
Genomics of Adaptation during Experimental Evolution of the Opportunistic Pathogen
Adaptation is likely to be an important determinant of the success of many pathogens, for example when colonizing a new host species, when challenged by antibiotic treatment, or in governing the establishment and progress of long-term chronic infection. Yet, the genomic basis of adaptation is poorly understood in general, and for pathogens in particular. We investigated the genetics of adaptation to cystic fibrosis-like culture conditions in the presence and absence of fluoroquinolone antibiotics using the opportunistic pathogen Pseudomonas aeruginosa. Whole-genome sequencing of experimentally evolved isolates revealed parallel evolution at a handful of known antibiotic resistance genes. While the level of antibiotic resistance was largely determined by these known resistance genes, the costs of resistance were instead attributable to a number of mutations that were specific to individual experimental isolates. Notably, stereotypical quinolone resistance mutations in DNA gyrase often co-occurred with other mutations that, together, conferred high levels of resistance but no consistent cost of resistance. This result may explain why these mutations are so prevalent in clinical quinolone-resistant isolates. In addition, genes involved in cyclic-di-GMP signalling were repeatedly mutated in populations evolved in viscous culture media, suggesting a shared mechanism of adaptation to this CF–like growth environment. Experimental evolutionary approaches to understanding pathogen adaptation should provide an important complement to studies of the evolution of clinical isolates.
Published in the journal:
. PLoS Genet 8(9): e32767. doi:10.1371/journal.pgen.1002928
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002928
Summary
Adaptation is likely to be an important determinant of the success of many pathogens, for example when colonizing a new host species, when challenged by antibiotic treatment, or in governing the establishment and progress of long-term chronic infection. Yet, the genomic basis of adaptation is poorly understood in general, and for pathogens in particular. We investigated the genetics of adaptation to cystic fibrosis-like culture conditions in the presence and absence of fluoroquinolone antibiotics using the opportunistic pathogen Pseudomonas aeruginosa. Whole-genome sequencing of experimentally evolved isolates revealed parallel evolution at a handful of known antibiotic resistance genes. While the level of antibiotic resistance was largely determined by these known resistance genes, the costs of resistance were instead attributable to a number of mutations that were specific to individual experimental isolates. Notably, stereotypical quinolone resistance mutations in DNA gyrase often co-occurred with other mutations that, together, conferred high levels of resistance but no consistent cost of resistance. This result may explain why these mutations are so prevalent in clinical quinolone-resistant isolates. In addition, genes involved in cyclic-di-GMP signalling were repeatedly mutated in populations evolved in viscous culture media, suggesting a shared mechanism of adaptation to this CF–like growth environment. Experimental evolutionary approaches to understanding pathogen adaptation should provide an important complement to studies of the evolution of clinical isolates.
Introduction
In the mid-1800's, Louis Pasteur advised microbiologists to think of the human body as a “culture vessel” for microbes, in the context of understanding immunity [1]. Pasteur's approach has been revised and updated several times [2], [3], with a recent review encouraging researchers to be attentive to the effects of different in vivo carbon sources on bacterial metabolism and physiology [2]. Pasteur's advice is particularly relevant for an understanding of the evolution of disease-causing microbes. Natural selection may be imposed by the particular nutritional and metabolic resources available in a given tissue, the innate and adaptive immune systems, and, in the past 80 or so years, by antibiotics or anti-virals. Many pathogens – particularly opportunistic pathogens, emerging pathogens, and microbes causing chronic disease – are faced with a novel and hostile growth environment to which they must adapt or face extinction. Colonization and establishment of an infection in a new host or host species can thus be interpreted as a specific instance of a more general process of adaptation to a novel environment.
Understanding adaptive processes in pathogen populations, and in particular characterizing the variety of genetic routes to adaptation, is important for developing effective treatment strategies. Take as an example the management of antibiotic resistance. Resistance is often thought to be costly, in the sense that resistant strains should be less fit than susceptible strains in the absence of antibiotic. If so, then attempts to reduce the frequency of resistance in patient populations by stopping the use of an antibiotic should afford sensitive strains an advantage, and so prolong the utility of an antibiotic for treatment. Antibiotic cessation has met with mixed success (e.g., [4]–[6]), however, either because some resistance mutations actually pay little or no cost, or because second site mutations that restore fitness without compromising resistance are common. The management of antibiotic resistance in patient populations depends crucially on which of these two mechanisms is more often responsible for the persistence of resistance.
The last 15 years have seen a number of studies of in vivo genome evolution in select pathogens, primarily viruses (e.g., [7], [8]) and bacteria (e.g., [9], [10]), that shed vital insight onto the genetic changes that occur during epidemics or chronic infections. The importance of these changes for pathogen fitness in a host can be difficult to ascertain, however, because it is rarely possible to establish with certainty that the observed mutations are adaptive, since some neutral or deleterious mutations may accumulate through drift or by hitchhiking with adaptive mutations. Moreover, it can be difficult to obtain sufficient in vivo samples to ask questions about the repeatability of in vivo evolution – that is, how often pathogens take the same adaptive routes in independent patients or populations.
For these reasons we have turned to a complementary approach, laboratory selection experiments, to provide an understanding of the broad patterns and principles of pathogen evolution. In a typical microbial experimental evolution protocol, many populations are founded from a single genotype, and are propagated serially or in a chemostat for tens, hundreds, or thousands of generations (reviewed in [11]). By maintaining multiple replicate populations in each of two or more environments (e.g., antibiotic treated vs. not antibiotic treated), the effects of a treatment can be systematically investigated in a manner that is often inaccessible with in vivo samples. Experimental evolution has by now a rich history in studying basic evolutionary processes (e.g., [11]–[14] for reviews), as well as more applied topics such as the evolution of antibiotic resistance [15], [16] and of virulence [7]. In addition, experimental evolution has significant potential as an investigative tool for elucidating basic biological processes [17], [18]. With the development of technologies that allow the rapid and affordable sequencing of entire bacterial genomes, an increasing number of studies have sought to describe the genomic basis of laboratory adaptation (reviewed in [19]).
Here we use a combination of experimental evolution and whole-genome sequencing (WGS) to investigate the initial stages of pathogen adaptation using the bacterium Pseudomonas aeruginosa. This gram-negative bacterium is widely distributed in nature [20], and is an important opportunistic pathogen. P. aeruginosa can cause acute infections of wounds, burns and of lungs, and is frequently implicated in nosocomial infections. Moreover, P. aeruginosa is an important pathogen of individuals with cystic fibrosis (CF), with approximately 60–70% of Canadian adults with CF harbouring this bacterium [21]. P. aeruginosa chronically infects the CF lung, and once the infection is established, it is virtually impossible to eradicate: Intensive antibiotic regimens are effective at reducing symptoms, but almost never succeed in clearing the infection entirely.
P. aeruginosa populations that have persisted for long periods of time in the lungs of individuals with CF show characteristic signatures of adaptation to this novel culture environment. Recent studies have documented patterns of parallel evolution at the level of phenotype, gene expression, and genotype [10], [22]–[25], indicating repeatable patterns of long-term adaptation to the CF lung. For example, CF lung sputum is highly viscous, and P. aeruginosa typically grows as an unattached biofilm, or microcolony, in this environment [26]. While environmental isolates of P. aeruginosa are motile, long-term CF colonists show evidence of adaptation to the sessile lifestyle of the microcolony, including reduced motility, and a morphological shift to small colony variants (SCVs) on agar plates [27], [28]. Increased intracellular levels of cyclic di-GMP are thought to be important for this adaptive shift [27]–[29], but the causative mutations have yet to be fully elucidated. Other characteristic changes include mutations associated with reduced virulence, presumably to avoid detection by the host immune system, and increased small molecule efflux that can afford resistance to antibiotics commonly used with CF patients [10].
Given evidence of long-term adaptation during chronic infection in P. aeruginosa, we have examined the genomic basis of adaptation to CF-like culture conditions and to fluoroquinolone antibiotics through WGS of experimentally evolved P. aeruginosa isolates. Our primary aim is to describe the genetic changes underlying adaptation to this novel environment, and to ask how repeatable these changes are. In addition, we also investigate the genetic architecture of the costs of resistance: When antibiotic resistance evolves, how often is it costly, and what mutations underlie those costs? Our data allow us to quantify the nature and extent of parallel genomic evolution and, in so doing, provide a unique view of the variety of genetic routes taken during adaptation to a medically relevant novel environment.
Results/Discussion
Adaptation to culture environments and to ciprofloxacin
In our selection experiment, we manipulated the bacterial growth environment so as to resemble the CF lung with respect to nutrition, viscosity, and antibiotic treatment. Populations of P. aeruginosa were evolved in synthetic cystic fibrosis sputum (scfm; [30]) for 8 days in the presence or absence of ciprofloxacin (Cip) and/or mucin. Scfm is a defined medium resembling the nutritional environment of the CF lung [30]. Ciprofloxacin was added at a concentration comparable to that found in the sputum of CF patients (1 ug/ml; [31]). Mucin increases the viscosity of the culture medium, and is meant to mimic the high viscosity of CF sputum [32], [33]. In vivo, viscous sputum is thought to support the growth of P. aeruginosa in unattached biofilms, called microcolonies [26], [34], and similar structures have been observed in mucin-supplemented media (e.g., [32], [33]). Mucin was added at 10 g/L. Mucin may also act as a source of nutrients. The selection experiment comprised a fully factorial design giving four selection environments: scfm alone, scfm+Cip, scfm+mucin, and scfm+mucin+Cip; 12 replicate populations were propagated in each environment. Populations were maintained in a 37°C shaking incubator in 1.5 ml of medium, with serial transfer at a 1∶61 dilution every 24 hours, with approximately 5.9 generations of growth per day (47.5 generations in total).
Since the CF lung – and by extension laboratory media designed to mimic aspects of the CF lung – is a unique growth environment for bacteria, our evolved P. aeruginosa populations are expected to adapt to this novel habitat. Adaptation is also expected to occur in response to ciprofloxacin through the selection of mutations conferring resistance. Our experimental design allows us to disentangle these two effects, with fitness in the absence of antibiotic serving as a measure of adaptation to the growth medium, and changes in resistance to ciprofloxacin indicating adaptation to the presence of this antibiotic. Since populations may harbour extensive genetic and phenotypic variation, we measured resistance and fitness for evolved populations, as well as for a single genotype isolated from each population.
As expected, antibiotic resistance evolved in the presence of ciprofloxacin at both the population and genotype levels (Figure 1). Populations evolved in the presence of Cip showed a 32-fold to 192-fold increase in minimal inhibitory concentration (MIC) over the ancestral genotype Pa14, whereas those evolved in the absence of Cip increased MIC by no more than 2-fold. Single genotypes isolated from each population gave similar results: genotypes evolved in Cip had MICs ranging from 32-fold to 192-fold greater than the ancestor.

To detect adaptation to the growth medium we assayed the fitness of evolved populations and genotypes in the absence of antibiotic using direct, head-to-head competitions against Pa14 (see Materials and Methods). We interpret the population-level assays as a measure of the extent of adaptation achieved, since these reflect the average increase in fitness of all genotypes present at the end of the experiment. The single-genotype assays provide a measure of adaptation for the same genotypes we have sequenced (see below). Note that there will be a close correspondence between measures of fitness at the population and single-genotype levels only if the population is genetically uniform, as expected under a model of periodic strong selection. If, however, the population is genetically polymorphic, perhaps because mutation supply rates (the product of population size, N, and mutation rate, u) are high or distinct genotypes are maintained by negative frequency dependent selection, then adaptation detected at the level of the population may not be accurately predicted by assays of fitness from single genotypes.
Our results are shown in Figure 2, where the dark bars represent the extent of adaptation by entire populations and the light bars adaptation by single genotypes. Evolved populations adapted to the growth medium without antibiotic only when mucin was present in the medium. In the absence of mucin, there was either no response to selection (scfm) or a significant cost to adaptation to Cip (scfm+Cip; ANOVA: P = 2.9×10−5; Table 1). Thus, the presence of mucin in the environment affords a greater opportunity for rapid adaptation.


The single genotype fitness data are more mixed and do not correspond well with the population-level fitness assays (Figure S1), suggesting the presence of substantial amounts of genetic diversity within populations. We saw no consistent effect of mucin or of antibiotic on adaptation to the growth medium, as indicated by a lack of main effect for either of these factors by ANOVA (Table 1). There was, however, a significant interaction between medium and antibiotic (ANOVA: P = 0.013; Table 1), reflecting the observation that scfm+Cip-evolved genotypes were on average more fit than the ancestor (mean relative fitness w = 1.09/generation), whereas the scfm+mucin+Cip-evolved genotypes were on average less fit than the ancestor (mean w = 0.86/generation). This interpretation is reinforced by a lack of correlation between genotypes and populations for MIC, for which there was little correspondence between the level of resistance (Figure S2).
Taken together, these results suggest two important conclusions about short-term adaptation to a CF lung-like environment: (1) adaptation does occur, and it is driven primarily by the presence of mucin; and (2) substantial genetic diversity is likely to be present in evolving populations shortly after colonization, a result consistent with the observation that P. aeruginosa isolates from CF patients can often be highly diverse [10], [35], [36].
Whole-genome sequencing of evolved genotypes
In order to gain insight into the genetic causes of adaptation, we sequenced the genomes of the pure genotypes assayed above, with one genotype sampled from each of the 48 evolved populations (that is, a single genotype from each population evolved in scfm alone, scfm+ciprofloxacin, scfm+mucin, and scfm+mucin+ciprofloxacin), as well as of our laboratory's isolate of the ancestral strain Pa14. We obtained a median coverage of ∼56-fold per genotype (mean = 55.5; range 31.8–85.4) on the Illumina platform, using 75-bp paired-end reads. Given that a previous study suggested that 15–20-fold coverage is sufficient for identifying a modest number of mutations in laboratory selected microbial strains [37], the depth of coverage we achieved should allow us to identify all SNPs and small indels throughout most of the genome. In addition, the sequenced genomes were surveyed for large insertion/deletion events, such as mobile element insertions or excisions. We were unable to survey ∼0.53% of the genome in each strain due to low coverage (defined as less than five reads covering a given nucleotide).
Across all 48 evolved strains, we identified 98 SNPs and small indels (mean 2.04/strain) not present in the ancestor (Table S1 lists all mutations and their predicted functional consequences). These mutations represented 77 unique changes, affecting a total of 44 genes and 4 putatively intergenic regions. No large insertion/deletion events were found using BRESEQ [38]. Two genotypes, both isolated from the scfm+mucin+Cip treatment, bore lesions in mutS and were thus likely mutator strains, an inference supported by the relatively high number of mutations found in these strains (one carried 30 mutations, and the other carried 4, representing the 1st and 3rd ranked genotypes in terms of number of mutations), as well as by an extreme transition∶transversion bias amongst point mutations (all 26 point mutations found in these two strains were transitions), which is characteristic of mutS mutants [39]. If these putative mutator strains are omitted, we found 64 mutations (44 unique changes) affecting 20 genes and 1 intergenic region (Figure S3). These mutations included 41 point mutations and 23 insertion/deletions (indels).
Genotypes evolved in the presence of ciprofloxacin or mucin carried more mutations on average than genotypes not evolved with antibiotic (Figure 3). Interestingly, genotypes from the most complex environment, containing both ciprofloxacin and mucin, carried more mutations than any other environment, on average. This result is broadly consistent with the idea that the number of mutations involved in adaptation increases with the number of distinct niche dimensions in the environment, an interpretation supported by both antibiotic and presence/absence of mucin being significant predictors of the number of mutations identified (ANOVA, mutators excluded; medium: F = 8.6, P = 0.005; antibiotic: F = 111.8, P = 2×10−13).
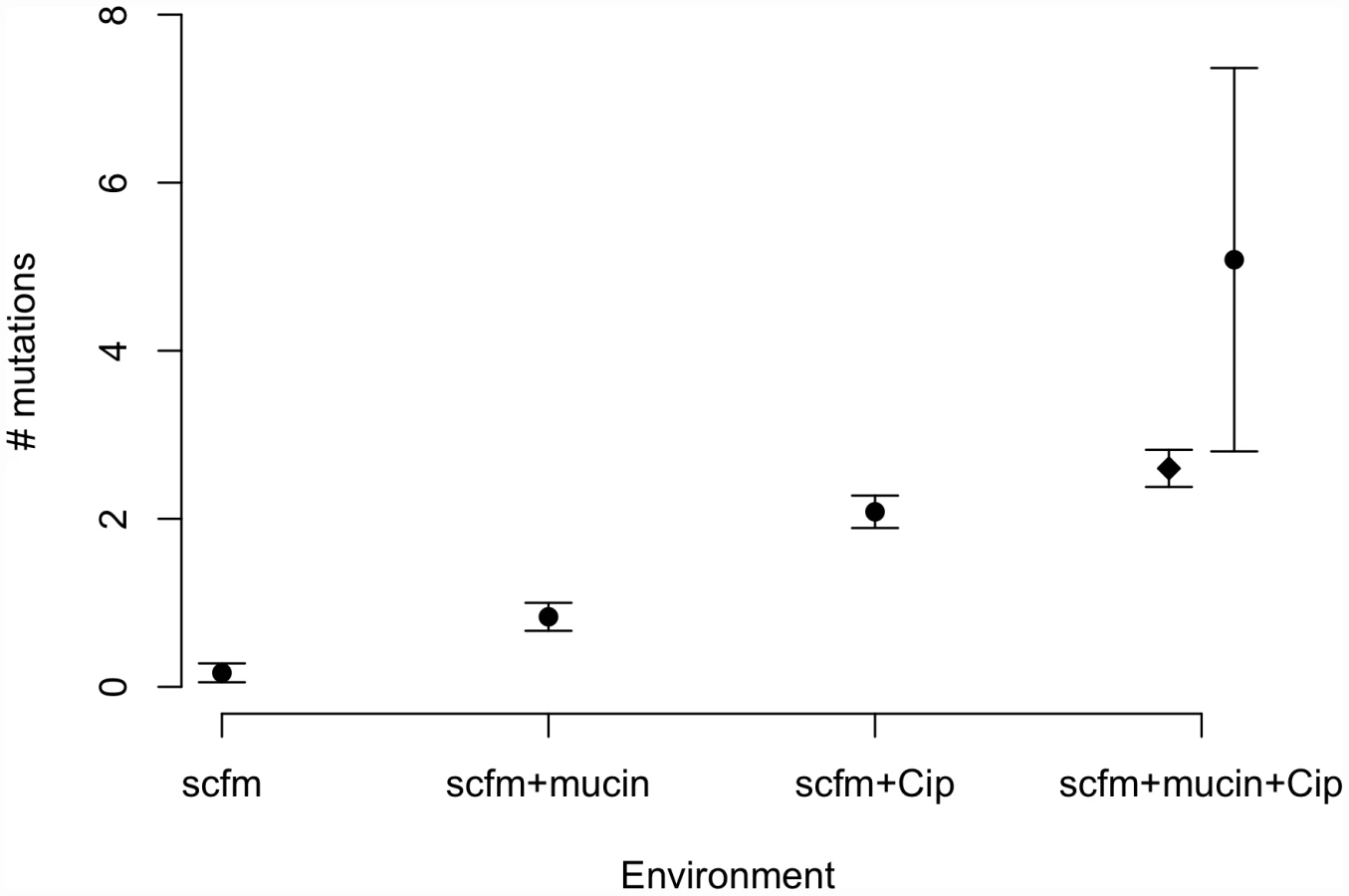
Previous studies of the genomic basis of adaptation in experimentally evolved bacterial populations have detected, on average, 1.07 mutations/100 generations (range: 0.09–3.94; [40]). The numbers of mutations observed after ∼48 generations in our antibiotic-evolved genotypes (mean 2.1 and 2.6 in scfm and scfm+mucin, respectively) are thus substantially higher than observed in previous studies. This difference probably reflects the strong selection imposed by antibiotic treatment, as opposed to the weaker selection commonly observed in resource-adaptation experiments, combined with sufficiently large population sizes to ensure the availability of multiple beneficial mutations in the same population or even the same genome [41]. Notably, the rate of accumulation of adaptive mutations observed here is consistent with theoretical models of substitution under strong selection that show expected fixation times of 50 generations or less for mutations with large selection coefficients (see Figure S4 from [42]). At the opposite end of the spectrum, very few mutations were detected in our scfm populations, with 10 genotypes bearing no mutations, and 2 genotypes carrying a single mutation each. This result is consistent with the lack of fitness response observed above (Figure 2) and is broadly consistent with the theoretical expectation under neutrality, whereby the expected fraction of 6.5 Mb genomes with zero mutations after 48 generations should be 0.73–0.97, depending on the per base pair mutation rate (taken as 1×10−10 to 1×10−9 for these estimates; [43]).
Broad patterns of nucleotide variability suggest that natural selection has played an important role in shaping the observed spectrum of mutations. Amongst the 41 point mutations observed in the non-mutator strains, 39 were nonsynonymous, 1 was synonymous, and 1 was putatively non-coding. Since approximately 1/3 of random coding changes are expected to be synonymous, the lack of synonymous mutations is consistent with natural selection favouring a substantial fraction of the observed mutations in the non-mutators. Using a randomization approach (see Materials and Methods), we find that both the excess of non-synonymous mutations, and the paucity of synonymous mutations, are highly significant (Figure S4; P<0.0005). By contrast, many more synonymous mutations were observed in the putative mutator strains, with 15 non-synonymous, 8 synonymous, 8 genic frame-shifts, and 3 intergenic mutations identified in the 2 putative mutators. The observed counts of non-synonymous and synonymous mutations in these mutators are not significantly different than expected by chance (non-synonymous: P = 0.30; synonymous: P = 0.43), suggesting that many more mutations are neutral and that these strains show a general and unbiased increase in mutation rate. The observed number of intergenic mutations (3) in the mutator strains is significantly higher than expected by chance, however (P = 0.011), suggesting that at least one of these mutations has been driven by selection.
Genetic basis of adaptation to ciprofloxacin
Observed changes in ciprofloxacin MIC and in fitness are attributable to some or all of the mutations identified by WGS. For example, in the ciprofloxacin-evolved strains, we observed multiple mutations in the known fluoroquinolone-resistance genes gyrA, gyrB, and nfxB. Amongst 24 genotypes from populations evolved in the presence of ciprofloxacin, 20 bore mutations in nfxB, 9 carried mutations in gyrB, and 4 genotypes bore gyrA mutations. Each of the gyrA mutations is a known resistance mutation affecting its quinolone-resistance determining region (QRDR; [44], [45]), with one strain carrying a T83I mutation, two with D87G, and one with a D87N mutation. The gyrB mutations were dispersed throughout this gene, with 6 different lesions amongst the 9 strains (Figure 4). In nfxB, loss of function mutations would be expected to be prevalent, since inactivation of this transcriptional repressor results in up-regulation of the MexCD-OprJ efflux pump (e.g., [46]). Concordant with this expectation, 8 distinct mutations were found in nfxB among the 20 genotypes bearing mutations (Figure 4). Interestingly, three sites were mutated in multiple strains (T39P in 3 strains, in a predicted helix-turn-helix DNA-binding domain; E146K in 5 strains; G180S in 8 strains), providing further evidence that these mutations are adaptive.

Additionally, 7 ciprofloxacin-resistant genotypes carried mutations in the gene orfN, 6 being isolated from populations evolved in scfm+Cip. orfN encodes a predicted glycosyl transferase, and is necessary for the glycosylation of type A flagellins [47]. 6 of the orfN mutants carried a single base pair deletion in a poly-G repeat, leading to the introduction of a premature stop codon. The predicted mutant protein is truncated after 53 amino acid residues (vs 338 for the wild-type protein). The seventh orfN mutant carries a single base-pair deletion in a poly-T repeat, leading to a truncated protein of 133 residues. The predicted mutant proteins are truncated before or in the glycosyl transferase domain, suggesting that the orfN mutations are likely to be loss-of-function mutations (Figure 4). While this gene has not previously been associated with fluoroquinolone resistance, this observation of extensive parallel evolution strongly suggests that orfN mutants have increased fitness in the presence of ciprofloxacin.
To obtain further evidence for an effect of orfN and other putative novel resistance mutations on Cip resistance, we surveyed isolates from evolving populations from early time points and assayed their MICs in the genetic backgrounds in which they arose. This approach allows us to sample candidate genes relatively quickly in the context in which they evolved. For orfN mutants, we sampled single colony isolates from early time points (days 3–5 of the evolution experiment) from populations where an orfN mutation was observed at day 8. Early time-point isolates were sequenced at all genes bearing a SNP at day 8, and clones bearing only an orfN mutation were selected. In this way, we identified several apparent single orfN mutants: 2 from population scfm-A5 at day 3, and 1 from population scfm-D6 at day 5. As expected each of these putative single mutants showed a 32-fold elevation in ciprofloxacin MIC in comparison to the ancestral Pa14 genotype, suggesting that orfN is a novel resistance gene.
While the observation of parallel evolution at nfxB, gyrA, gyrB, and orfN is indicative of natural selection acting on these genes, 12 of the mutations identified in the non-mutator strains appeared in only a single isolate each (Figure S5). Such mutations may represent adaptive mutations of minor effect, or they may be neutral mutations that are either segregating due to drift or have hitchhiked alongside other strongly adaptive mutations. In several cases, MIC analyses suggest a benefit to these mutations arising through increased levels of antibiotic resistance. Genotypes containing a single mutation in Pa14_32420 (encoding a putative oxidoreductase) isolated from an early time point (day 3) showed a 4-fold increase in ciprofloxacin MIC and a SNP in Pa14_46110 (encoding a predicted sodium∶solute symporter), which was the third mutation to arise in the population, had an 8-fold higher MIC than did genotypes carrying only the first two mutations (which occurred in nfxB and Pa14_23430). Thus, the evolution of quinolone resistance appears to have involved both highly parallel changes, as well as mutations specific to individual experimental populations.
Previously, Breidenstein et al. [48] conducted a screen of transposable-element insertions for novel ciprofloxacin resistance determinants. Interestingly, there is almost no overlap between between the 114 genes identified by Breidenstein et al. and the 44 genes bearing SNPs in this study. nfxB and mutS mutants were isolated in both experiments, but no other gene was found as a potential resistance factor in both studies. In addition, Breidenstein et al. identified a number of phage-related or phage-derived genes as resistance modifiers, and we found a non-coding mutation in a different cluster of phage-related genes (at position 1927375 of the Pa14 genome). The difference between these two studies is likely due to the different mechanisms that lead to resistance mutations in the two studies: transposon insertions were used by Breidenstein et al. paper, and spontaneous point mutations and indels in the current study. Importantly, the lack of overlap between the two studies is an indication that many genes potentially contribute to fluoroquinolone resistance in P. aeruginosa, and suggests that in general multiple approaches should be taken in the identification of genes underlying phenotypes of interest. Experimental evolutionary approaches, such as the one adopted here, differ from traditional mutational studies in that selection acts as an extra sieve that will weed out slow-growing mutants that, while they confer resistance, are out-competed on the way to fixation by other mutations conferring higher fitness (see [17], [18] for discussions of the use of experimental evolution as a tool for mutation discovery). While this effect of natural selection will likely eliminate some mutations of interest (especially for understanding underlying biological pathways), mutations observed under selection may be more clinically relevant due to their relatively high fitness.
What explains the prevalence of clinical resistance mutations?
Surveys of clinical samples of Pseudomonas aeruginosa often uncover a handful of genes with major effects on fluoroquinolone resistance. Most commonly, these genes are gyrA and gyrB, which encode the subunits of the fluoroquinolone target DNA gyrase, and the efflux pump regulators nfxB and mexR (e.g., [44], [46], [49], [50]). Given that mutational surveys have revealed many other genes that can confer resistance to fluoroquinolones, why is it that these four genes are repeatedly recovered from clinical samples?
One possibility is that these genes enjoy a large fitness advantage in the presence of antibiotic because they confer large increases in MIC. To test this prediction, we asked to what extent the presence or absence of mutations in classical resistance genes is a predictor of the level of ciprofloxacin resistance. As described above, many of the ciprofloxacin-evolved strains in this study bore mutations in one or several of gyrA, gyrB, and nfxB, although no mexR mutants were isolated. A linear model including selection medium (scfm+Cip or scfm+mucin+Cip) and presence or absence of mutations in gyrA, gyrB, and nfxB explains ∼87% of variation in MIC between genotypes (Table 2, Figure 5A, 5B). Under this model, mutations in nfxB, gyrA, and gyrB are associated with average MIC increases of 25.3, 3.2, and 10.9-fold, respectively. Thus, a substantial fraction of variation in the level of resistance is attributable to mutations in classical resistance genes. It should be noted that the genotypes indicated in Figure 5 are not exhaustive – for example, a given nfxB mutant on Figure 5 will also carry at least one additional mutation. Thus, variation within a genotype class (for example, the nfxB mutants) is attributable to these additional mutations.


An alternative, and not mutually exclusive, possibility is that these mutations pay little cost of resistance in the absence of antibiotic. Cost-free resistance may arise because the mutations themselves are not costly or because second-site mutations rapidly evolve that compensate for whatever cost they do incur. We tested this prediction by examining the fitness of strains bearing (or not) mutations in classical resistance genes in the absence of ciprofloxacin and found little relationship between genotype and fitness (Table 3, Figure 5C, 5D; see also Figure S6). Notably, only strains carrying nfxB mutations from the scfm+Cip environment show an increase in fitness in the absence of antibiotic (Table 3) and none of the gyrA, gyrB, or nfxB mutants from the scfm+mucin+Cip environment were significantly different from the ancestor. This result may be surprising, given that single mutations in gyrA and nfxB are typically costly [16], [51], [52] but we note that none of the strains examined here carried only a gyrA or nfxB mutation; all were at least double mutants. This result suggests that fitness in the absence of antibiotic appears to be determined or modulated by mutations in genes other than nfxB, gyrA, and gyrB. Thus cost-free resistance probably arises through second-site mutations that compensate for the costs incurred by these classical resistance genes, consistent with the results of previous studies [13], [53]–[56]. It is notable that these compensatory mutations would have to have arisen very quickly alongside or soon after resistance had evolved for them to be observed in the short time frame of our experiment.

What sorts of second-site mutations might be involved in compensating for the fitness costs of nfxB, gyrA, or gyrB resistance mutations? Our genome-wide survey of mutations provides some insight. We have found a wide range of mutations amongst the Cip-resistant genotypes sequenced in this study. These include mutations in the gene nusA encoding an elongation factor, a putative kinase encoding gene Pa14_28895, and ate1, which encodes an arginyl-trNA-protein transferase (see Table S2 for a full list).
While genotype at classical resistance genes predicts MIC (but generally not fitness), we find no evidence that the raw number of mutations present in a lineage predicts either MIC or fitness in the absence of antibiotic (data not shown). These data are consistent with a model in which classical resistance genes make particularly large contributions to MIC that can mask the smaller effects of other resistance mutations, even if these latter mutations occur first or provide additional increases to MIC or fitness.
Taken together, these results suggest that the prevalence of classical fluoroquinolone resistance mutations such as those in gyrA and nfxB in clinical isolates is due to the combination of high levels of resistance and apparent lack of costs due to second site mutations. These results are of clinical importance because they suggest that attempts to combat resistance in patient populations by stopping the use of the offending antibiotic in the hopes that drug sensitive types will replace resistant ones will often fail (e.g., [57]). Epidemiological evidence on the effectiveness of this strategy at controlling resistance is both limited and mixed [6], [58]: reducing the use of antibiotics often leads to a reduction in the frequency of resistant strains, but it rarely succeeds in eliminating them altogether [4], [5]. Our results suggest that the mechanistic reason for this failure is not that resistance mutations are cost-free but, rather, that their costs are rapidly compensated for by a diverse array of mutations elsewhere in the genome.
Genetic basis of adaptation to a CF–like culture environment
Our genomic analysis also sheds light on the genetic pathways to adaptation in CF-like conditions. Strains evolved in the most CF-like environment, scfm+mucin, often contained mutations in genes implicated in cyclic-di-GMP signalling. Elevated levels of intracellular cyclic-di-GMP are thought to induce a shift from a motile, planktonic lifestyle to a non-motile biofilm state in a variety of bacteria [27]–[29]. We suspect that increases in diguanylate cyclase activity may be adaptive in the presence of mucin, which encourages biofilm growth. Consistent with this hypothesis, three genes with putative roles in diguanylate cyclase signalling were repeatedly found mutated in the evolved strains. 9 of 24 populations (8 without ciprofloxacin, 1 with ciprofloxacin) contained isolates bearing mutations in the morA gene (Figure 4). morA encodes a predicted membrane-localized diguanylate cyclase, and serves as a negative regulator of flagellum formation [59]. In P. aeruginosa, expression of morA is required for the switch from wild-type colony morphology to the small-colony variant morphology [27], which is associated with biofilm formation in CF infections [25]. 7 distinct morA mutations – all missense point mutations - were identified in our evolved strains (Figure 4). Two scfm+mucin-evolved strains bore mutations in wspF, which encodes a regulator of the diguanylate cyclase WspR, with wspF loss-of-function mutants showing increased biofilm formation [28] and wrinkly colony morphologies in Pseudomonas fluorescens [60]. One of the wspF alleles recovered in this study is likely a loss-of-function mutation, since it encodes an early frame-shift. The second allele is a single in-frame codon deletion whose effects we cannot predict. Finally, the gene Pa14_56280, encoding another predicted diguanylate cyclase, was found to be mutated in two further scfm+mucin adapted strains.
In light of the role of cyclic-di-GMP signalling in biofilm formation [27]–[29], we predicted that our putative cyclic-di-GMP signalling mutants should show increased aggregation and biofilm formation. To test this prediction, we examined colony morphology on Coomassie blue/Congo red agar plates, which is a sensitive indicator of aggregation (e.g., [61]–[63]). Isolates bearing mutations in morA, wspF, or Pa14_56280 showed wrinkly, red morphologies in comparison to the ancestral Pa14 strain (Figure 6A–6E), consistent with increased aggregation and biofilm formation. Genotypes bearing mutations in different genes, and even different mutations in the same gene (e.g. for morA, compare Figure 6B and 6C), showed different colony morphologies, suggestive of different effects on the level, timing, and/or localization of aggregation signals, presumably cyclic-di-GMP.

The frequency with which cyclic-di-GMP signalling genes are mutated in our mucin evolved strains – with apparent consequences for aggregation and biofilm formation – strongly suggests a shared mode of adaptation towards a novel in vitro environment. This finding parallels data from clinical isolates of P. aeruginosa: Long-term adaptation of P. aeruginosa to the CF lung is characterized in part by extensive biofilm formation (e.g., [26], [64]) and the switch to a largely non-motile lifestyle is likely mediated by cyclic-di-GMP signalling (e.g., [25], [65]). Notably, wspF mutations have previously been documented in CF isolates (e.g., [10]); the current data suggest several other possible mediators of biofilm formation in clinical isolates.
Unexpectedly, all strains bearing mutations in the quinolone-resistance gene nfxB showed smooth colony morphologies (Figure 6F), a phenotype typically associated with impaired biofilm production (e.g., [61]–[63]). This observation suggests an effect of nfxB on biofilm formation and/or extracellular matrix production, which to our knowledge has not been previously reported.
Parallel evolution
The extent of parallel evolution during adaptation is of interest for a variety of reasons; evolution is in principle predictable (or not) to the extent that independent populations adapt to similar environments via the same (or different) mutations. The observation of substantial parallel evolution is also used as an indicator of strong positive selection. Previous experimental evolution studies have documented varying degrees of parallel evolution at both the phenotypic and genotypic levels [66]–[73]. We have already noted parallel evolution in response to ciprofloxacin and to mucin in our study, with multiple lineages bearing mutations in the quinolone resistance genes gyrA, gyrB, nfxB, orfN, and in the apparently mucin-adaptive genes morA, Pa14_56280, and wspF. These observations provide strong evidence that these mutations are beneficial.
How prevalent is parallel evolution in our study? To answer this we used the Jaccard index (J) to quantify the extent of within - and between-environment genic parallel evolution. For a given pair of evolved genotypes, J ranges from 0 to 1, with 0 indicating no parallel evolution and 1 indicating identity (see Materials and Methods for further details). We calculated the average Jaccard index for within - and between-environment comparisons, excluding genotypes with no SNPs, as well as mutS mutator strains (Figure S7). Within environments, was highest for the scfm+mucin genotypes, due to the high frequency of morA mutations in this environment. was intermediate for the scfm+Cip and scfm+mucin+Cip genotypes, reflecting parallel evolution at a handful of genes combined with a number of lineage-specific mutations. Between-environments, was 0, except for between the two ciprofloxacin treatments, indicative of some shared mechanisms of resistance. We rarely saw the exact same mutation evolving in parallel selection lines, suggesting that the bulk of parallel evolution in our experiment is through de novo mutations rather than the selection of rare, pre-existing variants. For the few cases where the same mutation was observed in multiple lineages, however, we note that the current study design cannot formally distinguish between these two alternatives since our experimental populations were started from a common founding culture.
We suspect that several different factors contribute to differences in the propensity for parallel evolution at different genes. Chevin et al. [74], analyzing an explicitly genomic model of trait evolution, show that the probability of parallel evolution at a given locus can depend on the locus specific mutation rate, the probability of a mutation being beneficial, and the probability of a mutation going to fixation. For some loci, e.g. nfxB, loss-of-function mutations are likely to be beneficial, and so the probability of a mutation being beneficial will be quite high (see [73] for a similar example). For other loci, such as gyrA, the probability of fixation for beneficial mutations may be high due to their large effects on MIC. Finally, in the case of orfN, where a slippage mutational mechanism is implicated by the observation of single base deletions in repeat regions, both the mutation rate and the probability of a mutation being beneficial are likely to be elevated. Thus, different genes may undergo parallel evolution for rather different reasons.
Summary and conclusions
We have studied the genomic basis of adaptation to CF-like culture conditions and to ciprofloxacin in experimentally evolved isolates of the opportunistic pathogen P. aeruginosa. Adaptation did occur to the most CF-like conditions and to the presence of ciprofloxacin, although our evolving populations are likely highly polymorphic. We observed parallel evolution at a handful of antibiotic resistance genes (gyrA, gyrB, nfxB, and orfN), as well as at putative cyclic-di-GMP signalling genes in the mucin environment. While the level of antibiotic resistance was determined largely by known resistance genes, fitness in the absence of antibiotic was not, such that there was no overall relationship between resistance and its associated costs.
These findings have several implications for understanding antibiotic resistance and pathogen evolution. First, we have identified a suite of novel ciprofloxacin resistance mutations. Our evolved antibiotic resistant isolates harbour mutations in 12 genes not previously implicated in fluoroquinolone resistance, and initial assays are consistent with effects on ciprofloxacin MIC for 3 of these genes (orfN, Pa14_46110, and Pa14_32420). Thus, experimental evolution, coupled with WGS, represents a powerful approach to identifying novel genes of interest.
Second, we find that the costs of resistance are not systematically determined by the same mutations that account for most of the variation in level of resistance (i.e., mutations in gyrA, gyrB, and nfxB). This finding suggests that whatever costs are associated with single resistance mutations are easily remediated by mutations at other loci. Moreover, these results suggest that the prevalence of these resistance mutations in clinical isolates are likely the result both of the high levels of resistance they confer and the rapid compensation of costs by second-site mutations.
Third, the finding of multiple cyclic-di-GMP mutations in the mucin environment underscores the importance of GMP-mediated biofilm formation in viscous environments, such as the CF lung.
Finally, our findings suggest that pathogen evolution has a partially repeatable genomic basis, insofar as some genes are repeatedly mutated in multiple replicate populations, while others are not. This observation has important implications for understanding pathogen evolution. Those genes that show highly parallel evolution may be particularly important in their influence on key adaptive traits governing infection or resistance to antibiotics. However, genes that are mutated only rarely are not necessarily unimportant: they often appear to have important phenotypic consequences, such as compensating for costs of resistance, and so cannot be ignored. In designing novel medical interventions, therefore, our results suggest that we would do well to focus attention first on these common targets of adaptation to the lung environment, while not losing sight of the potential importance of rare and sometimes idiosyncratic mutations that nevertheless play a major role in determining the overall fitness of the pathogen.
Materials and Methods
Experimental evolution
A single colony of P. aeruginosa strain Pa14 was grown overnight in minimal medium (NH4Cl 1 g/L, KH2PO4 3 g/L, NaCl 0.5 g/L, Na2HPO4 6.8 g/L; supplemented with CaCl2 15 mg/L, MgSO4 0.5 g/L; 0.8% dextrose as a carbon source). Forty-eight populations were founded from this progenitor by adding 25 µL overnight culture to 1.5 mL of fresh medium (media described below). An aliquot of progenitor was frozen at −80°C in glycerol. Populations were grown on an orbital shaker (150 rpm) at 37°C for 24 hours in 24-well plates. After 24 hours, each population was serially propagated by transferring 25 µL of overnight culture to 1.5 mL of fresh medium. Overnight cultures were frozen at −80°C in glycerol. Seven such transfers were conducted in total, such that approximately 50 generations of evolution occurred (∼5.9/day for 8 days).
Four selection environments were used, consisting of two different media with or without antibiotic. The media were chosen so as to examine the effects of CF sputum nutrition and viscosity on the evolution of antibiotic resistance in P. aeruginosa. Synthetic CF sputum (scfm) was prepared as described by [30]). In order to manipulate viscosity, we added 10 g/L porcine mucin (Sigma) to synthetic CF sputum (scfm+mucin)[32], [33]. For antibiotic treated populations, we used 1 µg/mL ciprofloxacin to mimic the concentration typically found in the sputum of CF patients [31].
Phenotypic analyses
For each evolved population, or for pure genotypes isolated from each population, level of resistance was assayed as the minimal inhibitory concentration (MIC) of ciprofloxacin. Overnight cultures were grown in Mueller-Hinton broth (MHB; Sigma), of which 5 µL was inoculated into 195 µL of fresh MHB with varying concentrations of ciprofloxacin in 96 well plates. MIC of the ancestor, i.e., the concentration at which growth was inhibited by 90%, was 0.05 µg/mL. For each evolved strain, we assayed growth at 0x, 0.5x, 1x, 2x, 4x, 8x, 16x, 32x, 64x, 128x, 192x, and 256x the ancestral MIC.
Fitness of each evolved population or genotype was assayed using a competitive fitness assay against a lacZ marked ancestral strain. Independent assays verified that the lacZ-marked strain did not bear a fitness cost in competitions with unmarked Pa14. Both competitors were grown for 24 hours in the competition medium. At time 0, 12.5 µL of marked ancestor and 12.5 µL of evolved strain were inoculated into 1.5 mL of fresh medium in a 24-well plate, and an aliquot was frozen at −80°C in glycerol. Following 24 hours of growth at 37°C at 150 rpm, a final aliquot was frozen at −80°C in glycerol. Serial dilutions of initial and final aliquots were grown on solid minimal media+X-gal, allowing us to determine the numbers of blue (ancestral) and white (evolved) individuals at the beginning and end of the competition. The selection coefficient s was calculated as:

Colony morphology was assayed according to [61]. Briefly, 10 µL of culture grown overnight in LB were spotted in triplicate onto tryptone plates (10 g/L) supplemented with 20 µg/ml Coomassie blue and 40 µg/ml Congo red. Plates were grown for 4 days at room temperature, after which digital photos were taken.
Whole-genome sequencing and analysis
For whole-genome sequencing, a single colony was picked from each evolved population, as well as for the ancestral Pa14 genotype. For each genotype, genomic DNA was extracted from an overnight culture using the Promega Wizard Genomic DNA Purification kit. 75-bp paired-end Illumina sequencing was performed by the Michael Smith Genome Sciences Centre, using DNA barcodes to sequence 10–12 isolates per lane. Mean coverage across all 49 genotypes was 55.5-fold at a quality score of 20 (range: 31.8–85.4).
We performed a pair-end mapping of reads on the Pa14 reference genome number NC_008463.1 using novoalign (http://novocraft.com/main/index.php). We used samtools [75] to call snps/indels, and filtered the resulting calls using the provided samtools.pl script, changing the window size for snps around indels at 5 base pairs, removing the limit on number of reads spanning a snp/indel position, and leaving the remaining parameters at their default values. We further filtered calls with quality scores below 60 for indels, and 20 for snps. To annotate the remaining snps/indels with respect to the reference genome, we used snpEff (http://snpeff.sourceforge.net/). We found results to be robust to performing a pre-mapping clipping of reads based on quality across cycles using FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/), and to performing local multiple sequence re-alignment around indels using the Genome Analysis ToolKit [76]. We also used the BRESEQ [38] pipeline as a further validation, and for its insertion/deletions detection capabilities.
Following removal of common assembly errors using custom perl scripts, a subset of SNPs was verified by Sanger sequencing of polymerase chain reaction (PCR) amplicons. For each of 31 mutations (out of 98 mutations identified in the 48 evolved strains), we amplified a 500–700 bp PCR product containing the putative SNP, and directly sequenced the PCR products (Genome Quebec, Montreal). All 31 mutations that we interrogated were successfully verified.
We used a randomization approach to determine the probability of observing by chance the distribution of non-synonymous, synonymous, and intergenic point mutations. This analysis was performed separately for putative mutator strains (two mutS mutants) and for putative non-mutator strains (the remaining 46 strains). 10 000 sets of point mutations were generated at random from the Pa14 genome sequence, maintaining the observed numbers of transitions and transversions (mutators: 30 transitions and 11 transversions; non-muators: 26 transitions and 0 transversions), and SNP effects were predicted using snpEff. Mean numbers of non-synonymous, synonymous, and intergenic mutations, as well as the 2.5% and 97.5% quantiles of the random distribution, were calculated in R [77].
Parallel evolution
The extent of parallel evolution was quantified using the Jaccard Index J. Given two sets G1 and G2 that list mutation-bearing genes found in genotypes 1 and 2, respectively,

J was calculated for all possible pairs of different genotypes amongst the 46 non-mutator strains. The average Jaccard Index was calculated within a treatment group as the mean J for all pairs of strains, where both strains were evolved under the same treatment. Similarly, was calculated between treatments A and B as the mean J for all pairs of strains, where one strain was evolved under treatment A and the second strain was evolved under treatment B.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. PasteurL (1878) La Theorie des Germes. Comptes Rendus l'Academie des Sciences 86 : 1037–1043.
2. BrownSA, PalmerKL, WhiteleyM (2008) Revisiting the host as a growth medium. Nat Rev Microbiol 6 : 657–666.
3. GarberED (1960) The host as a growth medium. Ann NY Acad Sci 88 : 1187–1194.
4. AnderssonDI (2003) Persistence of antibiotic resistant bacteria. Curr Opin Microbiol l6 : 452–456.
5. SalyersAA, Amábile-CuevasCF (1997) Why are antibiotic resistance genes so resistant to elimination? Antimicrob Agents Chemother 41 : 2321–2325.
6. LivermoreDM (2004) The need for new antibiotics. Clin Microbiol Infect 10 Suppl 4 : 1–9.
7. PingJ, KeletaL, ForbesNE, DankarS, StechoW, et al. (2011) Genomic and Protein Structural Maps of Adaptive Evolution of Human Influenza A Virus to Increased Virulence in the Mouse. PLoS ONE 6: e21740 doi:10.1371/journal.pone.0021740..
8. HolmesEC, ZhangLQ, SimmondsP, LudlamCA, BrownAJ (1992) Convergent and divergent sequence evolution in the surface envelope glycoprotein of human immunodeficiency virus type 1 within a single infected patient. Proc Natl Acad Sci USA 89 : 4835–4839.
9. MwangiMM, WuSW, ZhouY, SieradzkiK, de LencastreH, et al. (2007) Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc Natl Acad Sci USA 104 : 9451–9456.
10. SmithEE, BuckleyDG, WuZ, SaenphimmachakC, HoffmanLR, et al. (2006) Genetic adaptation by Pseudomonas aeruginosa to the airways of cystic fibrosis patients. Proc Natl Acad Sci USA 103 : 8487–8492.
11. ElenaSF, LenskiRE (2003) Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat Rev Genet 4 : 457–469.
12. BucklingA, Craig MacleanR, BrockhurstMA, ColegraveN (2009) The Beagle in a bottle. Nature 457 : 824–829.
13. HallAR, MacLeanRC (2011) Epistasis buffers the fitness effects of rifampicin - resistance mutations in Pseudomonas aeruginosa. Evolution 65 : 2370–2379.
14. KassenR (2009) Toward a general theory of adaptive radiation: insights from microbial experimental evolution. Ann NY Acad Sci 1168 : 3–22.
15. PerronGG, LeeAEG, WangY, HuangWE, BarracloughTG (2011) Bacterial recombination promotes the evolution of multi-drug-resistance in functionally diverse populations. Proc Biol Sci 279 : 1477–1484.
16. KugelbergE, LöfmarkS, WretlindB, AnderssonDI (2005) Reduction of the fitness burden of quinolone resistance in Pseudomonas aeruginosa. J Antimicrob Chemother 55 : 22–30.
17. CounagoR, ChenS, ShamooY (2006) In vivo molecular Evolution Reveals Biophysical Origins of Organismal Fitness. Mol Cell 22 : 441–449.
18. MarxCJ (2011) Evolution as an experimental tool in microbiology: ‘Bacterium, improve thyself!’. Environ Microbiol Rep 3 : 12–14.
19. BrockhurstMA, ColegraveN, RozenDE (2011) Next-generation sequencing as a tool to study microbial evolution. Mol Ecol 20 : 972–980.
20. SilbyMW, WinstanleyC, GodfreySAC, LevySB, JacksonRW (2011) Pseudomonas genomes: diverse and adaptable. FEMS Microbiol Lett 35 : 652–680.
21. Stephenson A (2008) Canadian Cystic Fibrosis Patient Data Registry Report 2008. Toronto.
22. HuseHK, KwonT, ZlosnikJEA, SpeertDP, MarcotteEM, et al. (2010) Parallel evolution in Pseudomonas aeruginosa over 39,000 generations in vivo. mBio 1: e00199–10.
23. WongA, KassenR (2011) Parallel evolution and local differentiation in quinolone resistance in Pseudomonas aeruginosa. Microbiology 157 : 937–944.
24. CiofuO, MandsbergLF, BjarnsholtT, WassermannT, HøibyN (2010) Genetic adaptation of Pseudomonas aeruginosa during chronic lung infection of patients with cystic fibrosis: strong and weak mutators with heterogeneous genetic backgrounds emerge in mucA and/or lasR mutants. Microbiology 156 : 1108–1119.
25. StarkeyM, HickmanJH, MaL, ZhangN, De LongS, et al. (2009) Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J Bacteriol 191 : 3492–3503.
26. WorlitzschD, TarranR, UlrichM, SchwabU, CekiciA, et al. (2002) Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest 109 : 317–325.
27. MeissnerA, WildV, SimmR, RohdeM, ErckC, et al. (2007) Pseudomonas aeruginosa cupA-encoded fimbriae expression is regulated by a GGDEF and EAL domain-dependent modulation of the intracellular level of cyclic diguanylate. Environ Microbiol 9 : 2475–2485.
28. HickmanJW, TifreaDF, HarwoodCS (2005) A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc Natl Acad Sci USA 102 : 14422–14427.
29. WolfeAJ, VisickKL (2008) Get the message out: cyclic-Di-GMP regulates multiple levels of flagellum-based motility. J Bacteriol 190 : 463–475.
30. PalmerKL, AyeLM, WhiteleyM (2007) Nutritional cues control Pseudomonas aeruginosa multicellular behavior in cystic fibrosis sputum. J Bacteriol 189 : 8079–8087.
31. PedersenSS, JensenT, HvidbergEF (1987) Comparative pharmacokinetics of ciprofloxacin and ofloxacin in cystic fibrosis patients. J Antimicrob Chemother 20 : 575–583.
32. FungC, NaughtonS, TurnbullL, TingpejP, RoseB, et al. (2010) Gene expression of Pseudomonas aeruginosa in a mucin-containing synthetic growth medium mimicking CF lung sputum. J Med Microbiol 1089–1100.
33. SriramuluDD, LünsdorfH, LamJS, RömlingU (2005) Microcolony formation: a novel biofilm model of Pseudomonas aeruginosa for the cystic fibrosis lung. J Med Microbiol 54 : 667–676.
34. HassettDJ, SuttonMD, SchurrMJ, HerrAB, CaldwellCC, et al. (2009) Pseudomonas aeruginosa hypoxic or anaerobic biofilm infections within cystic fibrosis airways. Trends Microbiol 17 : 130–138.
35. SibleyCD, ParkinsMD, RabinHR, DuanK, NorgaardJC, et al. (2008) A polymicrobial perspective of pulmonary infections exposes an enigmatic pathogen in cystic fibrosis patients. Proc Natl Acad Sci USA 105 : 15070–15075.
36. MowatE, PatersonS, FothergillJL, WrightEA, LedsonMJ, et al. (2011) Pseudomonas aeruginosa population diversity and turnover in cystic fibrosis chronic infections. Am J Respir Crit Care Med 183 : 1674–1679.
37. SmithDR, QuinlanAR, PeckhamHE, MakowskyK, TaoW, et al. (2008) Rapid whole-genome mutational profiling using next-generation sequencing technologies. Genome Res 18 : 1638–1642.
38. BarrickJE, YuDS, YoonSH, JeongH, OhTK, et al. (2009) Genome evolution and adaptation in a long-term experiment with Escherichia coli. Nature 461 : 1243–1247.
39. SchaaperRM, DunnRL (1987) Spectra of spontaneous mutations in Escherichia coli strains defective in mismatch correction: the nature of in vivo DNA replication errors. Proc Natl Acad Sci USA 84 : 6220–6224.
40. DettmanJR, RodrigueN, MelnykAH, WongA, BaileySF, et al. (2012) Evolutionary insight from whole-genome sequencing of experimentally evolved microbes. Mol Ecol epub Feb. 2012
41. DesaiMM, FisherDS (2007) Beneficial mutation selection balance and the effect of linkage on positive selection. Genetics 176 : 1759–1798.
42. SchoustraSE, BataillonT, GiffordDR, KassenR (2009) The properties of adaptive walks in evolving populations of fungus. PLoS Biol 7: e1000250 doi:10.1371/journal.pbio.1000250.
43. LynchM (2010) Evolution of the mutation rate. Trends Genet 26 : 345–352.
44. YoshidaH, BogakiM, NakamuraM, NakamuraS (1990) Quinolone resistance-determining region in the DNA gyrase gyrA gene of Escherichia coli. Antimicrob Agents Chemother 34 : 1271–1272.
45. YoshidaH, NakamuraM, BogakiM, NakamuraS (1990) Proportion of DNA gyrase mutants among quinolone-resistant strains of Pseudomonas aeruginosa. Antimicrob Agents Chemother 34 : 1273–1275.
46. PooleK (2005) Efflux-mediated antimicrobial resistance. J Antimicrob Chemother 56 : 20–51.
47. SchirmM, AroraSK, VermaA, VinogradovE, ThibaultP, et al. (2004) Structural and genetic characterization of glycosylation of type a flagellin in Pseudomonas aeruginosa. J Bacteriol 186 : 2523–2531.
48. BreidensteinEBM, KhairaBK, WiegandI, OverhageJ, HancockREW (2008) Complex ciprofloxacin resistome revealed by screening a Pseudomonas aeruginosa mutant library for altered susceptibility. Antimicrob Agents Chemother 52 : 4486–4491.
49. HooperDC (2001) Emerging Mechanisms of Fluoroquinolone Resistance. Emerg Infect Dis 7 : 337–341.
50. YoshidaH, BogakiM, NakamuraM, YamanakaLM, NakamuraS (1991) Quinolone resistance-determining region in the DNA gyrase gyrB gene of Escherichia coli. Antimicrob Agents Chemother 35 : 1647–1650.
51. SticklandHG, DavenportPW, LilleyKS, GriffinJL, WelchM (2010) Mutation of nfxB Causes Global Changes in the Physiology and Metabolism of Pseudomonas aeruginosa. Journal of Proteome Research 2957–2967.
52. BagelS, HüllenV, WiedemannB, HeisigP (1999) Impact of gyrA and parC mutations on quinolone resistance, doubling time, and supercoiling degree of Escherichia coli. Antimicrob Agents Chemother 43 : 868–875.
53. MarcussonLL, Frimodt-MøllerN, HughesD (2009) Interplay in the selection of fluoroquinolone resistance and bacterial fitness. PLoS Pathog 5: e1000541 doi:10.1371/journal.ppat.1000541..
54. TrindadeS, SousaA, XavierKB, DionisioF, FerreiraMG, et al. (2009) Positive epistasis drives the acquisition of multidrug resistance. PLoS Genet 5: e1000578 doi:10.1371/journal.pgen.1000578..
55. RozenDE, McGeeL, LevinBR, KlugmanKP (2007) Fitness costs of fluoroquinolone resistance in Streptococcus pneumoniae. Antimicrob Agents Chemother 51 : 412–416.
56. WardH, PerronGG, MacleanRC (2009) The cost of multiple drug resistance in Pseudomonas aeruginosa. J Evol Biol 22 : 997–1003.
57. GottesmanBS, CarmeliY, ShitritP, ChowersM (2009) Impact of quinolone restriction on resistance patterns of Escherichia coli isolated from urine by culture in a community setting. Clin Infect Dis 49 : 869–875.
58. BarbosaTM, LevySB (2000) The impact of antibiotic use on resistance development and persistence. Drug Resist Updat 3 : 303–311.
59. ChoyW-K, ZhouL, SynCK-C, ZhangL-H, SwarupS (2004) MorA defines a new class of regulators affecting flagellar development and biofilm formation in diverse Pseudomonas species. J Bacteriol 186 : 7221–7228.
60. BantinakiE, KassenR, KnightCG, RobinsonZ, SpiersAJ, et al. (2007) Adaptive divergence in experimental populations of Pseudomonas fluorescens. III. Mutational origins of wrinkly spreader diversity. Genetics 176 : 441–453.
61. FriedmanL, KolterR (2004) Genes involved in matrix formation in Pseudomonas aeruginosa PA14 biofilms. Mol Microbiol 51 : 675–690.
62. MerrittJH, HaD-G, CowlesKN, LuW, MoralesDK, et al. (2010) Specific control of Pseudomonas aeruginosa surface-associated behaviors by two c-di-GMP diguanylate cyclases. mBio 1: e00183–10.
63. DietrichLEP, TealTK, Price-WhelanA, NewmanDK (2008) Redox-active antibiotics control gene expression and community behavior in divergent bacteria. Science 321 : 1203–1206.
64. GovanJR, DereticV (1996) Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol Rev 60 : 539–574.
65. MaloneJG, JaegerT, SpanglerC, RitzD, SpangA, et al. (2010) YfiBNR mediates cyclic di-GMP dependent small colony variant formation and persistence in Pseudomonas aeruginosa. PLoS Pathog 6: e1000804 doi:10.1371/journal.ppat.1000804..
66. CrozatE, WinkworthC, GafféJ, HallinPF, RileyMA, et al. (2010) Parallel genetic and phenotypic evolution of DNA superhelicity in experimental populations of Escherichia coli. Mol Biol Evol 27 : 2113–2128.
67. WoodsR, SchneiderD, WinkworthCL, RileyMA, LenskiRE (2006) Tests of parallel molecular evolution in a long-term experiment with Escherichia coli. Proc Natl Acad Sci USA 103 : 9107–9112.
68. CunninghamCW, JengK, HustiJ, BadgettM, MolineuxIJ, et al. (1997) Parallel molecular evolution of deletions and nonsense mutations in bacteriophage T7. Mol Biol Evol 14 : 113–116.
69. BullJJ, BadgettMR, WichmanHA, HuelsenbeckJP, HillisDM, et al. (1997) Exceptional convergent evolution in a virus. Genetics 147 : 1497–1507.
70. WichmanHA, BrownCJ (2010) Experimental evolution of viruses: Microviridae as a model system. Philos Trans R Soc Lond B Biol Sci 365 : 2495–2501.
71. WichmanHA, MillsteinJ, BullJJ (2005) Adaptive molecular evolution for 13,000 phage generations: a possible arms race. Genetics 170 : 19–31.
72. RokytaDR, AbdoZ, WichmanHA (2009) The genetics of adaptation for eight microvirid bacteriophages. J Mol Evol 69 : 229–239.
73. CooperVS, SchneiderD, BlotM, LenskiRF (2001) Mechanisms causing rapid and parallel losses of ribose catabolism in evolving populations of Escherichia coli B. J Bacteriol 183 : 2834–2841.
74. ChevinL-M, MartinG, LenormandT (2010) Fisher's Model and the Genomics of Adaptation: Restricted Pleiotropy, Heterogenous Mutation, and Parallel Evolution. Evolution 64 : 3213–3231.
75. LiH, HandsakerB, WysokerA, FennellT, RuanJ, et al. (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25 : 2078–2079.
76. McKennaA, HannaM, BanksE, SivachenkoA, CibulskisK, et al. (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20 : 1297–1303.
77. R Core Development Team (2011) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Enrichment of HP1a on Drosophila Chromosome 4 Genes Creates an Alternate Chromatin Structure Critical for Regulation in this Heterochromatic Domain
- Normal DNA Methylation Dynamics in DICER1-Deficient Mouse Embryonic Stem Cells
- The NDR Kinase Scaffold HYM1/MO25 Is Essential for MAK2 MAP Kinase Signaling in
- Functional Variants in and Involved in Activation of the NF-κB Pathway Are Associated with Rheumatoid Arthritis in Japanese
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy