNEPs: how harsh a treatment is needed to reprogram them? If some ‘labile’ SNEPs are old, they have been maintained for a long time and one would expect them to be stable unless extreme environmental perturbations are experienced, like in our TSA-based assay. Likewise, it is possible that additional SNEPs could have been modified if we had applied a stronger or longer treatment. As mentioned above, class III HDACs such as Sir2p are not inhibited by TSA, and other SNEPs would probably be called ‘labile’ if an inhibitor of sirtuins was used instead of TSA. In contrast, some ‘labile’ SNEPs may be very unstable and might also disappear after a prolonged but unperturbed culture. It will therefore be interesting to monitor the dynamics of SNEP appearance and loss in unperturbed conditions. A time-course experiment tracking the H3K14ac epigenome of one strain over long culture periods would help determine its stability.
How and for how long were new SNEPs induced despite the fact that the treatment applied was the same for the two strains? As mentioned above, this can possibly result from a difference in the way the strains respond to the treatment, and this difference might or not be genetically encoded. Although our experiments were not designed to address this, it is also possible that epi-polymorphisms arise stochastically in particular environments regardless of the genetic background. This has been suggested by a recent study where the methylome of isogenic mice fed with high levels of methyl precursors was tracked over generations [23]. This treatment was shown to increase inter-individual epigenome diversity, although the diet itself was common to all animals. Thus, induced epi-polymorphisms may reflect not only differences in the history of past environmental exposures, but also genetic or stochastic differences in the way individuals reprogram their epigenome in response to specific environments.
We observed a clear correlation between environmental persistence and genetic control of acetylation variation. Importantly, the two datasets (reprogramming and QTL mapping) were generated and analysed independently: at different dates, by different experimenters, the former using the parental strains only and the latter using the segregants. Thus, we believe that this correlation truly reflects the robustness of DNA-encoded epi-polymorphisms to environmental reprogramming. However, our observations do not imply that all cases of epi-polymorphism persistence result from their anchoring in DNA. It remains entirely possible that specific cases have a purely epigenetic basis. For example, H3K14 acetylation may be more robust to environmental perturbation if it is accompanied by additional epigenetic marks that are commonly associated with it, such as H3K4 di - or tri-methylation, or H3K9 acetylation [40]. If such marks drive H3K14 acetylation and are not affected by the environmental change, then persistence is ensured without a DNA-encoded control.
Given our observation that both labile and persistent epi-polymorphisms coexist abundantly in natural epigenomes, we emphasize the importance of the stability of epi-polymorphism in the current debate on whether and how epigenotypes contribute to evolutionary mechanisms. As outlined by B. Turner, this question is fundamental because epi-polymorphisms potentially enable environmental conditions to reprogram molecular events for a durable time. This way, “epigenetic processes might contribute to evolutionary change, at least in part by expanding the range of phenotypic variants on which natural selection can act” [16]. A key factor for selection to act is then the amount of time during which the ‘novel’ phenotypic variants (those generated by chromatin changes) are exposed. If too short, individuals with beneficial traits may not have time to expand in the population, especially if the phenotypic variants consist of small quantitative differences. Although our results did not link SNEPs to phenotypic traits, they suggest that the amount of time for selection to act may differ if the phenotypic variants result from labile or from persistent epi-polymorphisms (Figure 5). This duration depends on the stability of the new epigenotype and on the probability to encounter environmental conditions that change its state. If the epigenotype is robust to environmental perturbations, then the phenotype is exposed as long as other genetic or epigenetic modifiers of it are acquired. Natural selection is therefore more likely to act on phenotypic variants resulting from persistent epi-polymorphisms. Note that such high persistence can sometimes result from a full genetic control. In this case, the fact that epi-polymorphisms are involved no longer matters: selection acts on the genetic determinant regardless of the mechanism leading to the phenotype.
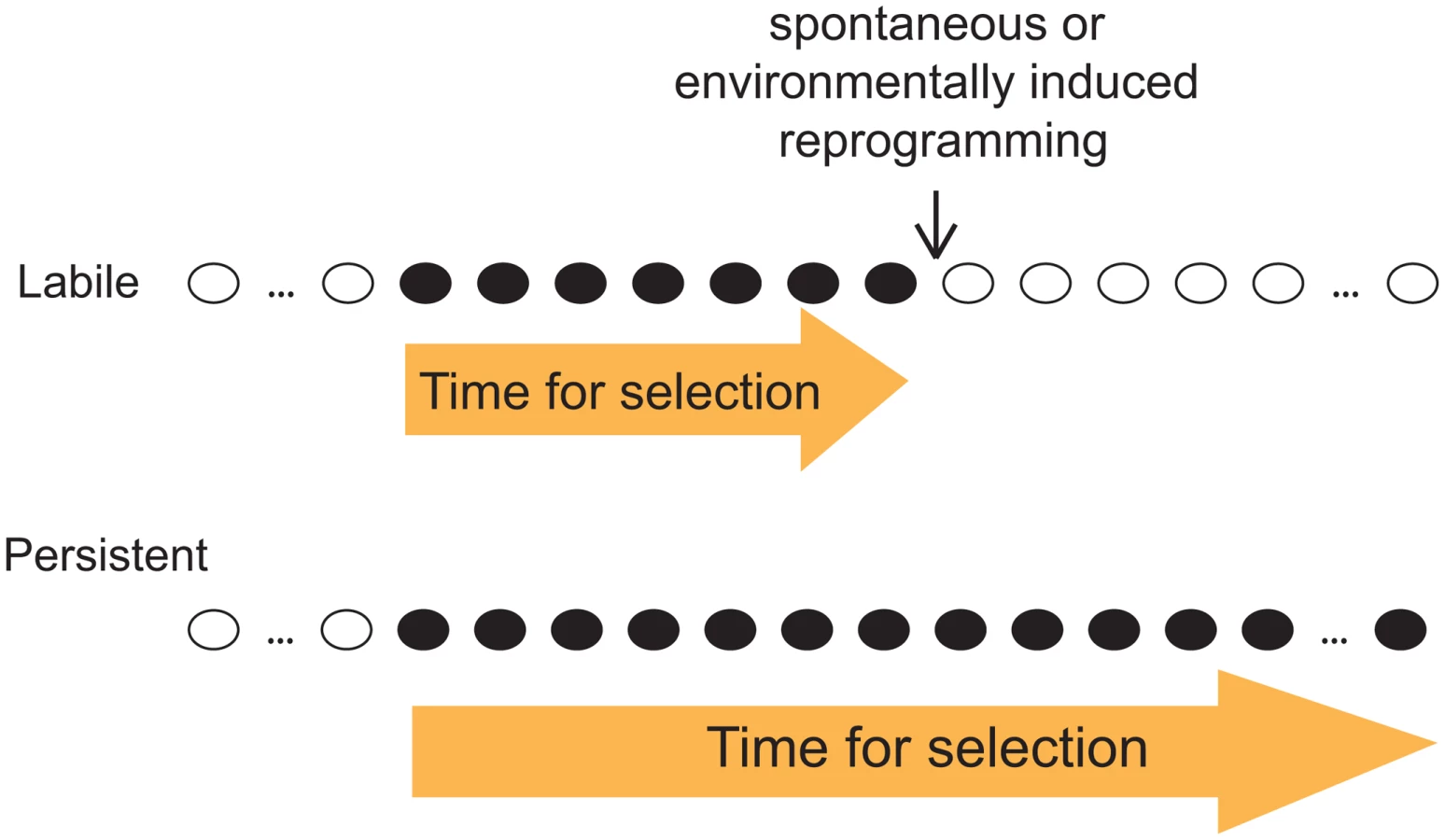
Importantly, loci harboring DNA-encoded epi-polymorphisms may remain highly susceptible to epigenetic regulations: as mentioned above, SNEPs represent small quantitative differences of molecular regulations, and it is likely that they do not prevent from switching between radically different epigenetic states. Thus, the prolonged duration of DNA-encoded epi-polymorphisms does not necessarily impair fitness in fluctuating environments, where adaptation requires rapid and profound chromatin remodelling at critical loci.
In addition, chromatin changes may reveal the effect of cryptic genetic variations. For example, a mutation occuring at a silenced locus can remain cryptic until silencing of the locus is alleviated. Such epigenetic alleviation might also be either labile or persistent, with different consequences on the cryptic variation: the phenotype and therefore the cryptic variation itself may be exposed to selection for a longer period of time if alleviation persists.
Altogether, our observations provide a necessary basis for the upcoming development of population epigenetics, where epi-polymorphisms of natural populations will be interpreted and possibly associated to the variation of common traits.
Methods
Strains and Culture Conditions
Strains used were BY4716 MATα lys2Δ0 (called ‘BY’ in text) and BY4715 MATa lys2Δ0 derivatives of S288c, RM11-1a MATa leu2Δ0 ura3Δ0 hoΔ::KanMX wine strain [38] (called ‘RM’ in text) and its GY689 MATα leu2Δ0 ura3Δ0 hoΔ::KanMX amn1-A1103T derivative (see Text S1), and 60 meiotic segregants from BYxRM previously used for eQTL mapping [38], [47]. Except for the TSA experiment, cells were grown to exponential phase in synthetic medium with 2% glucose (SDall) at 30°C as previously done for SNEP identification [12] and eQTL mapping [47].
ChIP–chip
ChIP–chip was performed at single-nucleosome resolution as previously described, using formaldehyde fixation followed by micrococcal nuclease digestion, anti-H3K14ac antibody (Upstate #07-353) precipitation and hybridization on Affymetrix Whole Genome Yeast Tiling 4-bp resolution microarrays [12].
Transient TSA Treatment
The following protocol was applied on 3 independent cultures for each strain. A 110 ml culture of SDall medium was inoculated at OD600 = 0.15 using an overnight starter culture and was grown at 30°C until OD600 reached 0.5–0.6. 45 ml of the culture was pelleted and frozen for later Western blot analysis (sample t1). TSA (Wako 204-11991) was resuspended in ethanol 50% at 45.5 mg/ml and 3.9 ml of this stock was added to the remaining of the culture (final TSA concentration: 2.6 mg/ml). The culture was kept at 30°C for 3–4 doubling times (OD600 = 3) and 20 ml was pelleted and frozen for Western Blot analysis (sample t2). Remaining cells were washed twice with 20 ml TBS1X [Tris 25 mM, NaCl 140 mM, KCl 2.5 mM, pH 7.4] and 1% of the suspension was added to 50 ml of SDall and incubated at 30°C. The next day, 10 microliters of the overnight culture was transfered to 50 ml fresh SDall medium and incubated at 30°C for 6 hours. This culture was used to inoculate 200 ml fresh SDall incubated at 30°C until OD = 0.9. This procedure corresponded to about 20 generations post-treatment. 25 ml of the final cell suspension was pelleted and frozen for Western blot analysis (sample t3) and the remaining of the culture was used for chromatin immunoprecipitation.
Statistical Test for Lability
A matrix of raw microarray hybridization intensities was considered that contained the BY (6 arrays), RM (6 arrays), BYart (3 arrays) and RMart (3 arrays) ChIP-chip values for all probes having a single perfect match on both BY and RM genomes. Here ‘Xart’ correspond to strain X after recovery from TSA treatment (time t3 on Figure S1). This matrix was quantile-quantile normalized using NucleoMiner [12] and for every probe, 6 independent values of LR = log(BY/RM) were derived as well as 3 independent values of LRart = log(BYart/RMart). For all 58,694 previously aligned nucleosomes [12], we extracted relevant probes according to their physical position on the genome. The window of extraction was defined by the overlapping region between nucleosomal position in BY and nucleosomal position in RM [12], trimmed at both extremities by 12 nucleotides to avoid possible artefactual border effects. All probes having mid-position within this window were used to test against the null hypothesis of similar inter-strain difference before TSA treatment and after recovery (LR = LRart). This was done by an analysis of variance (ANOVA) based on the model logratio∼tsa+probe, where tsa reflects whether LR or LRart is considered and probe reflects the probe index. Nominal P-values relevant to factor tsa were used to derive q-values that account for multiple testing, by using the QVALUE package [56]. 4,484 nucleosomes showed q<0.001. Among these nucleosomes, 1,076 belonged to the list carrying original SNEPs and these SNEPs were called ‘labile’. Note that testing for acetylation reprogramming for each strain separately would be difficult from our dataset because the two sets of experiments (before treatment and after recovery) were done at different dates, by different experimenters. We therefore preferred to use inter-strain log-ratios to ensure a consistent comparison of the inter-strain difference within each dataset.
Detection of Persistent SNEPs
We specifically searched for persistent SNEPs by running NucleoMiner on the BYart, RMart ChIP-chip dataset, together with the previously described BY and RM nucleosome mapping experiments [12]. 2,379 nucleosomes showed differential H3K14 acetylation levels at FDR<0.001 (nominal P-value<4.05×10−5) after recovery from TSA. Note that fewer biological replicates were used after recovery (3 for each strain) than before treatment (6 for each strain), which explains the detection of fewer SNEPs (2,379 instead of 5,442). The intersection between the two lists of SNEPs corresponded to 1,481 nucleosomes that were called ‘persistent’ SNEPs (magenta dots on Figure 2A). For every nucleosome, persistence was defined as 1−|log2(RM/BY)before tratment−log2(RM/BY)post recovery|.
aceQTL Mapping
We generated an extremely dense genetic map by inferring, at every SNP, a probabilistic genotype given the genotypes previously described at marker positions [47] (see Text S1). To avoid hybridization artefacts due to DNA polymorphisms, we considered only the microarray probes having a single perfect match on both BY and RM genomes. A dataset comprising 18 microarrays previously described (nucleosome mapping data and H3K14ac profiling on BY and RM replicates) [12] and the 60 ChIP-chip microarrays performed here on the BYxRM segregants was normalized by quantile-quantile normalization using the NMc2tab program of NucleoMiner with option -n lqq. We then computed estimates of nucleosome-level ChIP intensities. For every nucleosome, we considered signals from probes that were entirely contained in the overlap between nucleosome position in BY and nucleosome position in RM. These signals were further corrected by quantile normalization (to account for probe effects) and averaged using the eqmr-fdb command of eQTNminer [39] with option -m qnorm.
For every nucleosome, heritability of acetylation level was then computed as h = (varS−varE)/varS, where varS is the variance across all segregants and varE the environmental variance, estimated by the pooled variance of parental replicates. We noticed that one BY experiment had very high variation, therefore varE was estimated by the pooled variance of 5 BY and 6 RM ChIP-chip experiments.
Mapping of aceQTL was performed using the eqmr-ftr command of eQTNMiner [39] version 2.0 with default parameters on 59,936 nucleosomes (nucleosomes contained in translocated regions were not considered). A first scan was performed at a genome x epigenome level. To do so, we selected 36,558 nucleosomes with H3K14ac heritability higher than 0.2. This threshold was chosen arbitrarily in order to avoid multiplying tests on nucleosomes where linkage is unlikely to be discovered. For each of these, the entire genetic map was scanned for QTL using a Bayesian regression model, implemented in eQTNMiner [39],which follows the framework of Servin and Stephens [57]. The effect of individual i genotype at SNP j (gijk) on the acetylation level of the k-th nucleosome (yik) is assumed to follow a purely additive linear model: yik = μ+ajkg×gijk+εijk, where μ is the mean acetylation level of that nucleosome for individuals with g = 0, and where ajk is the additive effect of the minor allele at SNP j. The residual εijk is assumed to be normally distributed, with mean zero and variance 1/τ equal to the variance of acetylation levels within each genotype class. Let P0k denote the probability of the acetylation data Yk under the null hypothesis that there are no aceQTL controlling nucleosome k (i.e., ajk = 0 for all j). Similarly, let P1jk denote the probability of the acetylation data Yk under the hypothesis that SNP j is an aceQTL of nucleosome k. In this case, the effect size ajk is modelled as being drawn from mixtures of normal distributions centered on 0 (see below). The Bayes Factor reflecting genetic linkage between SNP j and nucleosome k is defined as BFjk = P1jk/P0k and measures the relative support for the hypothesis that SNP j is an aceQTL of nucleosome k, versus the null hypothesis. As suggested by Servin & Stephens [57], we assumed that the effect size ajk is drawn from mixtures of normal distributions centered on 0 with variance σa2/τ. Specifically, we assumed a mixture of 6 normal with σa2 = (0.05, 0.1, 0.2, 0.4, 0.8, 1.6), we computed a Bayes factor for each value of σa2, and considered the mean Bayes factor as our summary statistics. We controlled the False Discovery Rate empirically by re-scanning 100 permuted datasets. On average, only 20,288 linkages were obtained at a Bayes Factor threshold of 1000 from a permuted dataset, while 592,368 linkages were obtained at this level from the actual data (Table S2). The list passing this FDR = 0.034 threshold was used for further analysis. Note that many of the 592,368 linkages reflect redundant genetic information between adjacent DNA polymorphisms. In total, aceQTLs were found for 2,418 nucleosomes. To roughly see how many nucleosomes were controlled by two or more aceQTLs, we reduced the 592,368 linkages to account for linked markers: for each target nucleosome, the best QTL marker was recorded and all markers located within 100 Kb of it were discarded. This procedure was then repeated until no significant additional linkages remained. This way, 2341 nucleosomes were linked to a single aceQTL, 77 nucleosomes were linked to 2 distinct aceQTLs, and no nucleosome was linked to three or more loci.
A second scan was performed on all 59,936 nucleosomes to specifically detect cis-acting aceQTLs. Using the eqmr-expca command of eQTNminer [39], we applied a Principal Component Analysis and observed that the first 10 principal components represented significant general effects (as compared to eigenvalues obtained from permuted datasets) that could shade specific regulations. We therefore corrected for these effects by applying an elastic net regression on these 10 axes as implemented in eqmr-fenet of eQTNminer. The residuals were then used as the corrected traits. For each nucleosome, we used eqmr-fcr to search for linkages between the trait and any DNA polymorphism located within 5 Kb on each side. False Discovery Rate was controlled empirically by running 100 permutations (Table S3). We observed 235,942 linkages exceeding a Bayes Factor of 50 while only 160 were seen at this threshold from permuted datasets (FDR = 0.0007). In total, cis-aceQTLs were found for 4173 nucleosomes. 668 of these nucleosomes ( = 16%) were located within 20 Kb of a region known to affect nearby chromatin states (telomere, retrotransposon, rDNA, HML or HMR).
Finally, an aceQTL for nucleosome i and marker m found in the first scan was called trans-aceQTL if m was at least 50 Kb away from any cis-aceQTL found for i in the second scan.
Definition of Master Trans-aceQTLs
We determined which of the trans-aceQTLs affect the acetylation level of a significantly high number of nucleosomes. To do so, we first reduced the results to the best linkage scores in trans. For each nucleosome for which a trans-aceQTL was found, the marker M with highest linkage score was recorded and all significant linkages to markers close to M were discarded. If additional significant trans-aceQTLs remained for this nucleosome, the procedure was repeated. We then segmented the genome in 20 Kb bins and counted the number of reduced trans-aceQTLs in each bin (Figure 3B). We tested for enrichment by considering deviation from Poisson distribution, as done before for eQTLs [38], [47]. 25 bins were significantly enriched (at least 9 target nucleosomes, P<0.05 after Bonferroni correction), which could be concatenated to 17 non-consecutive bins. We then searched each bin for the best candidate polymorphism regulating the set of target nucleosomes: for every nucleosome i having a trans-aceQTL in the bin, we computed at every SNP k the posterior probability Pi(k) that the SNP is causal. This probability can be directly computed from the output of eQTNminer. Let Bi,m be the Bayes Factor for linkage between nucleosome i and SNP m, and Si the sum of Bi,m across all m of the genome, then the probability is simply Pi(k) = Bi,k/Si. These probabilities were then sumed across all nucleosomes in linkage to the bin, and the best candidate was identified as the SNP maximizing this sum (indicated as ‘Score’ in Table 1).
aceQTL Versus eQTL
To compare aceQTLs with eQTLs in a consistent way, we generated a set of eQTL results using the same method and same genetic map as for aceQTLs. Gene expression data was extracted for 4,464 genes from the “glucose condition” of Smith and Kruglyak [47]. eQTNminer was used to scan the genome x transcriptome space, without the hierarchical models previously described [39] and using data from 109 segregants previously generated under the same glucose medium as here [47]. This produced 2,159,456 linkages with Bayes Factor exceeding 50. One hundred permutations were run to control the FDR, which was 3.5% at this threshold. In total, eQTLs were found for 3,572 genes and the results were consistent with previous studies.
To then determine if aceQTLs could be considered as eQTLs, we considered all significant aceQTLs of the first scan. To remove redundant linkages supported by adjacent markers, we reduced the results to the best scores as described above for reducing trans-aceQTLs, leaving 2,530 aceQTL linkages out of the 592,368 original ones. For each one linking acetylation of a nucleosome i to a genetic marker m, we then recorded the best eQTL score between m and any gene located within 10 Kb of i. In several cases, no eQTL was found at a very relaxed threshold (Bayes Factor of 1) and this search was then labelled as ‘non-significant’ (Figure 3E). We considered that an aceQTL was not an eQTL if the best score found was not more significant than P<0.00125 (nominal value). This corresponds to the usual 0.01 threshold divided by 8 which is the average number of genes examined within 20 Kb of the yeast genome (4,464 * 20/12,000). Following this criterion, 790 of the 2,530 aceQTL linkages (31%) did not correspond to eQTL. Note that the detection power was much higher for eQTL than for aceQTL, as more segregants were used. Thus, it is unlikely that we missed relevant eQTLs at this relaxed threshold.
We also addressed the reciprocal question of whether eQTLs were aceQTLs. Redundant eQTLs supported by adjacent markers were removed as above. For each eQTL found at FDR = 0.035 between a marker m and a gene g, we considered all aceQTL scores between m and any nucleosome located with 10 Kb of g and recorded the best one. When no aceQTL was found at the relaxed threshold of BF = 1, then this search was called “non significant” (Figure S4).
To estimate whether master trans-aceQTLs correspond to master eQTLs (Table 1), we proceeded as follows. For each master trans-aceQTL controlling the acetylation levels of a set of nucleosomes νi, let m be the best candidate polymorphism as defined above. For each nucleosome νi we examined all genes located within 10 Kb and asked whether at least one of them was a significant eQTL target of m. The fraction of nucleosomes νi for which this was the case was called “Fraction of nucleosomes matching an eQTL target”. Because many target nucleosomes νi were located close to each other, we also examined them as distinct target loci: Target nucleosomes νi that were located within 1 Kb of each other were grouped into a “locus”. For each locus, we counted the fraction of target nucleosomes for which a relevant eQTL linkage was found (as above). This number was then averaged across all target loci to define the “Average fraction per locus” indicated in Table 1. Finally, master trans-aceQTLs were classified as being ‘also eQTL’, ‘aceQTL only’ or ‘partial’ based on whether this fraction was higher than 75%, lower than 25%, or in between, respectively.
ROC Analysis
Figure 4B was obtained by sorting the 5,442 original SNEPs by their persistence across the transient TSA treatment (defined above). A Receiver Operating Curve (ROC) was then built: ‘positive’ SNEPs were the ones for which at least one significant aceQTL was found, as this corresponds to the expectation of a genetic control underlying persistence; ‘negative’ SNEPs were those for which no aceQTL was found. Fraction of positives and negatives were computed at increasing persistence values.
Data Accession
All ChIP-chip raw data is available from ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) under accession numbers E-MTAB-575 and E-MTAB-1025. Additional processed data files are available from our web site: http://www.ens-lyon.fr/LBMC/gisv/snep/
Supporting Information
Zdroje
1. ZhangX, ShiuS, CalA, BorevitzJO (2008) Global analysis of genetic, epigenetic and transcriptional polymorphisms in Arabidopsis thaliana using whole genome tiling arrays. PLoS Genet 4: e1000032 doi:10.1371/journal.pgen.1000032.
2. VaughnMW, Tanurd IcM, LippmanZ, JiangH, CarrasquilloR, et al. (2007) Epigenetic Natural Variation in Arabidopsis thaliana. PLoS Biol 5: e174 doi:10.1371/journal.pbio.0050174.
3. FlanaganJM, PopendikyteV, PozdniakovaiteN, SobolevM, AssadzadehA, et al. (2006) Intra - and interindividual epigenetic variation in human germ cells. Am J Hum Genet 79 : 67–84.
4. ZhangD, ChengL, BadnerJA, ChenC, ChenQ, et al. (2010) Genetic control of individual differences in gene-specific methylation in human brain. Am J Hum Genet 86 : 411–419.
5. GibbsJR, van der BrugMP, HernandezDG, TraynorBJ, NallsMA, et al. (2010) Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet 6: e1000952 doi:10.1371/journal.pgen.1000952.
6. BellJT, PaiAA, PickrellJK, GaffneyDJ, Pique-RegiR, et al. (2011) DNA methylation patterns associate with genetic and gene expression variation in HapMap cell lines. Genome Biol 12: R10.
7. FragaMF, BallestarE, PazMF, RoperoS, SetienF, et al. (2005) Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A 102 : 10604–10609.
8. EnglerP, HaaschD, PinkertCA, DoglioL, GlymourM, et al. (1991) A strain-specific modifier on mouse chromosome 4 controls the methylation of independent transgene loci. Cell 65 : 939–947.
9. Valenza-SchaerlyP, PickardB, WalterJ, JungM, PourcelL, et al. (2001) A dominant modifier of transgene methylation is mapped by QTL analysis to mouse chromosome 13. Genome Res 11 : 382–388.
10. McDaniellR, LeeBK, SongL, LiuZ, BoyleAP, et al. (2010) Heritable individual-specific and allele-specific chromatin signatures in humans. Science 328 : 235–239.
11. TessadoriF, van ZantenM, PavlovaP, CliftonR, PontvianneF, et al. (2009) Phytochrome B and histone deacetylase 6 control light-induced chromatin compaction in Arabidopsis thaliana. PLoS Genet 5: e1000638 doi:10.1371/journal.pgen.1000638.
12. NagarajanM, VeyrierasJB, de DieuleveultM, BottinH, FehrmannS, et al. (2010) Natural single-nucleosome epi-polymorphisms in yeast. PLoS Genet 6: e1000913 doi:10.1371/journal.pgen.1000913.
13. RandoOJ, VerstrepenKJ (2007) Timescales of genetic and epigenetic inheritance. Cell 128 : 655–668.
14. BeckerC, HagmannJ, MullerJ, KoenigD, StegleO, et al. (2011) Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 480 : 245–249.
15. SchmitzRJ, SchultzMD, LewseyMG, O'MalleyRC, UrichMA, et al. (2011) Transgenerational epigenetic instability is a source of novel methylation variants. Science 334 : 369–373.
16. TurnerBM (2010) Environmental sensing by chromatin: An epigenetic contribution to evolutionary change. FEBS Lett
17. HeY (2009) Control of the transition to flowering by chromatin modifications. Mol Plant 2 : 554–564.
18. PecinkaA, DinhHQ, BaubecT, RosaM, LettnerN, et al. (2010) Epigenetic regulation of repetitive elements is attenuated by prolonged heat stress in Arabidopsis. Plant Cell 22 : 3118–3129.
19. Tittel-ElmerM, BucherE, BrogerL, MathieuO, PaszkowskiJ, et al. (2010) Stress-induced activation of heterochromatic transcription. PLoS Genet 6: e1001175 doi:10.1371/journal.pgen.1001175.
20. Lang-MladekC, PopovaO, KiokK, BerlingerM, RakicB, et al. (2010) Transgenerational inheritance and resetting of stress-induced loss of epigenetic gene silencing in Arabidopsis. Mol Plant 3 : 594–602.
21. KumarSV, WiggePA (2010) H2A.Z-containing nucleosomes mediate the thermosensory response in Arabidopsis. Cell 140 : 136–147.
22. MathersJC, MckayJA (2011) Diet induced epigenetic changes and their implications for health. Acta Physiologica 202 : 103–118.
23. LiCC, CropleyJE, CowleyMJ, PreissT, MartinDI, et al. (2011) A sustained dietary change increases epigenetic variation in isogenic mice. PLoS Genet 7: e1001380 doi:10.1371/journal.pgen.1001380.
24. CropleyJE, SuterCM, BeckmanKB, MartinDI (2006) Germ-line epigenetic modification of the murine A vy allele by nutritional supplementation. Proc Natl Acad Sci U S A 103 : 17308–17312.
25. CaroneBR, FauquierL, HabibN, SheaJM, HartCE, et al. (2010) Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 143 : 1084–1096.
26. FulcoM, SchiltzRL, IezziS, KingMT, ZhaoP, et al. (2003) Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell 12 : 51–62.
27. RodgersJT, LerinC, HaasW, GygiSP, SpiegelmanBM, et al. (2005) Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 434 : 113–118.
28. WellenKE, HatzivassiliouG, SachdevaUM, BuiTV, CrossJR, et al. (2009) ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324 : 1076–1080.
29. DoyleK, FitzpatrickFA (2010) Redox signaling, alkylation (carbonylation) of conserved cysteines inactivates class I histone deacetylases 1, 2, and 3 and antagonizes their transcriptional repressor function. J Biol Chem 285 : 17417–17424.
30. WaldeckerM, KautenburgerT, DaumannH, BuschC, SchrenkD (2008) Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem 19 : 587–593.
31. RichardsEJ (2006) Inherited epigenetic variation–revisiting soft inheritance. Nat Rev Genet 7 : 395–401.
32. YamaguchiY, NaritaT, InukaiN, WadaT, HandaH (2001) SPT genes: key players in the regulation of transcription, chromatin structure and other cellular processes. J Biochem 129 : 185–191.
33. LiuJ, HeY, AmasinoR, ChenX (2004) siRNAs targeting an intronic transposon in the regulation of natural flowering behavior in Arabidopsis. Genes Dev 18 : 2873–2878.
34. KumariD, UsdinK (2009) Chromatin remodeling in the noncoding repeat expansion diseases. J Biol Chem 284 : 7413–7417.
35. ToddPK, OhSY, KransA, PandeyUB, Di ProsperoNA, et al. (2010) Histone deacetylases suppress CGG repeat-induced neurodegeneration via transcriptional silencing in models of fragile X tremor ataxia syndrome. PLoS Genet 6: e1001240 doi:10.1371/journal.pgen.1001240.
36. O'DonnellWT, WarrenST (2002) A decade of molecular studies of fragile X syndrome. Annu Rev Neurosci 25 : 315–338.
37. DionMF, KaplanT, KimM, BuratowskiS, FriedmanN, et al. (2007) Dynamics of replication-independent histone turnover in budding yeast. Science 315 : 1405–1408.
38. YvertG, BremRB, WhittleJ, AkeyJM, FossE, et al. (2003) Trans-acting regulatory variation in Saccharomyces cerevisiae and the role of transcription factors. Nat Genet 35 : 57–64.
39. VeyrierasJB, KudaravalliS, KimSY, DermitzakisET, GiladY, et al. (2008) High-resolution mapping of expression-QTLs yields insight into human gene regulation. PLoS Genet 4: e1000214 doi:10.1371/journal.pgen.1000214.
40. LiuCL, KaplanT, KimM, BuratowskiS, SchreiberSL, et al. (2005) Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol 3: e328 doi:10.1371/journal.pbio.0030328.
41. WuJ, SukaN, CarlsonM, GrunsteinM (2001) TUP1 utilizes histone H3/H2B-specific HDA1 deacetylase to repress gene activity in yeast. Mol Cell 7 : 117–126.
42. WatsonAD, EdmondsonDG, BoneJR, MukaiY, YuY, et al. (2000) Ssn6-Tup1 interacts with class I histone deacetylases required for repression. Genes Dev 14 : 2737–2744.
43. KeleherCA, ReddMJ, SchultzJ, CarlsonM, JohnsonAD (1992) Ssn6-Tup1 is a general repressor of transcription in yeast. Cell 68 : 709–719.
44. ZillOA, RineJ (2008) Interspecies variation reveals a conserved repressor of alpha-specific genes in Saccharomyces yeasts. Genes Dev 22 : 1704–1716.
45. GammieAE, StewartBG, ScottCF, RoseMD (1999) The two forms of karyogamy transcription factor Kar4p are regulated by differential initiation of transcription, translation, and protein turnover. Mol Cell Biol 19 : 817–825.
46. UnnikrishnanA, GafkenPR, TsukiyamaT (2010) Dynamic changes in histone acetylation regulate origins of DNA replication. Nat Struct Mol Biol 17 : 430–437.
47. SmithEN, KruglyakL (2008) Gene-Environment Interaction in Yeast Gene Expression. PLoS Biol 6: e83 doi:10.1371/journal.pbio.0060083.
48. KuoMH, BrownellJE, SobelRE, RanalliTA, CookRG, et al. (1996) Transcription-linked acetylation by Gcn5p of histones H3 and H4 at specific lysines. Nature 383 : 269–272.
49. GrantPA, EberharterA, JohnS, CookRG, TurnerBM, et al. (1999) Expanded lysine acetylation specificity of Gcn5 in native complexes. J Biol Chem 274 : 5895–5900.
50. HoweL, AustonD, GrantP, JohnS, CookRG, et al. (2001) Histone H3 specific acetyltransferases are essential for cell cycle progression. Genes Dev 15 : 3144–3154.
51. Angus-HillML, DutnallRN, TafrovST, SternglanzR, RamakrishnanV (1999) Crystal structure of the histone acetyltransferase Hpa2: A tetrameric member of the Gcn5-related N-acetyltransferase superfamily. J Mol Biol 294 : 1311–1325.
52. CarmenAA, GriffinPR, CalaycayJR, RundlettSE, SukaY, et al. (1999) Yeast HOS3 forms a novel trichostatin A-insensitive homodimer with intrinsic histone deacetylase activity. Proc Natl Acad Sci U S A 96 : 12356–12361.
53. RundlettSE, CarmenAA, KobayashiR, BavykinS, TurnerBM, et al. (1996) HDA1 and RPD3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc Natl Acad Sci U S A 93 : 14503–14508.
54. ImaiS, ArmstrongCM, KaeberleinM, GuarenteL (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403 : 795–800.
55. JaroszDF, LindquistS (2010) Hsp90 and environmental stress transform the adaptive value of natural genetic variation. Science 330 : 1820–1824.
56. StoreyJD, TibshiraniR (2003) Statistical significance for genomewide studies. Proc Natl Acad Sci U S A 100 : 9440–9445.
57. ServinB, StephensM (2007) Imputation-based analysis of association studies: candidate regions and quantitative traits. PLoS Genet 3: e114 doi:10.1371/journal.pgen.0030114.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Enrichment of HP1a on Drosophila Chromosome 4 Genes Creates an Alternate Chromatin Structure Critical for Regulation in this Heterochromatic Domain
- Normal DNA Methylation Dynamics in DICER1-Deficient Mouse Embryonic Stem Cells
- The NDR Kinase Scaffold HYM1/MO25 Is Essential for MAK2 MAP Kinase Signaling in
- Functional Variants in and Involved in Activation of the NF-κB Pathway Are Associated with Rheumatoid Arthritis in Japanese
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy