
The Long Non-Coding RNA Affects Chromatin Conformation and Expression of , but Does Not Regulate Its Imprinting in the Developing Heart
Although many of the questions raised by the discovery of imprinting have been answered, we have not yet accounted for tissue - or stage-specific imprinting. The Kcnq1 imprinted domain exhibits complex tissue-specific expression patterns co-existing with a domain-wide cis-acting control element. Transcription of the paternally expressed antisense non-coding RNA Kcnq1ot1 silences some neighboring genes in the embryo, while others are unaffected. Kcnq1 is imprinted in early cardiac development but becomes biallelic after midgestation. To explore this phenomenon and the role of Kcnq1ot1, we used allele-specific assays and chromosome conformational studies in wild-type mice and mice with a premature termination mutation for Kcnq1ot1. We show that Kcnq1 imprinting in early heart is established and maintained independently of Kcnq1ot1 expression, thus excluding a role for Kcnq1ot1 in repressing Kcnq1, even while silencing other genes in the domain. The exact timing of the mono - to biallelic transition is strain-dependent, with the CAST/EiJ allele becoming activated earlier and acquiring higher levels than the C57BL/6J allele. Unexpectedly, Kcnq1ot1 itself also switches to biallelic expression specifically in the heart, suggesting that tissue-specific loss of imprinting may be common during embryogenesis. The maternal Kcnq1ot1 transcript is shorter than the paternal ncRNA, and its activation depends on an alternative transcriptional start site that bypasses the maternally methylated promoter. Production of Kcnq1ot1 on the maternal chromosome does not silence Cdkn1c. We find that in later developmental stages, however, Kcnq1ot1 has a role in modulating Kcnq1 levels, since its absence leads to overexpression of Kcnq1, an event accompanied by an aberrant three-dimensional structure of the chromatin. Thus, our studies reveal regulatory mechanisms within the Kcnq1 imprinted domain that operate exclusively in the heart on Kcnq1, a gene crucial for heart development and function. We also uncover a novel mechanism by which an antisense non-coding RNA affects transcription through regulating chromatin flexibility and access to enhancers.
Published in the journal:
. PLoS Genet 8(9): e32767. doi:10.1371/journal.pgen.1002956
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002956
Summary
Although many of the questions raised by the discovery of imprinting have been answered, we have not yet accounted for tissue - or stage-specific imprinting. The Kcnq1 imprinted domain exhibits complex tissue-specific expression patterns co-existing with a domain-wide cis-acting control element. Transcription of the paternally expressed antisense non-coding RNA Kcnq1ot1 silences some neighboring genes in the embryo, while others are unaffected. Kcnq1 is imprinted in early cardiac development but becomes biallelic after midgestation. To explore this phenomenon and the role of Kcnq1ot1, we used allele-specific assays and chromosome conformational studies in wild-type mice and mice with a premature termination mutation for Kcnq1ot1. We show that Kcnq1 imprinting in early heart is established and maintained independently of Kcnq1ot1 expression, thus excluding a role for Kcnq1ot1 in repressing Kcnq1, even while silencing other genes in the domain. The exact timing of the mono - to biallelic transition is strain-dependent, with the CAST/EiJ allele becoming activated earlier and acquiring higher levels than the C57BL/6J allele. Unexpectedly, Kcnq1ot1 itself also switches to biallelic expression specifically in the heart, suggesting that tissue-specific loss of imprinting may be common during embryogenesis. The maternal Kcnq1ot1 transcript is shorter than the paternal ncRNA, and its activation depends on an alternative transcriptional start site that bypasses the maternally methylated promoter. Production of Kcnq1ot1 on the maternal chromosome does not silence Cdkn1c. We find that in later developmental stages, however, Kcnq1ot1 has a role in modulating Kcnq1 levels, since its absence leads to overexpression of Kcnq1, an event accompanied by an aberrant three-dimensional structure of the chromatin. Thus, our studies reveal regulatory mechanisms within the Kcnq1 imprinted domain that operate exclusively in the heart on Kcnq1, a gene crucial for heart development and function. We also uncover a novel mechanism by which an antisense non-coding RNA affects transcription through regulating chromatin flexibility and access to enhancers.
Introduction
The genome is transcribed into a host of non-coding RNAs, some of which are designated as long intergenic (linc) RNAs. These have regulatory roles in gene activation and repression, and have been hypothesized to serve as guides for non-sequence-specific histone modifying enzymes [1]. One class of lincRNAs includes macroRNAs, which can range in size up to hundreds of kilobases. Several macroRNAs have been studied intensively in the fields of genomic imprinting and X-inactivation [2], [3], [4], [5]. The role of Xist in X-inactivation has been a long-standing paradigm for understanding how silencing of imprinted genes is achieved in domains that encode other macroRNAs. One prevalent model of imprinted gene silencing is that the non-coding (nc) RNA spreads along the chromatin, recruiting histone modifying repressive enzymes and rendering the region refractory to transcriptional activation.
The Kcnq1ot1 imprinted ncRNA is transcribed from the paternal allele, emerging from intron 11 of the Kcnq1 gene in antisense direction. In contrast to most long ncRNAs, Kcnq1ot1 is highly expressed and detectable in every tissue examined [3], [6]. Kcnq1ot1 transcription has a role in repressing several neighboring genes in cis [7], [8]. In both the embryo and the placenta, the polycomb group complex is reported to play a major role in silencing. In the placental lineage, repressive histone modifications are necessary to maintain the imprinting status of the genes surrounding Kcnq1ot1 in the absence of differentially methylated regions [9]. In the embryo, only genes with stable DNA methylation are silenced by the ncRNA [10], [11], [12]. Although the role of Kcnq1ot1 in the establishment of allelic repression is still unclear, evidence has emerged suggesting that maintenance of silencing results from the act of transcription through the locus rather than as a function of the RNA molecule per se, at least in cultured embryonic and trophoblastic cells [13].
The silencing capacity of Kcnq1ot1 across the Kcnq1 domain is not universal. At least one gene, Cdkn1c, is repressed by a mechanism independent of Kcnq1ot1 in certain tissues [14]. On the other hand, some genes in the domain escape silencing, either ubiquitously or in a tissue-specific manner [15], [16], [17]. Kcnq1, one such exception, is expressed from the maternal allele during early embryogenesis and transitions to biallelic expression during fetal heart development. This transition coincides with conformational changes involving the interaction between the Kcnq1 promoter and heart-specific enhancers [18]. Based on our previous studies, we hypothesized that paternal expression of Kcnq1ot1 represses Kcnq1 in cis, but this effect is countered when enhancer-driven activity of Kcnq1 becomes established during heart development.
Defects in KCNQ1 are responsible for congenital long QT syndrome, a cardiac disorder that can result in serious cardiac arrhythmias [19]. There is a wide range of phenotypes, with some individuals remaining mostly asymptomatic and others presenting severe symptoms. Understanding the molecular mechanisms underlying the phenotypic variability of this disease will require fully elucidating the epigenetic profile of Kcnq1 expression during cardiac development. We investigated function of Kcnq1ot1 transcription in regulating Kcnq1 in the heart in wild-type mice and in a mutant mouse model in which the Kcnq1ot1 transcript is truncated. Our studies show that imprinted Kcnq1 expression in early heart development is not dependent on the Kcnq1ot1 transcript or on its transcriptional status. In fact, Kcnq1ot1 itself suffers a loss of imprinting during cardiac development, although it remains monoallelic in other tissues. We also show that the ncRNA does have a role in later embryogenesis for proper regulation of Kcnq1 levels in the embryonic heart by way of modulating the three-dimensional chromatin structure of the locus.
Results
The Kcnq1ot1 ncRNA loses imprinted expression in the heart
The Kcnq1 domain exhibits complex expression and imprinting patterns, eliciting questions of how long-range regulatory mechanisms coexist with locally limited ones (Figure 1A). The Kcnq1 gene switches from mono - to biallelic expression during mid-gestation in a tissue-specific manner. In the embryonic heart the transition occurs at approximately E14.5, coinciding with enhancers contacting the Kcnq1 promoter [18]. The timing for this is very consistent, with the switch occurring at the earliest at E13.5 and no later than E15.5. We proposed that tissue-specific enhancers compete with Kcnq1ot1 and create a chromatin environment that is antagonistic to the ncRNA.

Activation of the paternal Kcnq1 allele could interfere with spreading of the antisense Kcnq1ot1 RNA, or it could impact its expression, thus limiting its role in silencing. To determine if the paternal Kcnq1ot1 allele is still expressed after paternal Kcnq1 activation, we carried out allele-specific RT-PCRs during normal heart development in progeny from reciprocal crosses between C57BL/6J and B6(CAST7) mice. B6(CAST7) are a consomic strain in which chromosome 7 is derived from Mus musculus castaneus [20]. Unexpectedly, we found that not only does the paternal Kcnq1ot1 remain active, but the maternal allele is now transcribed as well. This loss of imprinted expression for Kcnq1ot1 is independent of strain-specific effects, is specific to the heart and does not occur in other tissues (Figure 1B, Figure S1A).
Previous reports have shown that the paternal Kcnq1ot1 ncRNA acts in cis by coating the chromosome [12], [11]. We used dual DNA and RNA fluorescent in situ hybridization (FISH) to determine if the maternal Kcnq1ot1 transcript accumulates locally in the heart. RNA and DNA FISH was performed in primary cardiomyocytes that are biallelic for Kcnq1ot1 (Figure S2). Results show that the maternal transcript co-localizes with the DNA strand it is being transcribed from. This suggests that the reactivated maternal Kcnq1ot1 transcript does associate with the chromatin in cis.
As shown in Figure 1C, levels of Kcnq1ot1 mRNA emerging from the maternal allele almost equal the paternal levels, suggesting that maternal expression is not merely due to “leaky” transcription. In fact, the transition to biallelic expression results in an increase in total Kcnq1ot1 expression when compared to tissues in which Kcnq1ot1 remains monoallelic (Figure S1B), Thus, the paternal activation of Kcnq1 does not interfere with Kcnq1ot1 transcription in cis [18] and, furthermore, initiation of Kcnq1ot1 expression from the maternal allele does not silence the already active maternal Kcnq1 allele.
Our data clearly indicate that cardiac development is accompanied by regulatory events in the Kcnq1 domain that are very distinct from its expression and imprinting status in other tissues. To determine if the loss of imprinting in the heart affected genes in the region other than Kcnq1 and Kcnq1ot1, we tested the allele-specific expression of Cdk1nc and Slc22a18 using either RT-PCR followed by allele specific restriction digest or sequencing analysis (Figure S3). Interestingly, both Cdkn1c and Slc22a18 maintained their maternal expression patterns throughout heart development. This confirms that transcription of the maternal allele of Kcnq1ot1 that occurs in the heart does not have repressive capability. As anticipated, and as previously reported [14], Cdkn1c and Slc22a18 maintain their imprinting expression in both P2 brain and liver samples (Figure S2A, S2B).
The maternal Kcnq1ot1 ncRNA emerges from an alternative promoter and is shorter than its paternal counterpart
The promoter of the Kcnq1ot1 gene lies immediately upstream of two CG-rich regions that harbor a maternal-specific gametic methylation mark (KvDMR). To determine if activation of the maternal Kcnq1ot1 allele is due to loss of methylation at the KvDMR, we performed methylation-specific PCR (data not shown) and bisulfite mutagenesis sequencing (Figure S4). Our results show that the maternal KvDMR methylation mark is not lost in neonatal heart.
To determine if the maternal Kcnq1ot1 transcript is initiated at an alternative promoter, thus bypassing methylation on the maternal KvDMR, 5′RACE was performed in neonatal heart (Figure 2A). We find that in the neonatal heart, transcripts are initiated at previously reported transcriptional start sites (TSS) [21] and at two additional downstream sites (Figure 2A). Sequence analysis of an expressed single nucleotide polymorphism shows that although paternal transcripts are produced from both previously TSS and the newly identified TSS, the maternal transcripts are exclusively initiated at the cardiac-specific sites downstream of the CG-islands, thus bypassing the methylation mark.

To see if the maternal Kcnq1ot1 gene produces a full-length RNA, we scanned the region with allele-specific RT-PCR. As shown in Figure 2, the re-activated maternal transcript is truncated, producing an RNA of less than 44 kb long (Figure 2B and 2C, Figure S1B).
Kcnq1 expression levels and loss of imprinting are subject to genetic background effects
To determine the differences in genetic background effects on expression levels and the timing of the mono - to biallelic switch, we assayed relative mRNA abundance in B×C and C×B hearts during development (Figure S5). Early maternal expression of Kcnq1 is higher from the CAST/EiJ than from the C57BL/6J and continues to have a relatively higher expression level throughout development (Figure S5B). Also, the C×B mono - to biallelic transition occurs two days later than in the reciprocal cross (Figure S5A), with the C57BL/6J paternal activation beginning at 16.5 dpc. In addition, the Kcnq1 expression level attained by the paternal copy is 75% of the maternal allele in the C×B cross, whereas maternal and paternal expression is equalized in the reciprocal cross (Figure S5C). These results show that there is a strong strain-specific genetic component on chromosome 7 that determines levels of Kcnq1 and affects the timing of paternal allele reactivation.
Kcnq1 is imprinted in the absence of Kcnq1ot1 in the early stage heart
Our data show that in the context of the neonatal heart, Kcnq1ot1 does not silence Kcnq1. During early development, however, Kcnq1 exhibits monoallelic expression and it was assumed that Kcnq1ot1 was responsible for repressing Kcnq1 in cis (Figure S5A). To test this assumption, we examined a mutant mouse model in which the Kcnq1ot1 transcript is severely truncated due to the inclusion of a transcriptional termination site 1.5 kb downstream of the transcriptional start site [8]. In this mouse, designated as K-term, transmission of the mutation from males results in loss of paternal silencing of genes in the Kcnq1 domain in the placenta and late embryonic stages. We examined whether the absence of Kcnq1ot1 leads to biallelic Kcnq1 cardiac expression from embryonic day 10.5 to postnatal day 2.
Surprisingly, Kcnq1 did not lose imprinted expression and maintained the same profile in the K-term as in wild-type mice, i.e., expression was exclusively from the maternal allele in E10.5 hearts. Thereafter, expression became biallelic, as in wild-type hearts, although at a slightly earlier E12.5 dpc (Figure 3A). These results indicate that paternal-specific repression of Kcnq1 in early embryogenesis is independent of Kcnq1ot1 transcription and suggest that there are other mechanisms responsible for differential expression of the Kcnq1 gene.

To determine if the maintenance of imprinting at Kcnq1 was specific to cardiac tissue, we tested Kcnq1 allelic expression in heads and bodies of the matched mutant embryos. In these, biallelic expression of Kcnq1 was seen in the absence of full-length Kcnq1ot1 (Figure 3A, left), indicating that maintenance of imprinted expression in K-term mutants may be restricted to the heart. These results also show that while the ncRNA is not responsible for imprinted expression of Kcnq1 in the heart, it does have a role in silencing Kcnq1 in tissues other than heart.
While imprinted expression of Kcnq1 in the early heart is independent of Kcnq1ot1, paternal silencing of both Cdkn1c and Slc22a18 does depend on Kcnq1ot1 transcription: upon paternal transmission of the K-term mutation, these genes lose their imprinting pattern and are biallelically expressed (Figure S3).
Kcnq1 monoallelic expression is not due to methylation of its promoter
To determine if early silencing of the paternal Kcnq1 allele is due to methylation of the promoter, bisulfite mutagenesis sequencing of the promoter was performed on E10.5 and P2 wild type hearts. Results show that the Kcnq1 promoter is not methylated in the early embryonic heart and thus, imprinted expression cannot be attributed to differential methylation (Figure S6). These results confirm our published 5-methylcytosine ChIP data in E7.5 whole embryos confirm that the CG-island at the Kcnq1 promoter is not methylated at any stage during development [18], as well as previous reports from others [22], [23].
Absence of Kcnq1ot1 leads to Kcnq1 overexpression in the heart
Although Kcnq1ot1 is not responsible for Kcnq1 imprinted expression in the heart, we investigated if it had any repressive effect on Kcnq1 in later stages of heart development. To test this, we determined Kcnq1 mRNA abundance by qRT-PCR was performed on E10.5, E16.5 and P2 hearts in wild-type and K-term mice. Kcnq1 transcript levels were similar between wt and K-term hearts at E10.5. However, by E16.5, Kcnq1 abundance was significantly increased in K-term compared to wild type mice, indicating that Kcnq1ot1 transcription plays a role in regulating Kcnq1 levels (Figure 3B, Figure S5B).
To determine if the increase in mRNA levels of Kcnq1 in the mutant mice was due to increased transcription from one or both parental alleles, relative transcript abundance was determined in 14.5 dpc and neonatal hearts. Activation of the paternal Kcnq1 allele was progressive and levels reached 88% of the maternal allele RNA abundance (Figure 3C, Figure S5C). Thus, the overall increase in Kcnq1 expression follows the same dynamics as in the wild-type heart, i.e., the paternal allele becomes progressively activated and reaches levels similar to the maternal RNA. This activation coincides with tissue-specific enhancer-driven expression in the wild-type mouse.
Kcnq1ot1 modulates three-dimensional chromatin conformation
We have previously shown that the Kcnq1 promoter interacts with an upstream region in a cardiac-specific fashion [18]. Moreover, this interaction is established during the transition from mono - to biallelic expression of Kcnq1. Because the loss of the ncRNA results in a large increase in Kcnq1 transcript in the heart, we investigated whether the three-dimensional conformation of the locus was altered in the K-term mice.
Chromosome Conformation Capture (3C) assays were performed on P2 hearts of K-term mutant mice, with the anchor primer located at the Kcnq1 promoter (Figure 4A). In contrast to the tight interactions seen in the wild-type heart, the 3C assays showed that the Kcnq1 promoter interacts with many additional sites throughout the region in the K-term mutants (Figure 4B). These results implicate the transcription of the Kcnq1ot1 RNA in regulating the specificity of promoter interactions. Thus, although the Kcnq1ot1 RNA does not silence Kcnq1, its absence is permissive for ectopic promoter contacts that increase the overall levels of Kcnq1.
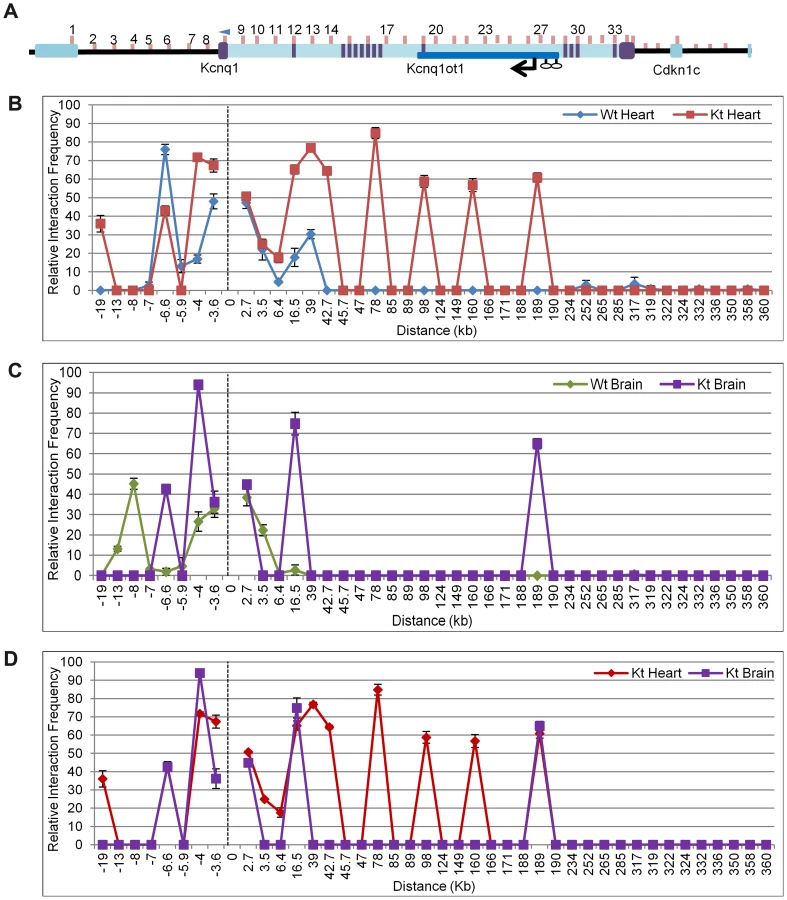
The 3C assays are targeted to regulatory elements and there is a paucity of polymorphisms in these regions. We were able to determine allele-specificity of contacts with one primer set (primer 27) by analyzing substrate prepared from heart from progeny of crosses between B6(CAST7) and K-term mice (Figure S7). PCR followed by allele-specific restriction digest showed ectopic contact between the Kcnq1 promoter and the Kcnq1ot1 promoter region only on the paternal chromosome, i.e., the one carrying the K-term mutation.
Analysis of brains from K-term mice showed a distinct pattern, with two associations that were not present in wild-type brains. These results suggest that the biological consequences of aberrant Kcnq1 promoter contacts are tissue-specific and reinforce that the Kcnq1 gene has multiple levels of regulation in the heart (Figure 4C).
To determine if the ectopic contact sites for the Kcnq1 promoter in K-term mice have the hallmarks of cis-acting regulatory sequences, we conducted chromatin immunoprecipitation (ChIP) assays (Figure 5A, 5B). Enhancers are typically occupied by p300 and have specific epigenetic marks, notably H3K4Me1 and H3K27Ac. In addition, some enhancers are highly conserved. Using ChIP analysis, the Kcnq1 region was scanned for p300, H3K27Ac and H3K4me1 occupancy in wild-type hearts, focusing on the sequences that exhibited interactions in the 3C assays.

Several of the ectopic contact sites showed epigenetic hallmarks of regulatory sequences (Figure 5B). Specifically, regions amplified with primer sets 13 and 17 (+39 kb and +78 kb relative to TSS, respectively) have p300 occupancy, with H3K4Me1 and H3K27Ac modifications. Although we did not observe these features with primers sets 23 and 25, H3K4Me1 cardiac-specific peaks are reported by the ENCODE consortium (annotated on UCSC), as are the primer 13 and 17 signals. Only regions 13, 21 and 23 are highly conserved (www.dcode.org), and these only between mouse, rat, rhesus and human.
Interestingly, the +189 kb peak lies within the Kcnq1ot1 promoter region. In fact, the Kcnq1 promoter does not make contacts beyond a CTCF binding site upstream (located in the Trpm5 gene) and two CTCF binding sites downstream in the Kcnq1ot1 promoter, suggesting that these may serve as boundaries to the movement of the Kcnq1 promoter, at least in the absence of Kcnq1ot1.
Discussion
The findings presented in this report underscore the value of tissue - and stage-specific studies of imprinted domains and have yielded several surprising insights. Our data and studies of other imprinted genes confirm that loss of imprinting in specific tissues is a normal developmental event. We find that strain-specific effects influence expression levels and timing of imprinting loss. We also find two co-existing mechanisms responsible for imprinting within the Kcnq1 domain in the embryo. Importantly, by mapping the higher order structure of a specific region, we reveal a novel mechanism by which an antisense non-coding RNA can regulate transcription.
Kcnq1ot1 is ubiquitously expressed and it had been assumed that it was imprinted in every tissue. In this study, we find that Kcnq1ot1 is biallelically expressed in the heart, with the transition from mono - to biallelic occurring in parallel with the Kcnq1 switch. This loss of imprinting is not seen in other tissues at that stage, indicating that there are mechanisms that shift the control of the region to meet tissue-specific needs. Perhaps the strong cardiac enhancers acting on Kcnq1 also exert influence on the Kcnq1ot1 promoter, activating the previously silent maternal allele.
The maternal methylation mark encompassing the Kcnq1ot1 promoter is not lost during the activation of transcription. This is in line with abundant data showing that gametic methylation is very stable [24], and suggests that the repressive effect of DNA methylation can be overcome. In fact, our data show that maternal transcription of Kcnq1ot1 ncRNA is initiated at an alternative promoter region downstream of the methylation mark. Several examples of alternative promoter usage bypassing DNA methylation have been reported [25], and we propose that this mechanism may be commonly deployed to fine-tune expression of imprinted genes for tissue-specific needs.
Interestingly, the Kcnq1ot1 molecule produced from the maternal chromosome is half the length of the paternal transcript. This could be because expression of maternal Kcnq1 from early stages blocks or competes with the Kcnq1ot1 in some way and impedes completion of the transcript. Another possibility is that an alternative transcriptional termination signal is used. These two mechanisms are not mutually exclusive. Surprisingly, the reactivated ncRNA does not repress Cdkn1c, even though it accumulates locally in a manner similar to the paternal Kcnq1ot1. It remains to be determined if this is because of the diminished length of the RNA or if cardiac cells do not provide repressive co-factors that are necessary for the spread of silencing.
Our data reveal a genetic background effect on the level of expression of Kcnq1, with the CAST/EiJ allele exhibiting higher abundance than the C57BL/6J allele at all stages in the developing heart. In addition to being expressed less abundantly, a C57BL/6J paternal allele is reactivated at a later time point than a CAST/EiJ one. Whether these two features are related needs to be determined, but our own analyses and publicly available genomic data lead us to hypothesize that strain-specific cis-regulatory polymorphisms may explain both phenomena. For example, sequence differences in transcription factor binding or affinity for enhancers could determine higher levels of Kcnq1 expression from the CAST/EiJ allele, and greater accessibility to reactivation when inherited paternally.
In exploring the cardiac regulation of the Kcnq1 gene, our findings delineate two phases, independently of strain-specific differences: one, in early development, when Kcnq1 exhibits imprinted expression independently of Kcnq1ot1; and two, in the neonate, when Kcnq1 is biallelic and its expression levels are modulated by Kcnq1ot1.
Our analysis of the K-term mutant mouse shows that Kcnq1 expression is imprinted in early cardiac development even in the absence of the ncRNA. This result excludes a role for Kcnq1ot1 in establishing or maintaining repression. Thus, there is an independent mechanism that silences the paternal Kcnq1 allele during early development. This mechanism is cardiac-specific, because absence of Kcnq1ot1 does release Kcnq1 from repression in tissues other than the heart. Although a secondary methylation mark at Cdkn1c is reportedly dependent on expression of Kcnq1ot1, the Kcnq1 CG-rich promoter is never methylated. Thus, monoallelic expression of Kcnq1 is likely due to chromatin conformation or trans-acting factors. Perhaps paternal expression of Kcnq1ot1 opens the chromatin and makes a tissue-specific silencer available to factors that repress Kcnq1. These factors would be present in early development and would disappear upon full maturation of the heart (Figure 6). Another possibility is that an inhibitory factor (IF) for Kcnq1 has a cognate bind site within a differentially methylated region. If the factor is methylation-sensitive, it would only be able to bind the unmethylated paternal allele, thus rendering the paternal Kcnq1 allele inactive. In fact, a silencing domain (SD) has been delimited downstream of the Kcnq1ot1 promoter, overlapping one of the differentially methylated CG-islands (Figure 2A). Interestingly, our bisulfite sequencing results for the paternal KvDMR show a trend towards acquisition and spreading of methylation that could be explained by loss of the inhibitory factor binding at the SD (Figure S3).

Early paternal repression of Kcnq1 may also be due to the presence of CTCF on the paternal KvDMR [21]. CTCF binding is methylation-sensitive. CTCF could negatively impact expression of Kcnq1 in early embryonic stages by repressing it directly or blocking it from access to enhancers required at that stage (Figure 6, inhibitory factor (IF) would be CTCF). Enhancer activity has been shown for a sequence immediately upstream of the Kcnq1ot1 promoter, a position that would be blocked by CTCF [21].
Comparison of Kcnq1 abundance and domain conformation between wild-type and mutant mice reveals what may be the key role for Kcnq1ot1 in later cardiac development. Our results show that absence of the ncRNA leads to much increased Kcnq1 levels, accompanied by a wider range of promoter contacts with sequences throughout the region. Although several of the regions contacted exhibit the marks of regulatory sequences, the Kcnq1 promoter is only associated with them if Kcnq1ot1 is prematurely terminated. The ectopic interactions reflect a greater flexibility of the chromatin fiber in the absence of Kcnq1ot1 transcription and may contribute to the increased Kcnq1 levels. It is imperative to further elucidate the molecular basis of the tissue-specificity of this effect. Subnuclear localization of the Kcnq1 domain in the presence or absence of the Kcnq1ot1 RNA will be an interesting avenue to pursue, to determine whether the ncRNA can act to tether the region to specific nuclear compartments [1], [26], . We also need to explore whether these mechanisms are more widely used by other ncRNA, especially those that are antisense to coding genes.
Tissue-specificity of Kcnq1ot1 repression has been previously reported, specifically for the neighboring Cdkn1c gene [14]. In fact, Cdkn1c imprinting is maintained in the kidney, liver and lung even with a truncated Kcnq1ot1. Several studies have also shown lineage-specific imprinting regulation in the placenta [11], [29]. Indeed, these observations collectively underscore the need to identify cis - and trans-acting factors and the dynamics of their interactions to test hypotheses relative to the tissue-specific effects of Kcnq1ot1.
Overall, our results are in accordance with the idea that the Kcnq1ot1 ncRNA itself is not responsible for directly silencing neighboring genes [13]. We posit that distinct cis-acting elements are made available for regulatory contacts depending on whether the act of transcription occurs or not, as previously suggested [30]. These associations have an additional layer of regulation that imposes the tissue-specificity of the patterns.
In conclusion, our results paint picture of the regulation at the Kcnq1 domain that is much more complex than previously assumed. Imprinting centers, which regulate extended domains of genes, are limited and counteracted by transcriptional mechanisms that may have evolved in response to the emergence of imprinting. Our studies on the Kcnq1 domain underscore that the expression pattern of a domain is ultimately determined by cooperative and competitive dialogues between enhancers, silencers and insulators [31], [32] and that understanding complex genetic loci will require identifying all the regulatory sequences and determining their linear order, tissue-specificity and three-dimensional structure.
Materials and Methods
Ethics statement
This study was carried out on mice in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Temple University Animal Care and Use Committee (Protocol 3294).
Mouse strains and crosses
All wild-type tissues used in the experiments were either C57BL/6J or F1 progeny of reciprocal crosses between C57BL/6J and B6(CAST7) (a consomic strain harboring a chromosome 7 derived from Mus musculus castaneus on a C57BL/6J background) (BxC or CxB for short). For the K-term mouse samples, mutant males were crossed with B6(CAST7) females and tissues of F1 progeny were obtained (CxK-term). To show the transition in allelic expression for Kcnq1 and Kcnq1ot1, 2–3 embryos were analyzed per litter and 3–5 litters were analyzed at each stage of development.
RNA purification
Tissues for total RNA extraction were collected at appropriate days of gestation from F1 progeny. Polymorphisms between strains were used to distinguish between parental alleles. RNA was extracted using either TRIzol Reagent (Invitrogen #15596-018) or the Roche High Pure RNA Isolation Kit (#11828665001), following the manufacturer's protocol. All RNA samples were subjected to DNAse treatment using Turbo DNA-free (Ambion #AM1907). Three to five biological samples were collected for each tissue.
Following the manufacturer's instructions, complementary DNA synthesis was performed on total RNA using SuperScript II Reverse Transcriptase (Invitrogen #18064-014). A Reverse Transcriptase negative control was used to ensure there was no DNA contamination.
Real-time qRT–PCR
Transcript levels of Kcnq1, Kcnq1ot1 and β-actin were analyzed on the ABI Step One Plus system. Reactions were conducted using the SYBR Green PCR Master Mix (ABI #4309155) in a final reaction volume of 20 µl. The PCR was performed under the following conditions: an initial denaturing step for 10 minutes at 95°C, an amplification step for 45 cycles of 95°C for 20 seconds, 55°C degrees for 30 seconds and 72°C for 30 seconds, and a final elongation step at 72°C for 2 minutes. The Kcnq1 transcript was detected using the following primers: 5′-CAAAGACCGTGGCAGTAAC-3′ and 5′-CCTTCATTGCTGGCTACAAC-3′. Kcnq1ot1 was detected using 5′-GGTCTGAGGTAGGGATCAGG-3′ and 5′-GGCACACGGTATGAGAAAAGATTG-3′. The Kcnq1 and Kcnq1ot1 levels were normalized to β-actin, using the primers 5′-TGTTACCAACTGGGACGACA-3′ and 5′-CCATCACAATGCCTGTGGTA-3′. Each qRT-PCR reaction was performed in triplicate with three biological replicates together with a negative control. The ΔΔCT values of the Kcnq1 transcript were normalized to the β-actin transcript. The standard deviation for each ratio was determined and the error bars represent one standard deviation from the average ratio. A t-test and p-value was obtained for all comparisons.
Allele-specific RT–PCR and quantification
The Kcnq1, Kcnq1ot1, Cdkn1c, and Slc22a18 transcripts were amplified using Ruby Taq Master Mix (Affymetrix - #71191) in a 15 µl reaction. PCR reactions were performed with experimental and control templates. Following the PCR, restriction digests were performed with NlaIII, StuI, and Taqα1 (New England Biolabs) respectively. PCR and digestion products were run on 7% polyacrylamide gels and quantified using the Kodak Gel Logic 2000 imaging system. The ratio of paternal to maternal band intensities was calculated for Kcnq1. The Slc22a18 product was run on a 2% agarose gel and extracted using the E.Z.N.A Gel Extraction Kit (Omega Bio-tek, Inc.) The purified PCR product was then sent for sequencing analysis by Europhins MWG Operon. Sequences were aligned with Geneious Pro 4.6.5. The following primers were used to amplify the transcripts:
Kcnq1 5′-CATCGGTGCCCGTCTGAACAGG-3′ and 5′-TTGCTGGGTAGGAAGAGCTCAG-3′ with NlaIII
Kcnq1ot1 5′-GGTCTGAGGTAGGGATCAGG-3′ and 5′ - GGCACACGGTATGAGAAAAGATTG-3′ with StuI
Cdkn1c 5′-CGGACGATGGAAGAACTCTGG-3′ and 5′-TACACCTTGGGACCAGCGTACTCC-3′ with Taqα1
Slc22a18 5′-GTGCTGCCTGTATTCCTGGT-3′ and 5′-CAGTCCCACAACAGCAAAGA-3′
Kcnq1ot1 gene scan
The following primers and associated restriction enzymes were used to scan the Kcnq1ot1 maternal transcript in P2 hearts from reciprocal crosses of C57BL/6J and B6(CAST7) mice:
2 kb Kcnq1ot1 5′-GGTCTGAGGTAGGGATCAGG-3′ and 5′ - GGCACACGGTATGAGAAAAGATTG-3′ with StuI
14 kb 5′-CTGGCCATCATTCACAGTTG-3′ and 5′ - CATTCCCGTAAGAGCTGGTG-3′ with HpyCH4IV
18 kb 5′-CAAAAAGCAGGTCCTGAAGC-3′ and 5′-TGATGCCTTGTGATGGAAAA-3′ with MnlI
22 kb 5′-AGGCACAAATGAACTGAAAGA-3′ and 5′-GCACCTCCTGTGAAGTGAGA-3′ with BceAI
27 kb 5′-CAGTGGCTACGGACCATCTT-3′ and 5′-TGGATACCTGTCCACATTTGC-3′ with SmlI
33 kb 5′-TATCCCCCTGATCACTCACC-3′ and 5′-CCTCGTTGGTGCTCACAGTA-3′ with NcoI
44 kb 5′ - TCAACTTTTGGAGAAGATAGTGCTT-3′ and 5′ - AGGACAGGCAATGACAGAGC-3′ with SpeI
60 kb 5′-GATCAGCATGGGTTATTGGA-3′ and 5′ –ATTAAGGGACCACAGCAAGG-3′ with HpaI
5′ RACE assays
5′ RACE assays to determine the Kcnq1ot1 start site were conducted using the 5′ RACE system (Invitrogen), with RNA from three independent samples of neonatal hearts and livers. Primers for cDNA synthesis and nested PCR amplification (combined with a universal adapter primer) were as follows:
A: 5′-ACTGTATTAAAGGGTCAAAGCACAA;
An: 5′-GCAACCACTGCGGCCTCCACCCCAAGTTCCCAATC
B: 5′-ATGACGAAACAAGATAAGACCTCAC
Bn: 5′-GGACAAACACTGAGGAGGATCGCGTTGAGCAAAGC
C: 5′-CTGCTCTTCCTTACTCTAAGCACTGT
Cn: 5′-TAAGCCTTTGTTTGCCTCCTCTGGCCTAGAAGGCC
Relative positions of the primers are shown in Figure 2A. Bands of interest were recovered after gel electrophoresis, cloned (TOPO-TA Cloning Kit, Life Technologies #K4500-01) and transformed into competent E.coli cells. Plasmid DNA was prepared from bacteria (Spin Mini Prep Kit (QIAGEN #30020)and sequenced (Eurofins MWG Operon). Sequences were aligned with Geneious Pro 4.6.5 [34].
Chromosome conformation capture (3C) assays
3C was performed as described previously [18]. Digestion efficiency at each restriction site was determined by comparing amplification with primers spanning the restriction site to amplification with primers immediately downstream on the digested template. No restriction bias was observed across the region assayed by 3C [18].
PCR efficiency of each combination of primers was assessed with a control template prepared from three BACs encompassing the Kcnq1ot1 region analyzed, as described previously. All PCR reactions were performed with RubyTaqTM (USB) as follows: 67° 20″, 72° 20″ for 15 cycles, with a decrease in the annealing temperature of 2° every 3 cycles, followed by 25 cycles of 55° 20″, 72° 20″ and a final extension step of 72° 1′30″. The linear range for each primer pair was determined by serial dilution. PCR reactions were performed with experimental and control templates in parallel and PCR products were run on 7% polyacrylamide gels and quantified using the Kodak Gel Logic 2000 imaging system. Crosslinking frequencies were calculated from duplicate PCR analyses of three independent 3C preparations. Interaction frequencies for Kcnq1 and Kcnq1ot1 were normalized to a control interaction to allow comparisons between different tissues and cell types. A full list of primers can be found in Table S1.
ChIP assays
Primary tissues were collected at neonatal stage P2, chopped into 1 mm pieces and cross-linked in 1% formaldehyde for 10 minutes. The formaldehyde was quenched with 0.125 M glycine for 5 minutes. The cells were spun down and the cell pellet was washed twice in PBS before re-suspension in lysis buffer (10 mM Tris pH 7.5, 10 mM NaCl, 3 mM MgCl2, 0.5% NP-40). Nuclei were extracted from the cells by incubating on ice for 10 minutes for brain tissue and 30 minutes for heart tissue, followed by 15 strokes in a dounce homogenizer. Nuclei were washed twice with PBS and resuspended in digestion buffer (10 mM Tris pH 7.5, 10 mM NaCl, 3 mM MgCl2, 1 mM CaCl2 and 4% NP-40). 107 nuclei were sonicated and 8 ug of chromatin was used for each IP, with 10% saved as the input. Protein A beads were primed with their respective antibody in IP buffer (20 mM Tris pH 8, 2 mM EDTA, 1% Triton X-100 and 150 mM NaCl) for 2 hours at 4°C. Chromatin was pre-cleared with IgG in IP buffer at 4°C for 1 hour. Chromatin and antibody/bead complexes were incubated overnight and washed 3 times with buffer #1 (20 mM Tris pH 8, 2 mM EDTA, 0.1% SDS and 150 mM NaCl), twice with buffer #2 (20 mM Tris pH 7.5, 2 mM EDTA, 1% Triton X-100, 0.1% SDS and 500 mM NaCl), once with buffer #3 (10 mM Tris pH 8, 1 mM EDTA, 0.25 M LiCl and 1% NP-40) and twice with TE. The chromatin was eluted by incubating chromatin/antibody/bead complex at 65°C for 15 minutes in a buffer containing 25 mM Tris pH 7.5, 10 mM EDTA and 0.5% SDS. A final concentration of 0.5% SDS and 1.5 ug/ul of Proteinase K was added to all samples including the input. The samples were incubated for 1 hour at 42°C and de-crosslinked overnight at 65°C in a shaking water bath, followed by phenol∶chloroform extraction and ethanol precipitation. Pellets were re-suspended in 10 mM Tris.
The ChIP substrates were amplified using the ABI step-one plus machine and Sybr green reagents using the same conditions as the qRT-PCR assays. The data was analyzed using the percent input method. A full list of primers can be found in Table S2.
Bisulfite mutagenesis sequencing
1 ug of DNA was mutagenized using the EZ DNA Methylation-Gold Kit (Zymo Research #5005) following the manufacturer's protocol. Amplification of the Kcnq1ot1 CpG island was performed as previously described [33]. Primers for the Kcnq1 promoter were:
KBS-F: 5′-GGTTGGTGTATTGTAAGTGT
KBS-R: 5′ - CTAACAACAATAACTACCC
PCR products were cloned into the pCR2.1-TOPO vector using a TOPO-TA Cloning Kit (Life Technologies #K4500-01). The bacterial colonies were cultured and plasmids purified using a Spin Mini Prep Kit (QIAGEN #30020) and plasmids were sent to Eurofins MWG Operon for sequencing. Sequences were analyzed with Geneious Pro 4.6.5. Data were obtained from three independent biological samples, with at least 10 clones sequenced from each.
Primary cardiomyocyte collection and culture
Collection of cardiomyoctes was performed as described (Sreejit, et. al., 2008) with the following modifications. The hearts were incubated in Hanks Balanced Salt Solution (HBSS) for 10 minutes on ice and the cells were cultured in DMEM supplemented with 10% FBS and 2 mM L-glutamine, penicillin (100 u/mL) and streptomycin (100 mg/mL). After the fibroblasts settled, the cardiomyocytes were collected and plated directly onto 4 well #1.5 sterile chambered glass coverslips (Thermo Fisher Scientific).
RNA and DNA FISH
Cells were fixed and permeabilized using Life Technology's FIX AND PERM kit following the manufacturer's protocol. RNA FISH was performed as previously described (Golding, et. al., 2011). The probes for hybridization were made using Life Technology's BioPrime DNA Labeling kit with fosmids WI1-2505B3 and WI1-2753P18 (CHORI) following the manufacturer's protocol. Cy3-dCTP (GE) was used for RNA detection and FITC-dUTP (Roche) was used for DNA detection. After the washes for the RNA probe hybridization, DNA FISH was performed incubating the cells with 0.01 N HCl with 0.1 mg/ml (0.01%) pepsin for 3 minutes at room temperature. The cells were rinsed with PBS and dehydrated with a series of ethanol washes. The DNA was denatured for 30 minutes at 85°C with 70% formamide in 2× SSC. The cells were rinsed with 2× SSC and the probe was hybridized overnight. The following day, excess probe was washed with SSC, mounted and stained with Vectashield augmented with DAPI (Vector Laboratories). Cells were imaged on a Leica Sp5 Confocal Microscope with Z-stacking.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. LeeJT (2009) Lessons from X-chromosome inactivation: long ncRNA as guides and tethers to the epigenome. Genes Dev 23 : 1831–1842.
2. SleutelsF, ZwartR, BarlowDP (2002) The non-coding Air RNA is required for silencing autosomal imprinted genes. Nature 415 : 810–813.
3. SmilinichNJ, DayCD, FitzpatrickGV, CaldwellGM, LossieAC, et al. (1999) A maternally methylated CpG island in KvLQT1 is associated with an antisense paternal transcript and loss of imprinting in Beckwith-Wiedemann syndrome. Proc Natl Acad Sci U S A 96 : 8064–8069.
4. BorsoniG, TonlorenziR, SimmlerMC, DandoloL, ArnaudD, et al. (1991) Characterization of a murine gene expressed from the inactive X chromosome. Nature 351 : 325–328.
5. LeeJT, LuN (1999) Targeted mutagenesis of Tsix leads to nonrandom X inactivation. Cell 99 : 47–57.
6. LeeMP, HuR, JohnsonLA, FeinbergAP (1997) Human KVLQT1 gene shows tissue-specific imprinting and encompasses Beckwith-Wiedemann syndrome chromosomal rearrangements. Nature Genet 15 : 181–185.
7. FitzpatrickGV, SolowayPD, HigginsMJ (2002) Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat Genet 32 : 426–431.
8. Mancini-DinardoD, SteeleSJ, LevorseJM, IngramRS, TilghmanSM (2006) Elongation of the Kcnq1ot1 transcript is required for genomic imprinting of neighboring genes. Genes Dev 20 : 1268–1282.
9. LewisA, MitsuyaK, UmlaufD, SmithP, DeanW, et al. (2004) Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat Genet 36 : 1291–1295.
10. UmlaufD, GotoY, CaoR, CerqueiraF, WagschalA, et al. (2004) Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat Genet 36 : 1296–1300.
11. PandeyRR, MondalT, MohammadF, EnrothS, RedrupL, et al. (2008) Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol Cell 32 : 232–246.
12. TerranovaR, YokobayashiS, StadlerMB, OtteAP, van LohuizenM, et al. (2008) Polycomb Group Proteins Ezh2 and Rnf2 Direct Genomic Contraction and Imprinted Repression in Early Mouse Embryos. Dev Cell 15 : 668–679.
13. GoldingMC, MagriLS, ZhangL, LaloneSA, HigginsMJ, et al. Depletion of Kcnq1ot1 non-coding RNA does not affect imprinting maintenance in stem cells. Development 138 : 3667–3678.
14. ShinJY, FitzpatrickGV, HigginsMJ (2008) Two distinct mechanisms of silencing by the KvDMR1 imprinting control region. Embo J 27 : 168–178.
15. GouldTD, PfeiferK (1998) Imprinting of mouse Kvlqt1 is developmentally regulated. Hum Mol Genet 7 : 483–487.
16. LatosPA, BarlowDP (2009) Regulation of imprinted expression by macro non-coding RNAs. RNA Biol 6 : 100–106.
17. PaulsenM, DaviesKR, BowdenLM, VillarAJ, FranckO, et al. (1998) Syntenic organization of the mouse distal chromosome 7 imprinting cluster and the Beckwith-Wiedemann syndrome region in chromosome 11p15.5. Hum Mol Genet 7 : 1149–1159.
18. KorostowskiL, RavalA, BreuerG, EngelN (2011) Enhancer-driven chromatin interactions during development promote escape from silencing by a long non-coding RNA. Epigenetics Chromatin 4 : 21–32.
19. BokilNJ, BaisdenJM, RadfordDJ, SummersKM (2010) Molecular genetics of long QT syndrome. Mol Genet Metab 101 : 1–8.
20. MannMR, ChungYG, NolenLD, VeronaRI, LathamKE, et al. (2003) Disruption of imprinted gene methylation and expression in cloned preimplantation stage mouse embryos. Biol Reprod 69 : 902–914.
21. FitzpatrickGV, PugachevaEM, ShinJY, AbdullaevZ, YangY, et al. (2007) Allele-specific binding of CTCF to the multipartite imprinting control region KvDMR1. Mol Cell Biol 27 : 2636–2647.
22. YatsukiH, JohK, HigashimotoK, SoejimaH, AraiY, et al. (2002) Domain regulation of imprinting cluster in Kip2/Lit1 subdomain on mouse chromosome 7F4/F5: large-scale DNA methylation analysis reveals that DMR-Lit1 is a putative imprinting control region. Genome Res 12 : 1860–1870.
23. MohammadF, MondalT, GusevaN, PandeyGK, KanduriC (2010) Kcnq1ot1 noncoding RNA mediates transcriptional gene silencing by interacting with Dnmt1. Development 137 : 2493–2499.
24. WoodfineK, HuddlestonJE, MurrellA (2011) Quantitative analysis of DNA methylation at all human imprinted regions reveals preservation of epigenetic stability in adult somatic tissue. Epigenetics Chromatin 4 : 1–13.
25. ChotaliaM, SmallwoodSA, RufN, DawsonC, LuciferoD, et al. (2009) Transcription is required for establishment of germline methylation marks at imprinted genes. Genes Dev 23 : 105–117.
26. MondalT, KanduriC (2012) Noncoding RNA scaffolds in pluripotency. Circ Res 110 : 1162–1165.
27. LaurentGS, SavvaYA, KapranovP (2012) Dark matter RNA: an intelligent scaffold for the dynamic regulation of the nuclear information landscape. Front Genet 3 : 1–11.
28. WangKC, ChangHY (2011) Molecular mechanisms of long noncoding RNAs. Mol Cell 43 : 904–914.
29. RedrupL, BrancoMR, PerdeauxER, KruegerC, LewisA, et al. (2009) The long noncoding RNA Kcnq1ot1 organises a lineage-specific nuclear domain for epigenetic gene silencing. Development 136 : 525–530.
30. PaulerFM, KoernerMV, BarlowDP (2007) Silencing by imprinted noncoding RNAs: is transcription the answer? Trends Genet 23 : 284–292.
31. BoniferC (2000) Developmental regulation of eukaryotic gene loci: which cis-regulatory information is required? Trends Genet 16 : 310–315.
32. EngelN, BartolomeiMS (2003) Mechanisms of insulator function in gene regulation and genomic imprinting. Int Rev Cytol 232 : 89–127.
33. RiveraRM, SteinP, WeaverJR, MagerJ, SchultzRM, et al. (2008) Manipulations of mouse embryos prior to implantation result in aberrant expression of imprinted genes on day 9.5 of development. Hum Mol Genet 17 : 1–14.
34. Drummond AJ, Ashton B, Buxton S, Cheung M, Cooper A, Duran C, Field M, Heled J, Kearse M, Markowitz S, Moir R, Stones-Havas S, Sturrock S, Thierer T, Wilson A (2011) Geneious v5.4. http://www.geneious.com
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Enrichment of HP1a on Drosophila Chromosome 4 Genes Creates an Alternate Chromatin Structure Critical for Regulation in this Heterochromatic Domain
- Normal DNA Methylation Dynamics in DICER1-Deficient Mouse Embryonic Stem Cells
- The NDR Kinase Scaffold HYM1/MO25 Is Essential for MAK2 MAP Kinase Signaling in
- Functional Variants in and Involved in Activation of the NF-κB Pathway Are Associated with Rheumatoid Arthritis in Japanese
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy