
SWI/SNF-Like Chromatin Remodeling Factor Fun30 Supports Point Centromere Function in
Budding yeast centromeres are sequence-defined point centromeres and are, unlike in many other organisms, not embedded in heterochromatin. Here we show that Fun30, a poorly understood SWI/SNF-like chromatin remodeling factor conserved in humans, promotes point centromere function through the formation of correct chromatin architecture at centromeres. Our determination of the genome-wide binding and nucleosome positioning properties of Fun30 shows that this enzyme is consistently enriched over centromeres and that a majority of CENs show Fun30-dependent changes in flanking nucleosome position and/or CEN core micrococcal nuclease accessibility. Fun30 deletion leads to defects in histone variant Htz1 occupancy genome-wide, including at and around most centromeres. FUN30 genetically interacts with CSE4, coding for the centromere-specific variant of histone H3, and counteracts the detrimental effect of transcription through centromeres on chromosome segregation and suppresses transcriptional noise over centromere CEN3. Previous work has shown a requirement for fission yeast and mammalian homologs of Fun30 in heterochromatin assembly. As centromeres in budding yeast are not embedded in heterochromatin, our findings indicate a direct role of Fun30 in centromere chromatin by promoting correct chromatin architecture.
Published in the journal:
. PLoS Genet 8(9): e32767. doi:10.1371/journal.pgen.1002974
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002974
Summary
Budding yeast centromeres are sequence-defined point centromeres and are, unlike in many other organisms, not embedded in heterochromatin. Here we show that Fun30, a poorly understood SWI/SNF-like chromatin remodeling factor conserved in humans, promotes point centromere function through the formation of correct chromatin architecture at centromeres. Our determination of the genome-wide binding and nucleosome positioning properties of Fun30 shows that this enzyme is consistently enriched over centromeres and that a majority of CENs show Fun30-dependent changes in flanking nucleosome position and/or CEN core micrococcal nuclease accessibility. Fun30 deletion leads to defects in histone variant Htz1 occupancy genome-wide, including at and around most centromeres. FUN30 genetically interacts with CSE4, coding for the centromere-specific variant of histone H3, and counteracts the detrimental effect of transcription through centromeres on chromosome segregation and suppresses transcriptional noise over centromere CEN3. Previous work has shown a requirement for fission yeast and mammalian homologs of Fun30 in heterochromatin assembly. As centromeres in budding yeast are not embedded in heterochromatin, our findings indicate a direct role of Fun30 in centromere chromatin by promoting correct chromatin architecture.
Introduction
The functional state of chromatin domains results from the action of multiple determinants, including histone modifications, histone variants, nonhistone proteins and nucleosome remodeling factors. The inclusion of specific histone variants is essential for the organisation of chromatin to delineate specific domains [1]. For example, histone H3 variant CENP-A (CENH3) and its orthologs characterize centromeric regions [2]. The histone H2A variant H2AZ (Htz1 in budding yeast) demarcates many promoters and boundary elements in yeast and other organisms [3]. The distribution of histones and specific histone variants, in turn, is regulated by SWI/SNF2-like ATP-dependent remodeling activities. The Fun30/SMARCAD1/Etl1 family is a poorly characterized class of SWI/SNF-like factors [4]. In mice SMARCAD1 (also referred to as ETL1) is important for normal development [5] and is implicated in pluripotency and self renewal in embryonic stem cells [6]. SMARCAD1 has a role in maintenance of silent chromatin through replication in mammalian cells [7]. In Saccharomyces cerevisiae the unique SMARCAD1 homolog Fun30 is required for silencing in heterochromatin loci [8], [9]. Fun30 has nucleosome remodeling activity in vitro and affects chromatin structure in vivo [8]–[10]. Fission yeast has three genes coding for Fun30-like factors, one of which, FFT3, has been shown to function at boundary elements, protecting heterochromatin from euchromatin invasion [11].
The analysis of centromere establishment and maintenance has provided many important insights into how various chromatin factors cooperate to assemble a very specific and essential chromatin configuration. Centromeres serve as attachment anchors for kinetochore proteins, which, in turn, interact with microtubules of the mitotic spindle (reviewed in: [12], [13]). A hallmark of all eukaryotic centromeres is the centromeric histone H3 variant CENP-A (termed Cse4 in budding yeast) that provides an essential platform for kinetochore assembly and subsequent chromosome segregation [13], [14]. In budding yeast centromeres are well defined and composed of a single Cse4-containing variant nucleosome for each chromosome, each occupying approximately 125 bases pairs comprising three regions (CDE I, CDE II, CDE III) [13], [15]–[21]. Multiple mechanisms contribute to the specific localization of Cse4, including Scm3, a Cse4-specific histone chaperone [22], [23], and the regulated Cse4 removal and degradation at extra-centromeric sites [24]–[26]. Each single point centromere is essential for viability. Therefore, these point centromere nucleosomes provide a unique model system to explore how specific chromatin configurations are assembled and maintained.
To gain insight into Fun30 function, we mapped Fun30 binding sites genome-wide and found that it bound chromatin at specific loci, particularly at the centromeres. We found that Fun30 supports chromosome segregation, and determines nucleosome positioning at many sites, including the majority of centromeres. Fun30 has a major impact on Htz1 occupancy genome-wide, including around centromeres. We propose that Fun30 assists faithful chromosome segregation by promoting a correct chromatin infrastructure at and around centromeres and limits perturbation of centromeres through transcription.
Results
Fun30 Is Recruited to Specific Genomic Loci including Centromeres
To obtain insights into the biological role of Fun30, we performed chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-Seq) to obtain a genome-wide binding profile for Fun30. We obtained more than 4.8×10+6 sequence reads - i.e. 14-fold coverage of the genome - giving us a comprehensive insight into Fun30 enrichment across the chromatin. A browser overview showed that peaks of Fun30 enrichment are found predominantly at intergenic regions (Figure 1A). Fun30 is relatively depleted within ORFs compared to intergenic sites and Fun30 peaks are predominantly over the 3′ end of genes compared to the 5′ start site (Figure 1B, Figure S1).

To further explore the binding of Fun30 to intergenic sites, we analyzed its association with genomic features (Figure 1B). Several genomic elements show preferential enrichment of Fun30 including tRNAs genes, small nuclear RNA (snRNA) genes, small nucleolar RNA (snoRNAs) genes, Long Terminal Repeats and Autonomous Replicating Sequence regions (ARSs) (Figure 1B). We also noticed telomeric repeats are enriched in Fun30 whereas subtelomeric elements X and Y′ are not (Figure 1B, left panel). Interestingly centromeric regions show the greatest enrichment of Fun30 compared to other sites (Figure 1B, right panel, Figure 1C). The enrichment of Fun30 at centromeres was confirmed by chromatin immunoprecipitation experiments followed by quantification using PCR (Figure S2).
FUN30 Deletion Leads to Upregulation of Genes Involved in Chromosome Segregation
To further understand how Fun30 might have a role in chromosome segregation and to test direct versus indirect roles, we performed expression profiling of mRNA using RNA-seq in Δfun30 deletion and control (‘wildtype’) cells. A ‘global expression profile’ analysis [27] indicated that Fun30 activity is largely required to silence genes (Figure S3). We employed a 1.5-fold cutoff value to define lists of FUN30-deletion affected genes, identifying 255 genes which were downregulated and 573 genes which were upregulated (Table S1). To investigate if genes involved in specific cellular processes were affected by FUN30 deletion, the up or down-regulated genes were submitted to Gene Ontology (GO) analysis [28] by applying a p-value cutoff (P<0.05). This analysis did not revealed significant GO terms in the group of downregulated genes. The upregulated gene group showed several genes involved in chromosome segregation (pvalue−log10 = 2.8) and meiosis (pvalue−log10 = 1.9) (Figure 2). The deletion of FUN30 caused the upregulation of genes belonging to the anaphase promoting complex (AMA1, APC1, APC2, APC4, APC2, APC9, CDC26) which is required for sister chromatid separation and exit from mitosis [29]. Other upregulated genes are components of the kinetochore or involved in its assembly, such as IML3/MCM19 [30], [31]), CNN1 [32], [33]), DAM1 [34], [35]), TID3 [36]). The hypergeometric distribution analysis revealed only a poor relationship between Fun30 recruitment and upregulated (P = 0.042) or downregulated genes (P = 0.012). Thus, the upregulation of genes involved in these specific pathways appears to be a cellular response to the absence of Fun30 function. To explore this further, we investigated a quantitative genetic interaction profile database containing 75% of all genes in S. cerevisiae [37] (http://drygin.ccbr.utoronto.ca/) and found 147 genes (SGA 0.04, P<0.05 cutoff) that have a significantly similar genetic interaction profile as Fun30. Interestingly analysis of this list of genes by Gene Ontology also reveals roles in meiosis and chromosome segregation (Figure 2). This analysis shows significant negative genetic interactions with several genes involved in the spindle checkpoint (MAD3, BUB1, BUB3) (p-values: 3.01×10−18, 1.27×10−3 and 2×10−4) and kinetochore formation (NDC10, AME1) (p-values: 8.45×10−18 and 6.25×10−3) [38], [39]. The analysis also indicates a genetic similarity to components of the TRAMP complex, which, in turn, has been linked to chromosome segregation [40]. Together, these results suggest a role of Fun30 in chromosome segregation.

FUN30 Genetically Interacts with CSE4
Deletion of Fun30 alone does not significantly affect viability, whereas overexpression of Fun30 results in chromosome segregation defects [41]. To explore the role of Fun30 in centromere function, we used the conditional cse4-1ts mutant with an amino acid substitution [42] which leads to reduced Ctf3, Ctf19, Ndc10 and Scm3 binding over the centromere at the nonpermissive temperature (38°C) and causes cell cycle arrest in G2 phase accompanied by short bipolar mitotic spindles at 38°C [23], [42], [43]. At the permissive temperature all strains grew well (Figure 3, left panels, 30°C). At a semi-permissive temperature, control (wildtype) and Δfun30 single mutant cells did not show growth defects, but growth of the cse4-1 mutant was reduced, as expected (Figure 3, right panels, 37°C, 35°C). However, the double mutant Δfun30 cse4-1 was significantly more inhibited (Figure 3, right panels, 37°C, 35°C). Growth could be rescued by expression of wildtype Fun30 in trans, but not by an ATP-binding site mutant Fun30 (Figure 3, lower right panel). These results therefore suggest a link between chromatin remodeling by Fun30 to centromeric function.

Fun30 Counteracts Segregation Defects Mediated by Transcription through Centromeres
Inhibition of transcription through centromeres is required for the de novo establishment and maintenance of centromere function [44]–[46]. Consequently, forcing transcription through a centromere disrupts its normal function [47]–[49]. To test if Fun30 has a role in chromosome segregation when centromere function is perturbed, we employed a yeast strain where transcription through CEN3 can be induced from a centromere proximal GAL1 promoter by addition of galactose, and segregation can be monitored using live cell marking of chromosome III [45] (Figure 4A). Deletion of FUN30 did not have a noticeable effect on transcription driven from the GAL1 promoter at the CEN3 (Figure S4). We examined chromosome segregation by determining if the GFP dots, marking chromosome III, are segregated into both mother and daughter cells, or remain in the mother cell or are both found in the daughter cell. In the absence of transcription through CEN3, ∼1% of control cells showed some segregation defect. Deletion of FUN30 increased the number of cells with both copies of chromosome III remaining in the mother cell ∼3 fold, indicative of a segregation defect or delay (Figure 4B). When transcription was induced, segregation defects increased dramatically in the control cells and this was further increased when FUN30 was deleted (Figure 4B). Importantly, persistent transcription over days led to a substantial loss of viability when FUN30 was deleted (Figure 4C). Therefore, it is possible that Fun30 affects events downstream of the transcription process, e.g., re-establishment of centromeric and pericentromeric chromatin.

A minichromosome loss assay with a plasmid bearing the centromere sequence of chromosome VI showed that FUN30 was required for maintenance of this chromosome through multiple cell generations (Figure 5A). Defects in various pathways, including DNA replication, could explain this. However, given the centromeric localization of Fun30, one plausible explanation is that Fun30 is involved in the formation of a functional centromere de novo on naked centromere DNA. To explore this further, we employed a conditional galactose-regulated dicentric chromosome and the fact that multiple centromeres are deleterious in yeast [46] (Figure 5B, left panel). Activation of the second centromere by suppression of transcription through it results in chromosome breakage and loss of viability [46]. Mutations affecting centromere establishment, such as deletion of CHL4, an outer kinetochore component, result in a effective suppression of this dicentric chromosome breakage [46]. We found that deletion of FUN30 promoted cell viability to almost the same extent as deletion of CHL4 (Figure 5B, right panel), suggesting that Fun30 might assist activation of a functional dicentric chromosome, and, therefore, the establishment of a centromere de novo.

Because Fun30 has been linked to gene silencing, we asked if it might be required to silence transcription within the centromeres. We tested how centromere silencing was affected in the Δfun30 mutant by measuring transcript levels for the CEN3 region where a cryptic unstable transcript has been detected upon deletion of PAP2, the gene for Trf4, a component of the TRAMP complex involved in RNA surveillance and noncoding RNA degradation [50]–[53]. We found that deletion of FUN30 increased the amount of transcript over the centromere compared to control to the same amount as seen when TRF4 was deleted (Figure 6, wt, Δfun30, Δtrf4). Double deletion of FUN30 and TRF4 increased the amount of detectable transcript even further (Figure 6, Δfun30 Δtrf4).

Together, our data suggest that Fun30 promotes faithful chromosome segregation when centromere structure is challenged and this may be linked to Fun30's role in gene silencing.
Fun30 Is Required for Normal CEN-Flanking Nucleosome Positioning and/or CEN Core Particle Structure
Next, we tried to elucidate if Fun30 promotes chromosome segregation by ensuring a correct centromere chromatin structure, in line with its binding to centromeres.
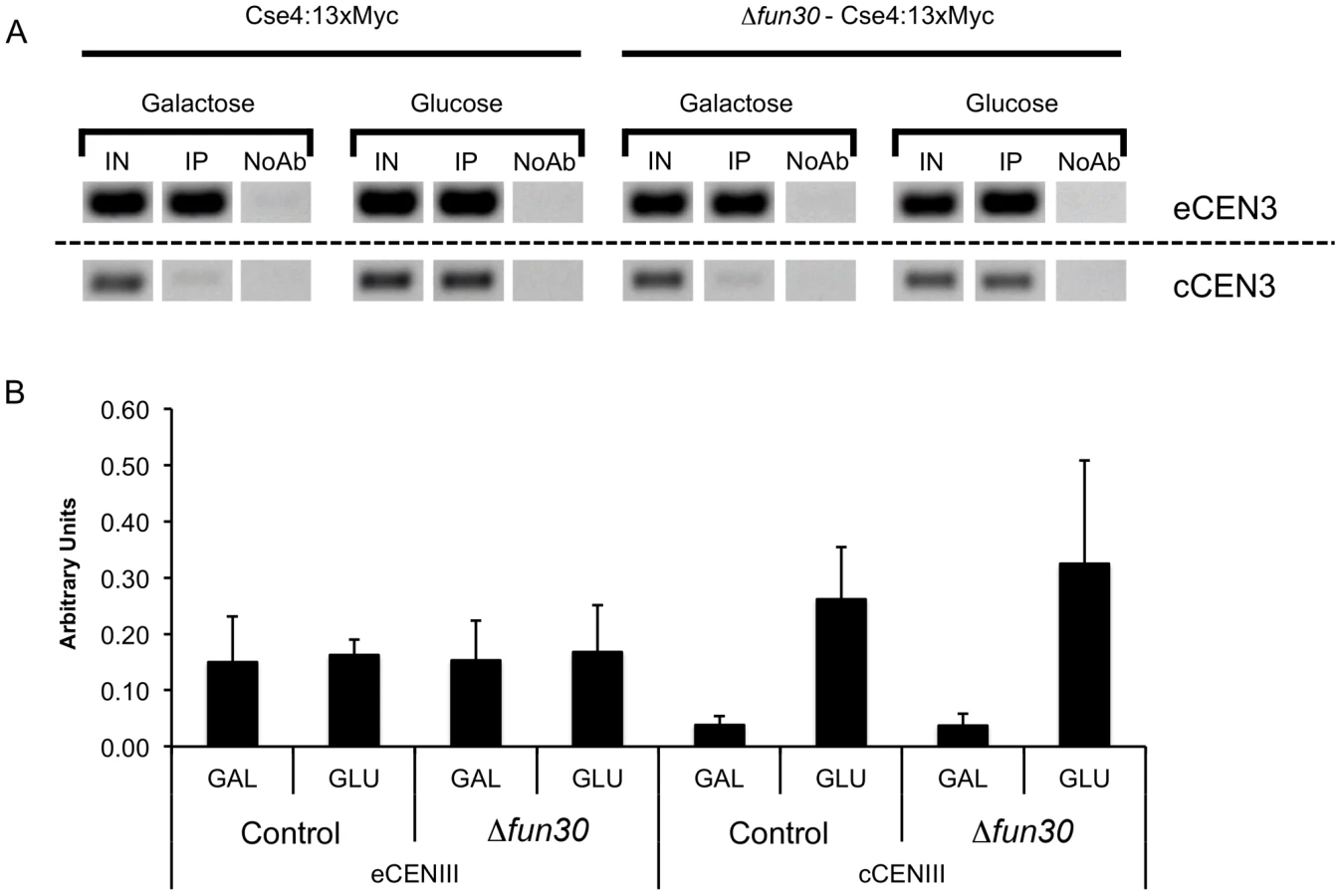
Our genome-wide analysis of Fun30 binding and the impact of FUN30 deletion on histone H3 occupancy indicates that Fun30 binds preferentially at the terminator region of genes and that its binding there is linked to a loss of histone H3 occupancy at this region (Figures S1, S5). Thus, our data indicate that Fun30 is involved in nucleosome removal in intergenic regions and suggest that Fun30 may promote occupancy of the centromeric Cse4 containing nucleosome by favoring removal of canonical nucleosomes over the centromere. To test this idea, we measured Cse4 occupancy over the endogenous, constitutive CEN3 centromere and over an induced centromere in control and fun30 deleted cells. This analysis did not show significant changes in Cse4 occupancy upon fun30-deletion (Figure 7).
Because Fun30 has previously been shown to influence nucleosome positions at the HMR and HML boundaries [8], [9], we determined if Fun30 affects nucleosome positions at the centromere. In order to examine the role of Fun30 at all CENs, we used Illumina paired-end-mode technology to sequence micrococcal nuclease (MNase)-digested chromatin samples from wildtype and Δfun30 mutant cells. Nucleosome-like particle positions were mapped as distributions of the center points of paired-end reads with an end-to-end distance of ∼150 bp. This class of size-selected paired-end reads largely defines the DNA entry - and exit-points on nucleosomes exposed by MNase digestion in the original chromatin sample. The frequency distributions of paired-read centre points, therefore, effectively estimate the frequency of nucleosome dyads, and peaks in the distributions correspond to translationally positioned nucleosome-like chromatin particles in the original genome [54], [55]. Figure 8 shows these data for areas around CEN1, CEN10, CEN11, and CEN12. At these sites both CEN flanking nucleosome positions, and/or the MNase accessibility of the CEN core particles themself are altered in the Δfun30 mutant confirming that Fun30 is required for normal CEN chromatin structure. Figure S6 shows that such changes are seen at a majority of centromeres. Our analysis indicates a broad distribution of Fun30 over centromeres with peaks of several 100 bps or more, encompassing the central centromeric nucleosome (Figure 1, Figure S6), consistent with a role of Fun30 in regulating pericentromeric and centromeric chromatin. A similar localised alteration in nucleosome positioning in Δfun30 cells was also observed at the other sites identified by Fun30 ChIP sequencing (Figure S7 shows Fun30-dependent nucleosome positioning at ARS elements). These results therefore suggest that Fun30 plays a major role in defining local nucleosome positioning at a variety of structural loci, particularly those with boundary and silencing functions, in a manner similar to its S. pombe ortholog Fft3 [11]. In conclusion, we show that Fun30 not only binds at centromeres but also affects their structure.

FUN30 Deletion Perturbs Htz1 Binding Genome-Wide, including at Centromeres
Positioned nucleosomes at yeast promoters and other genomic sites, including areas in the vicinity of centromeres, telomeric elements and ARS, are often specifically enriched for Htz1 [56]–[60]. Fun30 has been shown to be able to catalyze histone H2A/H2B dimer exchange from nucleosomes in vitro including Htz1/H2B dimers [10]. Preliminary experiments suggested that FUN30 deletion led to an increase of Htz1 at telomeric sites and within and around the silent mating locus HMR (ATV, WRW, PVW, data not shown). Therefore, we tested if FUN30 deletion affected Htz1 occupancy genome-wide, including at centromeres. An analysis of Htz1 occupancy at divergent promoters allows focusing on the effect on promoters as opposed on terminator sites from adjacent genes. This shows a dramatic loss of Htz1 around that 5′ transcription start sites upon FUN30 deletion and a corresponding increase within gene bodies (Figure 9A). A corresponding analysis of Htz1 occupancy surrounding terminator sites of convergent genes also demonstrates a drastic increase of Htz1 within the coding regions up to the 3′ terminator sites (Figure 9B). FUN30 deletion does not affect the expression from any of the histone genes that we tested, including HTZ1 (Figure S8) and did not affect total Htz1 protein levels relative to histone H3 (data not shown). We found changes in Htz1 occupancy around several centromeres, as shown for CEN10 and CEN11 in Figure 9C. While such changes are not seen at all centromeres they are evident at a majority of them (Figure S9). We see both loss of Htz1 occupancy at promoters in the vicinity of the core centromere and increased Htz1 binding at other, e.g., downstream sites. This is well illustrated with CEN10, but also evident with other centromeres, such as in the vicinity of CEN2, CEN4, CEN7, CEN11, CEN15, and CEN16. Thus, Fun30 affects not only nucleosome positioning but also Htz1 occupancy at centromeres and this may be linked to defects in centromeric silencing that we observed upon FUN30 deletion.

Discussion
Fun30 is one of the most highly conserved members of the SWI/SNF-like enzymes and homologues appear to be present in all eukaryotes [4], [8]. However, its biological role and mode of function remained poorly characterized. In this study we employed genome-wide chromatin analysis to obtain insights into how Fun30 shapes the chromatin landscape. We show that loss of FUN30 leads to alterations in nucleosome positions and occupancy at several sites that are normally occupied by Fun30, including centromeric and pericentromeric sites. Furthermore, deletion of FUN30 leads to a substantial perturbation of the binding of Htz1, a key H2A-variant histone, and this is also observed around centromeres. We provide evidence that Fun30 is involved in supporting faithful chromosome segregation through its role in determining centromeric and pericentromeric chromatin. This role of Fun30 is required when centromeric function is perturbed, e.g., by mutation of Cse4 or forcing transcription through centromeres. A recent study on a fission yeast homolog of Fun30, FFT3, shows a role for this protein in chromosome segregation and the regulation of CENP-A occupancy by promoting the formation of centromeric heterochromatin [11]. Unlike in fission yeast, budding yeast centromeres are not embedded in heterochromatin, but are surrounded by genes that are actively transcribed at some of the centromeres. Thus Fun30 has a role at centromeres that can be separated from a role in heterochromatin.
We found that Fun30 is required for normal nucleosome positioning and occupancy surrounding the centromeric nucleosome. There is also loss of nuclease protection over the centromeric nucleosomes at CEN5, CEN9 and CEN10 upon FUN30 deletion. This loss may indicate a structural alteration of the centric nucleosome, maybe because of loss of a centromeric component, or a change in the overall chromatin configuration at this site that renders chromatin more accessible. In addition, and possibly linked to its role in determining nucleosome positioning, Fun30 is required for the correct occupancy of Htz1 genome-wide, including at centromeres. It is possible that the correct chromatin structure around the core centromeric nucleosome, including fine-tuned nucleosome spacing and correct Htz1 occupancy, is required for the optimal presentation of the centromere to the kinetochore. Thus, Fun30 may support centromere function by ensuring a correct chromatin environment. Because we detect an increase of transcription through CEN3 upon Fun30 deletion, we believe that Fun30 may be involved in establishing a chromatin environment around the centromere that represses transcription over it, possibly by buffering against transcription emanating from surrounding genes. Both negative and positive roles for transcription have been reported at yeast centromeres [47], [49], [61]. The role of Fun30 in mediating correct Htz1 occupancy may therefore be linked to its role in suppressing transcriptional noise or in fine-tuning the precise level of transcriptional activity.
Fun30 appears to have a profound role in regulating Htz1 occupancy and this may be connected to its reported role in mediating silencing [8], [9]. Presently, we do not know if this is the result of direct chromatin remodeling by Fun30 or by a more indirect mechanism. For example, Fun30 may interact with and regulate components of the SWR1 complex that deposits Htz1 [62]–[64]. A direct role of Fun30 in regulating Htz1 occupancy would be consistent with its previously demonstrated in vitro histone dimer exchange activity, including H2AZ-H2B dimers [10]. It is intriguing that deletion of FUN30 has a very similar outcome with respect to Htz1 occupancy as the deletion of chromatin remodeling factor complex INO80, which also results in a loss of Htz1 over promoters and gain of Htz1 occupancy downstream in the body of genes [65]. While there is evidence that Ino80 can regulate removal of Htz1 from nucleosomes directly [65], miss-incorporation of Htz1 on deletion of FUN30 or INO80 might be a common outcome of stress on the yeast cells. What could be the connection between Htz1 occupancy and centromere function? In fission yeast H2AZ mediates suppression of antisense transcripts [66]. It is possible that in budding yeast Htz1 also controls antisense transcripts, such as cryptic un-translated transcripts emanating from promoters and that this functions limits transcription into and over centromeres. Remarkably, H2A.Z has a role in mitosis in mammalian cells and is a structural component of mammalian centromeres [67], [68].
The recent study of fission yeast Fun30 homologue FFT3 showed a role of this factor at boundary elements by evicting nucleosomes and preventing the spread of euchromatin into heterochromatin. We also found that Fun30 accumulates at putative boundary elements, such as tRNA genes (this study, [8]). Therefore, it is likely that budding yeast Fun30 has a similar role as proposed for FFT3, and this may, at least in part, be linked to the silencing defects in fun30-deleted cells that we observed previously [8].
While we did not find that deletion of FUN30 affected binding of Cse4 over the endogenous CEN3 and an inducible CEN, we show that Fun30 affects centromeric and pericentromeric chromatin (Figure 8, Figure 9) in line with its role in supporting chromosome segregation (Figure 3, Figure 4, Figure 5). Even the relatively simple centromere of budding yeast is a very complex, multi-subunit structure that, on top of this, is highly dynamic. While Cse4 is an essential component of the centromere, centromere function can be compromised at several levels, including the pericentromeric chromatin. The studies of the diverse roles of ATP-dependent nucleosome remodeling factors in supporting centromere function, as described below, make this point very clearly. Several other ATP-dependent nucleosome remodeling factors have been implicated in chromosome segregation and centromere maintenance or function in budding yeast, including the RSC complex [69]–[71] and the SWI/SNF complex [26]. RSC has been proposed to act following Cse4 recruitment and SWI/SNF has been shown to support segregation by preventing Cse4 binding to extra-centromeric sites [26], [70]. The budding yeast Ino80 complex also binds centromeres and is involved in sister chromatid cohesion, but is not required for centromeric association of kinetochore components including Cse4 [72]. In fission yeast, HRP1, a homolog of the budding yeast Chd1 protein, is required for faithful chromosome segregation and full CENP-A (CNP1) occupancy [73], [74]. Similar conclusions have been made for chicken and human Chd1 [75], but it has also been reported that Chd1 has no role in CENP-A binding in Drosophila [76]. It is not known if the budding yeast Chd1 fulfills a centromere function. Overall, a picture emerges where several remodeling factors, including Fun30, have complementary and overlapping roles in assuring correct centromere and pericentromeric chromatin structure and faithful chromosome segregation. Whether a remodeling factor exists in budding yeast that is actively involved in depositing Cse4 is an open question. Recent work from the Bloom laboratory highlights the importance of regulated histone dynamics of the pericentromeric chromatin for chromosome segregation by maintaining kinetochore structure during mitosis and implicates remodeling factors in this process [77].
ISW2 is a nucleosome remodeling factor that prevents noncoding transcription away from promoters and other nucleosome depleted regions, by limiting nucleosome free region size [78]. Fun30 may be another remodeling factor that regulates noncoding transcription. We detected an increase of nongenic transcripts by qPCR over centromeres on deletion of FUN30. However, using northern blotting upon FUN30 deletion we did not detect an increase of cryptic unstable transcripts (CUTs) at several other sites including at sites between convergent genes where we find peaks of Fun30 binding (JRM, unpublished results). Given the pronounced Fun30 binding over intergenic regions, especially between convergent genes and its link to loss of histone H3, it will be interesting to examine what is the biological role of Fun30 at these sites. A clue may be given by the fact that Fun30 also binds intergenic sites, tRNA elements, ARS sequences, snoRNA genes and centromeres. All these sites have been shown to also bind cohesin and condensin [79], [80]. Future studies will examine if Fun30 collaborates with these chromosome-organizing factors and elucidate how Fun30 identifies its specific binding sites, such as centromeres.
Materials and Methods
Cell Cultures, Plasmids
Yeast strains used in this study are listed in Table 1. Strains SC138 and SC140 were generated by integrating CSE4-myc13 driven by the CSE4 promoter into the LEU2 locus of strains KBY4001B and SC110 using BstX1 cleaved integration vector SB500, kind gift from Dr Sue Biggins. Standard budding yeast genetic techniques and media were used according to Guthrie et al. [81]. Cells were usually grown in YPD media at 30°C. For spotting and serial dilution experiments, cells were grown to mid-log phase and counted by haemocytometer. Cultures were diluted to 2.5×106 cells/ml with sterile H2O, than 1∶5 serial dilutions were performed five times. For the dicentric chromosome assay, strains containing GALCEN3 were plated for single colonies on YP galactose or glucose at 30°C as described in [46]. For the mitotic stability assay, cells transformed with pUG25 centromere plasmid (Gueldener and Hegemann, unpublished, [82]) were grown in nonselective minimal media for 12 generations and then plated on −leu or + leu plates. Plasmids used in this study are listed in Table 2.


RNA Extraction and Reverse Transcription (RT)
For RT-qPCR analysis, total RNA was extracted from mid-logarithmic phase cells (O.D.600: 0.7) in YPD media using the hot acidic phenol standard extraction protocol [83]. Total RNA was treated by DNAse I amplification grade (Invitrogen). For analysis by RT-qPCR, RNA was reverse transcribed and amplified in one-step using specific primers and iScriptTM One-Step RT-PCR Kit with SYBR Green (Bio-Rad Laboratories). Each sample was prepared in duplicate and a control without the Reverse Transcriptase was included to control for contaminating DNA.
Chromosome Segregation Assay and Microscopy
Cells were grown overnight in minimal media without uracil and leucine containing 2% glucose. Cells were then collected, washed three times in minimal media without glucose and grown in minimal media with either 2% glucose or 3% galactose for 4 h. To count the number and location of GFP dots of the LacO array proximal to CEN3 [45], cells were fixed at room temperature with 2% paraformaldehyde, 10 min directly in the media and then washed once with PBS. A Nikon Eclipse E600 equipped with a ×100 1.4 NA lens (Nikon), GFP filter, a Cascade 512B digital camera (Photometrics) and MetaMorph software (Universal Imaging Corporation) was used to determine the number of GFP dots per cell by moving the focal plane through the sample and analyzing the live digital image on the computer screen.
Chromatin Immunoprecipitation
ChIP was carried out essentially as described [8]. Overnight cultures grown in YPD at 30°C were diluted to 0.2 OD595, then grown to 0.7 OD595 at 30°C before crosslinking. Samples were crosslinked 15 min for H3, 3Myc-Htz1 and Cse4-Myc or 30 min for Fun30 detection with 1% final formaldehyde and chromatin extracts were sonicated to ∼500 bp. Triplicates or duplicate ChIP samples were validated by qPCR. Chromatin extracts were then immunoprecitated with 5 µg the Rabbit polyclonal anti-H3 (Ab1791, Abcam) for histone H3; 2 µg affinity-purified rabbit polyclonal anti-Fun30 for Fun30 [8] or with 2 µg of mouse monoclonal anti-myc (9E10, Ab32, Abcam) for Cse4-Myc and 3Myc-Htz1.
Quantitative PCR (qPCR) Analysis
Immunoprecipitated and Input DNAs were analysed by qPCR using the SYBR Green PCR Master Mix (Applied Biosystems). For immunoprecipitated DNA a 8-fold dilution was performed, input DNA was diluted 500 times; primers used are listed in Table 3. The enrichment of the protein in a specific locus was calculated as percentage of input DNA. The background binding was calculated form the no-antibody control and subtracted from the respective sample.

ChIP–Seq Library Preparation
Illumina sequencing was performed using protocols derived from [84]–[86] and the standard Illumina protocol according to the manufacturer. ChIP DNA fragments were purified and concentrated using MinElute columns (QIAGEN). Eluted DNAs from two pooled ChIP reactions of biological replicas (equal amount of DNA) were separated by electrophoresis through 2% agarose in TAE and DNA fragments with size range 150–450 bp were excised. Excised DNA fragment were purified using the QIAGEN Gel Extraction Kit and eluted in 30 µl of EB buffer (10 mM Tris-HCL, pH 8). The entire size selected ChIP reaction was then used in the end-filling and A-tailing reactions, essentially as described in the standard Illumina protocol, using standard molecular biology reagents purchased from New England Biolabs. The adapter ligation step was performed using barcoded single-end adapters synthesized by Sigma-Genosys described in [86]. Briefly, forward and reverse adapters were mixed in equimolar ratios, incubated at 95°C for 5 minutes, and allowed to anneal by using a ramp of −1°C/10 seconds until the sample reached 4°C. The ligation of adapters with DNA fragments was performed using T4 DNA ligase from Enzymatics, with incubation for 30 minute at 16°C followed by an additional 30 minutes at 22°C. Next the library was purified using the QIAGEN MinElute kit and separated in a 2% agarose/TAE gel for 1 hour at 90 V. Libraries were excised from the gel between 150 bp and 500 bp. Amplification was performed using Pfx Platinum polymerase (Invitrogen) for 15 cycles as described by Quail et al. [84]. The libraries were concentrated with QIAGEN MinElute kits and electrophoresed in a 1.9% agarose/TAE gel. Samples were quantified with SYBR Green qPCR Master Mix (Applied Biosystems) and the primers SYBR FP4 and SYBR RP7 and compared to a standard curve of phiX174 library, as described [84]. The libraries were diluted to 10 nM in EB buffer.
RNA–Seq Library Preparation
Overnight cultures were grown in YPD at 30°C were diluted to 0.2 OD595, then grown to 0.7 OD595 at 30°C. Total RNA was isolated using the hot acidic phenol method [83]. Next 10 µg aliquots were treated with DNase I amplification grade (Invitrogen) for 30 minutes at 37°C, purified by ethanol precipitation and quality-checked by 8 M Urea 6% polyacrylamide gel electrophoresis in 0.5× TBE. PolyA RNA were purified using Oligo-dT Dynabeads (Invitrogen). Purified polyA RNA samples were concentrated by ethanol precipitation and then fragmented using the Ambion RNA fragmentation kit. Samples were ethanol precipitated and the RNA was used in first strand and second strand cDNA synthesis with random hexamers at 150 ng/µl. The entire reaction was used in library generation. Libraries are summarized in Table 4.

ChIP–Seq Data Analysis
To increase the sequence yields, the Illumina sequence reads, carrying custom barcodes at the start, were re-analyzed using bareback-processing [87]. Barcodes were used to sort files and were subsequently stripped off. Alignments to the yeast genome (genome build SGD1.01, Dec 2006) were performed with Bowtie [88] using default options and ‘–best’. Next, data were loaded into Seqmonk (http://www.bioinformatics.bbsrc.ac.uk/projects/seqmonk/). Read quantification of probe regions were designed depending on purpose. For analysis of 5′ Transcription Start Site (5TSS) and 3′ Transcription Termination Site (3′TTS) regions, UTRs length were obtained from [89]. Tiled probes of 25 bp resolution were generated from −1000 to +2000 bp relative to the 5′TSS region of the gene and from −2000 to +1000 bp relative the 3′TTS regions. Quantification of the reads were corrected for the total read count and for probe length. Normalizations were performed using an input DNA library. For the analysis of 5′ intergenic regions (5′IGRs), genomic elements and 3′ intergenic regions (3′IGR) we performed gene annotations according to Figure S10. For coding genes having identified 5′UTRs and 3′UTRs according to [89], 5′IGR and 3′IGR regions comprised UTR regions plus an extended regions of 150 bases to integrate the promoter or terminator regions. Each segment was then averaged vertically for each subgroup of expression to create the average binding values along each position. Three expression categories were assigned according to their log2 signal intensities. Visualization of data was performed using the Affymetrix Integrated Genome Browser (IGB) (http://www.affymetrix.com/) and Mochiview [90]. The resulting ratio ChIP versus DNA input (from chromatin) were extracted for each probe position, defined as the center (6th) base coordinate for each 13-nucleotide probe. High-resolution cluster visualization of 5′TSS and 3′TTS were performed using MultiExperiment Viewer MeV4.5.1 [91], [92]. Correlations were performed using Venn Mapper software (http://www.gatcplatform.nl/vennmapper/) [93]. GO analysis was performed using Mochiview with multiple testing correction [90]. Annotation features were downloaded from SGD database (http://www.yeastgenome.org/), genome version SGD01.01 (Dec 2006). For determining gene orientation we also considered noncoding genomic elements, i.e. snoRNAs, snRNAs and tRNAs
Nucleosome Sequencing
S. cerevisiae used for nucleosome sequencing were EUROSCARF collection wild-type reference strain BY4742 (MATα; his3Δ1; leu2Δ0; lys2Δ0; ura3Δ0) and mutant Y10389 (MATα; leu2Δ0; lys2Δ0; ura3Δ0; Δfun30::KanMX4). Cells were grown in YPD rich medium (1% peptone, 1% yeast extract, 2% D-glucose) at 29°C to 2.6–2.8×107 nucleated cells per ml (determined by haemocytometry). Chromatin digestion and DNA preparation was performed exactly as described [55]. Briefly, un-fixed detergent-permeabilised yeast spheroplasts were incubated with MNase, and then a DNA fraction containing all MNase-digested DNA species <1 Kb (including sequences protected by sequence-specific DNA binding proteins, mono-nucleosomes and poly-nucleosomes) was released and purified. 10 µg pooled triplicate samples of DNA (Figure S11A) were processed for library preparation, size-selected on polyacrylamide gels (to preserve the size distribution of DNA fragments) and sequenced using the 76 base Illumina GAIIx paired end mode process exactly as described [55]. Raw paired sequence reads are deposited at the NCBI short read archive under accession number SRA039099.2. Paired reads were aligned to the NCBI S. cerevisiae reference genome using Bowtie 0.12.7 [88] with command line flags: -n 0 –trim3 40 –solexa.3-quals –maxins 5000 –fr -k 1 –sam. Sequences were, therefore, clipped from the 3′ end to 36 bp allowing Bowtie to return overlapping read pairs resulting from sequencing of relatively short input DNA species. 13,626,902 and 13,362,948 perfectly-aligned reads pairs were obtained from the wild-type and Δfun30 samples respectively. The paired reads were sorted into a range of classes based on the SAM format ISIZE value (difference between 5′ end of the mate read and the 5′ end of the first mapped read) plus or minus a window value of 0.2 times ISIZE as described [55]. Figure S11B shows the frequency distributions of aligned paired sequence reads from both yeast strains, and confirms that the ISIZE distributions reflect the ∼150 bp nucleosomal periodicity of the input chromatin samples. To specifically map nucleosomes, aligned paired sequence reads with an ISIZE of 150 bp±30 bp were selected, the assumption being that the DNA species falling into this size class would have been generated by protection of DNA from MNase digestion in chromatin by mono-nucleosome binding. The center value of each read-pair was calculated to represent the map position of the putative nucleosome dyad, and a genome-wide frequency distribution of the dyad positions determined and binned to 15 bp. The frequency distributions were smoothed by taking a 3 bin moving average and output in a zero-referenced, chromosome base, three-column format (chromosome number, genomic bin position, dyad frequency value) as described [55]. The frequency-distribution files for wild-type and Δfun30 mutant cells were given an .sgr file ending and rendered using the Integrated Genome Browser [94] to produce the nucleosome dyad frequency traces presented in this work.
Accession Code
ChIP-seq and RNA-seq read data has been deposited in the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress/) under accession codes E-MTAB-955, E-MTAB-956 and E-MTAB-759. Raw paired sequence reads of nucleosome mapping are deposited at the NCBI short read archive, accession number SRA039099.2.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. SarmaK, ReinbergD (2005) Histone variants meet their match. Nat Rev Mol Cell Biol 6 : 139–149.
2. Torras-LlortM, Moreno-MorenoO, AzorinF (2009) Focus on the centre: the role of chromatin on the regulation of centromere identity and function. EMBO J 28 : 2337–2348.
3. Eirin-LopezJ, AusioJ (2007) H2A.Z-Mediated Genome-Wide Chromatin Specialization. Curr Genomics 8 : 59–66.
4. FlausA, MartinDM, BartonGJ, Owen-HughesT (2006) Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res 34 : 2887–2905.
5. SchoorM, Schuster-GosslerK, RoopenianD, GosslerA (1999) Skeletal dysplasias, growth retardation, reduced postnatal survival, and impaired fertility in mice lacking the SNF2/SWI2 family member ETL1. Mech Dev 85 : 73–83.
6. HongF, FangF, HeX, CaoX, ChipperfieldH, et al. (2009) Dissecting early differentially expressed genes in a mixture of differentiating embryonic stem cells. PLoS Comput Biol 5: e1000607 doi:10.1371/journal.pcbi.1000607.
7. RowbothamSP, BarkiL, Neves-CostaA, SantosF, DeanW, et al. (2011) Maintenance of silent chromatin through replication requires SWI/SNF-like chromatin remodeler SMARCAD1. Mol Cell 42 : 285–296.
8. Neves-CostaA, WillWR, VetterAT, MillerJR, Varga-WeiszP (2009) The SNF2-family member Fun30 promotes gene silencing in heterochromatic loci. PLoS ONE 4: e8111 doi:10.1371/journal.pone.0008111.
9. YuQ, ZhangX, BiX (2011) Roles of chromatin remodeling factors in the formation and maintenance of heterochromatin structure. J Biol Chem 286 : 14659–14669.
10. AwadS, RyanD, ProchassonP, Owen-HughesT, HassanAH (2010) The Snf2 homolog Fun30 acts as a homodimeric ATP-dependent chromatin-remodeling enzyme. J Biol Chem 285 : 9477–9484.
11. StralforsA, WalfridssonJ, BhuiyanH, EkwallK (2011) The FUN30 chromatin remodeler, Fft3, protects centromeric and subtelomeric domains from euchromatin formation. PLoS Genet 7: e1001334 doi: 10.1371/journal.pgen.1001334.
12. VerdaasdonkJS, BloomK (2011) Centromeres: unique chromatin structures that drive chromosome segregation. Nat Rev Mol Cell Biol 12 : 320–332.
13. EkwallK (2007) Epigenetic control of centromere behavior. Annu Rev Genet 41 : 63–81.
14. AllshireRC, KarpenGH (2008) Epigenetic regulation of centromeric chromatin: old dogs, new tricks? Nat Rev Genet 9 : 923–937.
15. Fitzgerald-HayesM, ClarkeL, CarbonJ (1982) Nucleotide sequence comparisons and functional analysis of yeast centromere DNAs. Cell 29 : 235–244.
16. CollinsKA, CastilloAR, TatsutaniSY, BigginsS (2005) De novo kinetochore assembly requires the centromeric histone H3 variant. Mol Biol Cell 16 : 5649–5660.
17. KeithKC, BakerRE, ChenY, HarrisK, StolerS, et al. (1999) Analysis of primary structural determinants that distinguish the centromere-specific function of histone variant Cse4p from histone H3. Mol Cell Biol 19 : 6130–6139.
18. OrtizJ, StemmannO, RankS, LechnerJ (1999) A putative protein complex consisting of Ctf19, Mcm21, and Okp1 represents a missing link in the budding yeast kinetochore. Genes Dev 13 : 1140–1155.
19. ChenY, BakerRE, KeithKC, HarrisK, StolerS, et al. (2000) The N terminus of the centromere H3-like protein Cse4p performs an essential function distinct from that of the histone fold domain. Mol Cell Biol 20 : 7037–7048.
20. MeluhPB, KoshlandD (1997) Budding yeast centromere composition and assembly as revealed by in vivo cross-linking. Genes Dev 11 : 3401–3412.
21. FuruyamaS, BigginsS (2007) Centromere identity is specified by a single centromeric nucleosome in budding yeast. Proc Natl Acad Sci U S A 104 : 14706–14711.
22. MizuguchiG, XiaoH, WisniewskiJ, SmithMM, WuC (2007) Nonhistone Scm3 and histones CenH3-H4 assemble the core of centromere-specific nucleosomes. Cell 129 : 1153–1164.
23. CamahortR, LiB, FlorensL, SwansonSK, WashburnMP, et al. (2007) Scm3 is essential to recruit the histone h3 variant cse4 to centromeres and to maintain a functional kinetochore. Mol Cell 26 : 853–865.
24. HewawasamG, ShivarajuM, MattinglyM, VenkateshS, Martin-BrownS, et al. Psh1 Is an E3 Ubiquitin Ligase that Targets the Centromeric Histone Variant Cse4. Mol Cell 40 : 444–454.
25. RanjitkarP, PressMO, YiX, BakerR, MacCossMJ, et al. An E3 ubiquitin ligase prevents ectopic localization of the centromeric histone H3 variant via the centromere targeting domain. Mol Cell 40 : 455–464.
26. GkikopoulosT, SinghV, TsuiK, AwadS, RenshawMJ, et al. (2011) The SWI/SNF complex acts to constrain distribution of the centromeric histone variant Cse4. EMBO J 30 : 1919–1927.
27. Durand-DubiefM, SinhaI, Fagerstrom-BillaiF, BonillaC, WrightA, et al. (2007) Specific functions for the fission yeast Sirtuins Hst2 and Hst4 in gene regulation and retrotransposon silencing. EMBO J 26 : 2477–2488.
28. AshburnerM, BallCA, BlakeJA, BotsteinD, ButlerH, et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25 : 25–29.
29. PetersJM (2006) The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol 7 : 644–656.
30. GhoshSK, PoddarA, HajraS, SanyalK, SinhaP (2001) The IML3/MCM19 gene of Saccharomyces cerevisiae is required for a kinetochore-related process during chromosome segregation. Mol Genet Genomics 265 : 249–257.
31. PotI, MeasdayV, SnydsmanB, CagneyG, FieldsS, et al. (2003) Chl4p and iml3p are two new members of the budding yeast outer kinetochore. Mol Biol Cell 14 : 460–476.
32. BockLJ, PagliucaC, KobayashiN, GroveRA, OkuY, et al. (2012) Cnn1 inhibits the interactions between the KMN complexes of the yeast kinetochore. Nat Cell Biol 14 : 614–624.
33. SchleifferA, MaierM, LitosG, LampertF, HornungP, et al. (2012) CENP-T proteins are conserved centromere receptors of the Ndc80 complex. Nat Cell Biol 14 : 604–613.
34. LiY, BachantJ, AlcasabasAA, WangY, QinJ, et al. (2002) The mitotic spindle is required for loading of the DASH complex onto the kinetochore. Genes Dev 16 : 183–197.
35. GrishchukEL, EfremovAK, VolkovVA, SpiridonovIS, GudimchukN, et al. (2008) The Dam1 ring binds microtubules strongly enough to be a processive as well as energy-efficient coupler for chromosome motion. Proc Natl Acad Sci U S A 105 : 15423–15428.
36. WestermannS, DrubinDG, BarnesG (2007) Structures and functions of yeast kinetochore complexes. Annu Rev Biochem 76 : 563–591.
37. CostanzoM, BaryshnikovaA, BellayJ, KimY, SpearED, et al. The genetic landscape of a cell. Science 327 : 425–431.
38. EspelinCW, SimonsKT, HarrisonSC, SorgerPK (2003) Binding of the essential Saccharomyces cerevisiae kinetochore protein Ndc10p to CDEII. Mol Biol Cell 14 : 4557–4568.
39. PotI, KnocklebyJ, AneliunasV, NguyenT, Ah-KyeS, et al. (2005) Spindle checkpoint maintenance requires Ame1 and Okp1. Cell Cycle 4 : 1448–1456.
40. CastanoIB, Heath-PagliusoS, SadoffBU, FitzhughDJ, ChristmanMF (1996) A novel family of TRF (DNA topoisomerase I-related function) genes required for proper nuclear segregation. Nucleic Acids Res 24 : 2404–2410.
41. OuspenskiII, ElledgeSJ, BrinkleyBR (1999) New yeast genes important for chromosome integrity and segregation identified by dosage effects on genome stability. Nucleic Acids Res 27 : 3001–3008.
42. StolerS, KeithKC, CurnickKE, Fitzgerald-HayesM (1995) A mutation in CSE4, an essential gene encoding a novel chromatin-associated protein in yeast, causes chromosome nondisjunction and cell cycle arrest at mitosis. Genes Dev 9 : 573–586.
43. MeasdayV, HaileyDW, PotI, GivanSA, HylandKM, et al. (2002) Ctf3p, the Mis6 budding yeast homolog, interacts with Mcm22p and Mcm16p at the yeast outer kinetochore. Genes Dev 16 : 101–113.
44. GrewalSI (2010) RNAi-dependent formation of heterochromatin and its diverse functions. Curr Opin Genet Dev 20 : 134–141.
45. LacefieldS, LauDT, MurrayAW (2009) Recruiting a microtubule-binding complex to DNA directs chromosome segregation in budding yeast. Nat Cell Biol 11 : 1116–1120.
46. MythreyeK, BloomKS (2003) Differential kinetochore protein requirements for establishment versus propagation of centromere activity in Saccharomyces cerevisiae. J Cell Biol 160 : 833–843.
47. HillA, BloomK (1987) Genetic manipulation of centromere function. Mol Cell Biol 7 : 2397–2405.
48. DohenyKF, SorgerPK, HymanAA, TugendreichS, SpencerF, et al. (1993) Identification of essential components of the S. cerevisiae kinetochore. Cell 73 : 761–774.
49. OhkuniK, KitagawaK (2011) Endogenous transcription at the centromere facilitates centromere activity in budding yeast. Curr Biol 21 : 1695–1703.
50. LaCavaJ, HouseleyJ, SaveanuC, PetfalskiE, ThompsonE, et al. (2005) RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell 121 : 713–724.
51. HouseleyJ, TollerveyD (2008) The nuclear RNA surveillance machinery: the link between ncRNAs and genome structure in budding yeast? Biochim Biophys Acta 1779 : 239–246.
52. HouseleyJ, TollerveyD (2009) The many pathways of RNA degradation. Cell 136 : 763–776.
53. HouseleyJ, KotovicK, El HageA, TollerveyD (2007) Trf4 targets ncRNAs from telomeric and rDNA spacer regions and functions in rDNA copy number control. EMBO J 26 : 4996–5006.
54. FloerM, WangX, PrabhuV, BerrozpeG, NarayanS, et al. (2010) A RSC/nucleosome complex determines chromatin architecture and facilitates activator binding. Cell 141 : 407–418.
55. KentNA, AdamsS, MoorhouseA, PaszkiewiczK (2011) Chromatin particle spectrum analysis: a method for comparative chromatin structure analysis using paired-end mode next-generation DNA sequencing. Nucleic Acids Res 39: e26.
56. AlbertI, MavrichTN, TomshoLP, QiJ, ZantonSJ, et al. (2007) Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature 446 : 572–576.
57. GuillemetteB, BatailleAR, GevryN, AdamM, BlanchetteM, et al. (2005) Variant histone H2A.Z is globally localized to the promoters of inactive yeast genes and regulates nucleosome positioning. PLoS Biol 3: e384 doi:10.1371/journal.pbio.0030384.
58. ZhangH, RobertsDN, CairnsBR (2005) Genome-wide dynamics of Htz1, a histone H2A variant that poises repressed/basal promoters for activation through histone loss. Cell 123 : 219–231.
59. RaisnerRM, HartleyPD, MeneghiniMD, BaoMZ, LiuCL, et al. (2005) Histone variant H2A.Z marks the 5′ ends of both active and inactive genes in euchromatin. Cell 123 : 233–248.
60. LiB, PattendenSG, LeeD, GutierrezJ, ChenJ, et al. (2005) Preferential occupancy of histone variant H2AZ at inactive promoters influences local histone modifications and chromatin remodeling. Proc Natl Acad Sci U S A 102 : 18385–18390.
61. GrewalSI, JiaS (2007) Heterochromatin revisited. Nat Rev Genet 8 : 35–46.
62. KoborMS, VenkatasubrahmanyamS, MeneghiniMD, GinJW, JenningsJL, et al. (2004) A protein complex containing the conserved Swi2/Snf2-related ATPase Swr1p deposits histone variant H2A.Z into euchromatin. PLoS Biol 2: e131 doi:10.1371/journal.pbio.0020131.
63. KroganNJ, KeoghMC, DattaN, SawaC, RyanOW, et al. (2003) A Snf2 family ATPase complex required for recruitment of the histone H2A variant Htz1. Mol Cell 12 : 1565–1576.
64. MizuguchiG, ShenX, LandryJ, WuWH, SenS, et al. (2004) ATP-driven exchange of histone H2AZ variant catalyzed by SWR1 chromatin remodeling complex. Science 303 : 343–348.
65. Papamichos-ChronakisM, WatanabeS, RandoOJ, PetersonCL (2011) Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell 144 : 200–213.
66. ZofallM, FischerT, ZhangK, ZhouM, CuiB, et al. (2009) Histone H2A.Z cooperates with RNAi and heterochromatin factors to suppress antisense RNAs. Nature 461 : 419–422.
67. RangasamyD, GreavesI, TremethickDJ (2004) RNA interference demonstrates a novel role for H2A.Z in chromosome segregation. Nat Struct Mol Biol 11 : 650–655.
68. GreavesIK, RangasamyD, RidgwayP, TremethickDJ (2007) H2A.Z contributes to the unique 3D structure of the centromere. Proc Natl Acad Sci U S A 104 : 525–530.
69. TsuchiyaE, UnoM, KiguchiA, MasuokaK, KanemoriY, et al. (1992) The Saccharomyces cerevisiae NPS1 gene, a novel CDC gene which encodes a 160 kDa nuclear protein involved in G2 phase control. EMBO J 11 : 4017–4026.
70. HsuJM, HuangJ, MeluhPB, LaurentBC (2003) The yeast RSC chromatin-remodeling complex is required for kinetochore function in chromosome segregation. Mol Cell Biol 23 : 3202–3215.
71. TsuchiyaE, HosotaniT, MiyakawaT (1998) A mutation in NPS1/STH1, an essential gene encoding a component of a novel chromatin-remodeling complex RSC, alters the chromatin structure of Saccharomyces cerevisiae centromeres. Nucleic Acids Res 26 : 3286–3292.
72. OgiwaraH, EnomotoT, SekiM (2007) The INO80 chromatin remodeling complex functions in sister chromatid cohesion. Cell Cycle 6 : 1090–1095.
73. YooEJ, JinYH, JangYK, BjerlingP, TabishM, et al. (2000) Fission yeast hrp1, a chromodomain ATPase, is required for proper chromosome segregation and its overexpression interferes with chromatin condensation. Nucleic Acids Res 28 : 2004–2011.
74. WalfridssonJ, BjerlingP, ThalenM, YooEJ, ParkSD, et al. (2005) The CHD remodeling factor Hrp1 stimulates CENP-A loading to centromeres. Nucleic Acids Res 33 : 2868–2879.
75. OkadaM, OkawaK, IsobeT, FukagawaT (2009) CENP-H-containing complex facilitates centromere deposition of CENP-A in cooperation with FACT and CHD1. Mol Biol Cell 20 : 3986–3995.
76. PodhraskiV, Campo-FernandezB, WorleH, PiattiP, NiedereggerH, et al. (2010) CenH3/CID incorporation is not dependent on the chromatin assembly factor CHD1 in Drosophila. PLoS ONE 5: e10120 doi:10.1371/journal.pone.0010120.
77. VerdaasdonkJS, GardnerR, StephensAD, YehE, BloomK (2012) Tension-dependent nucleosome remodeling at the pericentromere in yeast. Mol Biol Cell 23 : 2560–2570.
78. WhitehouseI, RandoOJ, DelrowJ, TsukiyamaT (2007) Chromatin remodelling at promoters suppresses antisense transcription. Nature 450 : 1031–1035.
79. D'AmbrosioC, SchmidtCK, KatouY, KellyG, ItohT, et al. (2008) Identification of cis-acting sites for condensin loading onto budding yeast chromosomes. Genes Dev 22 : 2215–2227.
80. LengronneA, KatouY, MoriS, YokobayashiS, KellyGP, et al. (2004) Cohesin relocation from sites of chromosomal loading to places of convergent transcription. Nature 430 : 573–578.
81. Guthrie C, Fink GR (1991) Guide to Yeast genetics and Molecular Biology; Abelson JN, Simon MI, eds Pasadena: ACADEMIC PRESS, INC.
82. HegemannJH, KleinS, HeckS, GuldenerU, NiedenthalRK, et al. (1999) A fast method to diagnose chromosome and plasmid loss in Saccharomyces cerevisiae strains. Yeast 15 : 1009–1019.
83. CollartMA, OlivieroS (2001) Preparation of yeast RNA. Curr Protoc Mol Biol Chapter 13: Unit13 12.
84. QuailMA, SwerdlowH, TurnerDJ (2009) Improved protocols for the illumina genome analyzer sequencing system. Curr Protoc Hum Genet Chapter 18: Unit 18 12.
85. MarioniJC, MasonCE, ManeSM, StephensM, GiladY (2008) RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18 : 1509–1517.
86. LefrancoisP, EuskirchenGM, AuerbachRK, RozowskyJ, GibsonT, et al. (2009) Efficient yeast ChIP-Seq using multiplex short-read DNA sequencing. BMC Genomics 10 : 37.
87. KruegerF, AndrewsSR, OsborneCS (2011) Large scale loss of data in low-diversity illumina sequencing libraries can be recovered by deferred cluster calling. PLoS ONE 6: e16607 doi:10.1371/journal.pone.0016607.
88. LangmeadB, TrapnellC, PopM, SalzbergSL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25.
89. NagalakshmiU, WangZ, WaernK, ShouC, RahaD, et al. (2008) The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320 : 1344–1349.
90. HomannOR, JohnsonAD MochiView: versatile software for genome browsing and DNA motif analysis. BMC Biol 8 : 49.
91. SaeedAI, BhagabatiNK, BraistedJC, LiangW, SharovV, et al. (2006) TM4 microarray software suite. Methods Enzymol 411 : 134–193.
92. SaeedAI, SharovV, WhiteJ, LiJ, LiangW, et al. (2003) TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34 : 374–378.
93. SmidM, DorssersLC (2004) GO-Mapper: functional analysis of gene expression data using the expression level as a score to evaluate Gene Ontology terms. Bioinformatics 20 : 2618–2625.
94. NicolJW, HeltGA, BlanchardSGJr, RajaA, LoraineAE (2009) The Integrated Genome Browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics 25 : 2730–2731.
95. CapiaghiC, HoTV, ThomaF (2004) Kinetochores prevent repair of UV damage in Saccharomyces cerevisiae centromeres. Mol Cell Biol 24 : 6907–6918.
96. NieduszynskiCA, KnoxY, DonaldsonAD (2006) Genome-wide identification of replication origins in yeast by comparative genomics. Genes Dev 20 : 1874–1879.
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 9
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Enrichment of HP1a on Drosophila Chromosome 4 Genes Creates an Alternate Chromatin Structure Critical for Regulation in this Heterochromatic Domain
- Normal DNA Methylation Dynamics in DICER1-Deficient Mouse Embryonic Stem Cells
- The NDR Kinase Scaffold HYM1/MO25 Is Essential for MAK2 MAP Kinase Signaling in
- Functional Variants in and Involved in Activation of the NF-κB Pathway Are Associated with Rheumatoid Arthritis in Japanese
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy