
Balancing Selection at the Tomato Guardee Gene Family Maintains Variation in Strength of Pathogen Defense
Coevolution between hosts and pathogens is thought to occur between interacting molecules of both species. This results in the maintenance of genetic diversity at pathogen antigens (or so-called effectors) and host resistance genes such as the major histocompatibility complex (MHC) in mammals or resistance (R) genes in plants. In plant–pathogen interactions, the current paradigm posits that a specific defense response is activated upon recognition of pathogen effectors via interaction with their corresponding R proteins. According to the “Guard-Hypothesis,” R proteins (the “guards”) can sense modification of target molecules in the host (the “guardees”) by pathogen effectors and subsequently trigger the defense response. Multiple studies have reported high genetic diversity at R genes maintained by balancing selection. In contrast, little is known about the evolutionary mechanisms shaping the guardee, which may be subject to contrasting evolutionary forces. Here we show that the evolution of the guardee RCR3 is characterized by gene duplication, frequent gene conversion, and balancing selection in the wild tomato species Solanum peruvianum. Investigating the functional characteristics of 54 natural variants through in vitro and in planta assays, we detected differences in recognition of the pathogen effector through interaction with the guardee, as well as substantial variation in the strength of the defense response. This variation is maintained by balancing selection at each copy of the RCR3 gene. Our analyses pinpoint three amino acid polymorphisms with key functional consequences for the coevolution between the guardee (RCR3) and its guard (Cf-2). We conclude that, in addition to coevolution at the “guardee-effector” interface for pathogen recognition, natural selection acts on the “guard-guardee” interface. Guardee evolution may be governed by a counterbalance between improved activation in the presence and prevention of auto-immune responses in the absence of the corresponding pathogen.
Published in the journal:
. PLoS Genet 8(7): e32767. doi:10.1371/journal.pgen.1002813
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1002813
Summary
Coevolution between hosts and pathogens is thought to occur between interacting molecules of both species. This results in the maintenance of genetic diversity at pathogen antigens (or so-called effectors) and host resistance genes such as the major histocompatibility complex (MHC) in mammals or resistance (R) genes in plants. In plant–pathogen interactions, the current paradigm posits that a specific defense response is activated upon recognition of pathogen effectors via interaction with their corresponding R proteins. According to the “Guard-Hypothesis,” R proteins (the “guards”) can sense modification of target molecules in the host (the “guardees”) by pathogen effectors and subsequently trigger the defense response. Multiple studies have reported high genetic diversity at R genes maintained by balancing selection. In contrast, little is known about the evolutionary mechanisms shaping the guardee, which may be subject to contrasting evolutionary forces. Here we show that the evolution of the guardee RCR3 is characterized by gene duplication, frequent gene conversion, and balancing selection in the wild tomato species Solanum peruvianum. Investigating the functional characteristics of 54 natural variants through in vitro and in planta assays, we detected differences in recognition of the pathogen effector through interaction with the guardee, as well as substantial variation in the strength of the defense response. This variation is maintained by balancing selection at each copy of the RCR3 gene. Our analyses pinpoint three amino acid polymorphisms with key functional consequences for the coevolution between the guardee (RCR3) and its guard (Cf-2). We conclude that, in addition to coevolution at the “guardee-effector” interface for pathogen recognition, natural selection acts on the “guard-guardee” interface. Guardee evolution may be governed by a counterbalance between improved activation in the presence and prevention of auto-immune responses in the absence of the corresponding pathogen.
Introduction
The coevolutionary arms race between hosts and pathogens is often described as a recurrent struggle for increased resistance in hosts and evasion of recognition by pathogens [1]–[3]. The coevolutionary dynamics can be driven by negative frequency-dependent selection, leading to the maintenance of allelic diversity at genes involved in interactions between hosts and pathogens [4]–[7]. In plants, the molecular perception of pathogens and activation of defense are well understood (reviewed in [8]–[10]) and provide an ideal means to study coevolutionary processes. For interactions of plants with biotrophic pathogens, two layers of pathogen recognition and defense are commonly described: 1) the basal defense is initiated following recognition of common pathogen-associated molecular patterns (PAMPs), such as bacterial flagellin or LPS (reviewed in [11]), and 2) a specific defense response that is activated upon pathogen recognition of host-specific pathogens via gene-for-gene interactions of pathogen effectors with their corresponding resistance (R) proteins [2], [9], [12], [13]. The specific defense response typically involves a localized cell death response, called the hypersensitive response (HR), which stops the course of infection [10], [14].
The latter specific interaction between effector and R protein can be direct or indirect. Direct interactions between pathogen effectors and R proteins have been demonstrated in remarkably few cases, for example between Pita and AvrPita in the rice-Magnaporthe pathosystem [15] and between L and AvrL567 proteins in the flax-flax rust pathosystem [16]. However, the majority of interactions appear to be indirect, following the ‘Guard-Hypothesis’ [17], [18]. In this scenario, the pathogen effector is recognized through detection of its activity in the host. Specific target molecules in the host plant, the ‘guardees’, are modified by the activity of secreted pathogen effector molecules. This modification is detected by the R protein, which serves as the so-called ‘guard’, triggering downstream defense responses including HR.
Due to the complex interaction between the guardee, its guard and the pathogen effector, the guardee is expected to be subject to contrasting evolutionary forces [19], [20]. For example if pathogen pressure is high, positive selection on the effector-guardee interface could act to improve the detection of the effector in presence of the guard, or curtail damage caused by the effector. Alternatively, positive selection on the guard-guardee interface may improve pathogen triggered activation and/or prevent auto-activation of the defense response resulting in auto-immune response [21]. Balancing selection may act on the guardee-effector interface (due to frequency-dependent selection for pathogen recognition [22]) or guard-guardee interface (due to selection for defense activation [22], [23]), if pathogen pressure or the allele frequency of the corresponding effector vary in time or space. Although it has been shown that guardees exhibit high inter - and intraspecific diversity [20], [24], it is still unknown which evolutionary forces shape their genetic diversity and genomic structure.
To decipher the role of the guardee in the evolution of the plant immune system, we quantified the natural genetic variation and investigated the functional consequences of this variation at RCR3, a secreted papain-like cysteine protease, which is thought to be guarded by the R protein Cf-2 in tomato. Cf-2 confers resistance to the leaf mold pathogen Cladosporium fulvum through recognition of the fungal protease inhibitor AVR2, which physically interacts with and inhibits RCR3 ([25], reviewed in [26]). The AVR2-RCR3 interaction is thought to cause conformational changes in RCR3, which are detected by the Cf-2 protein, leading to activation of the Cf-2-mediated defense response [25], which typically involves HR.
C. fulvum is a host specific pathogen of the tomato clade [27]. Tomatoes (Solanum section Lycopersicon) form a monophyletic clade within the Solanaceae family. The section Lycopersicon includes a total of 13 species representing all described wild tomato species and the cultivated tomato S. lycopersicum, which diverged within the last 6 million years [28]. The native geographical distribution of wild tomato species ranges from Ecuador to northern Chile and these species are found across a range of diverse habitats including temperate deserts, Andean highlands and tropical rainforests in the Amazon basin [29]. Each species displays a characteristic geographical distribution pattern, which is defined by its habitat preference [30]. Hence, wild tomatoes are suitable model organisms to study adaptation to biotic and abiotic stress. Within the tomato clade, the obligate outcrossing species Solanum peruvianum exhibits the highest level of morphological and genetic diversity and has the largest and most variable habitat range including both arid and mesic habitats. Since this species harbors the greatest variation of all species in the clade of Lycopersicon, it is an ideal starting point to understand the interplay of functional diversity and natural selection. S. peruvianum diverged from its closest relatives at least 500,000 years ago [31], [32]. Adaptation to biotic factors plays an important role in evolution of this species [30], [33]–[35]. Furthermore, since the habitat range of this plant species is large and infection and transmission of pathogenic fungi such as C. fulvum are likely affected by climatic conditions, pathogen pressure may be variable in time and space.
Even though documentation of C. fulvum in wild populations of tomato is lacking, empirical studies suggest that this fungus is a natural, coevolving pathogen of wild tomato species. Wild tomato species (S. peruvianum, S. pimpinellifolium and S. habrochaites) can be infected by C. fulvum and respond with different levels of resistance and susceptibility to pathogen challenge [36]. The observed differences vary within and between these three species suggesting variability in historical pathogen pressure. Furthermore, resistance genes to C. fulvum are present and functional in these wild tomato species [37] and have been introgressed from resistant accessions into the cultivated tomato [38]. A previous study reported high diversity at the RCR3 gene among different (wild) tomato species and suggested that the elevated amino acid variation at this locus might translate into functional diversity upon pathogen challenge [24].
Here we describe the natural variation occurring at the RCR3 locus in several wild tomato species with particular focus on a set of individuals originating from a population of the species S. peruvianum. Previous studies of other resistance genes in this species indicate that pathogen pressure is a significant evolutionary force, at least in some parts of the species range [33]–[35]. Moreover, high levels of polymorphism in this species provide sufficient power for population genetic analyses. In fact, we show that nucleotide and amino acid diversity at the RCR3 locus present in the Tarapaca population of S. peruvianum reflect the total diversity observed in interspecific comparisons across the whole tomato clade [24].
Combining a population genetic with a four-pronged functional approach, we show that the evolutionary history of the RCR3 locus is characterized by balancing selection, recent gene duplication and frequent gene conversion in S. peruvianum. The RCR3 gene forms a young gene family in this species and a closely related sister species. Two differentiated sequence types are maintained within and across RCR3 loci. In contrast to other studies that find variation in pathogen recognition segregating at resistance loci [33], [39], we find evidence for variation in the activation of the defense response. Our results suggest that coevolution between the guardee and its guard rather than with the pathogen effector is the major force in the evolution of the RCR3 locus.
Results/Discussion
The RCR3 locus is duplicated in S. peruvianum and its sister species
To investigate the evolutionary history of the RCR3 locus, we cloned and sequenced RCR3 alleles from 28 individuals of multiple wild tomato species (S. chilense, S. chmielewskii, S. corneliomulleri, S. habrochaites, S. lycopersicoides, S. pennellii, S. peruvianum and S. pimpinellifolium) and the cultivated tomato S. lycopersicum (Table S2). This approach revealed that RCR3 forms a gene family with at least two paralogs in S. peruvianum and its sister species S. corneliomulleri. These paralogs are more closely related to RCR3 than to other cysteine proteases, including PIP1, which cluster in the same genomic region [40]. The duplication of the RCR3 locus appears to be restricted to S. peruvianum and S. corneliomulleri, since no evidence for a duplication event was found in the draft genome of the cultivated tomato or in the other tomato species investigated in this study. However, we cannot exclude the existence of more diverged paralogs in the other tomato species studied, which may not have been detected through our sequencing approach. The RCR3 paralogs detected in this study could not be unambiguously distinguished from one another based on sequence divergence in the RCR3 open reading frame (ORF). Therefore, we cloned and sequenced the flanking regions (FLRs) of 43 alleles from one S. peruvianum population (LA2744 from Tarapaca, Chile, described also in [34], [35]) and defined their genomic origin relative to the cultivated tomato through BLAST and phylogenetic analyses. These analyses showed consistent results: FLRs that corresponded to the orthologous RCR3 containing region of the cultivated tomato based on significant BLAST hits clustered together in the phylogenetic tree. FLRs that mapped to other genomic locations in S. lycopersicum formed distinct clusters (Figure S1). The analyses of the RCR3 FLRs revealed that the RCR3 gene was duplicated at least twice in S. peruvianum – the duplicates are named Locus A, Locus B and Locus C hereafter. All 5′ flanking regions matched the RCR3 locus from S. lycopersicum over the full sequenced length reaching 400 to 900 bp upstream of the gene. This indicates that the duplicated region extends further upstream of the RCR3 gene. In contrast, based on BLAST hits, only a portion of the 3′FLRs matched the RCR3 locus from S. lycopersicum. At approximately 580 bp downstream of the stop codon, Locus B diverges from both Locus A and the S. lycopersicum sequence (Figure 1C). This marks the likely insertion point of the duplicated RCR3 segment into a novel genomic location at the time of origin of this new duplicate. BLAST hits for the 3′FLR of Locus B alleles beyond this breakpoint mapped to a genomic region located approximately 8.2 kb downstream of the RCR3 locus in the tomato genome. Locus C is characterized by a large deletion in the 3′FLR relative to the S. lycopersicum sequence and it was not possible to map it using the draft tomato genome. The phylogenetic and BLAST analyses of the flanking sequences indicated that alleles from Locus A have the highest sequence similarity to RCR3 from other Solanum species (S. lycopersicum and S. pimpinellifolium) and sequence divergence lies within the range of the overall sequence divergence observed between S. lycopersicum and S. peruvianum (which ranges from 0.0039 to 0.0589 across 50 loci, [41]). It is therefore likely that alleles from Locus A are orthologous to the RCR3 gene in the other species, in which the RCR3 gene is not duplicated. This implies that Locus B and Locus C are more recently derived duplicates of Locus A in S. peruvianum.
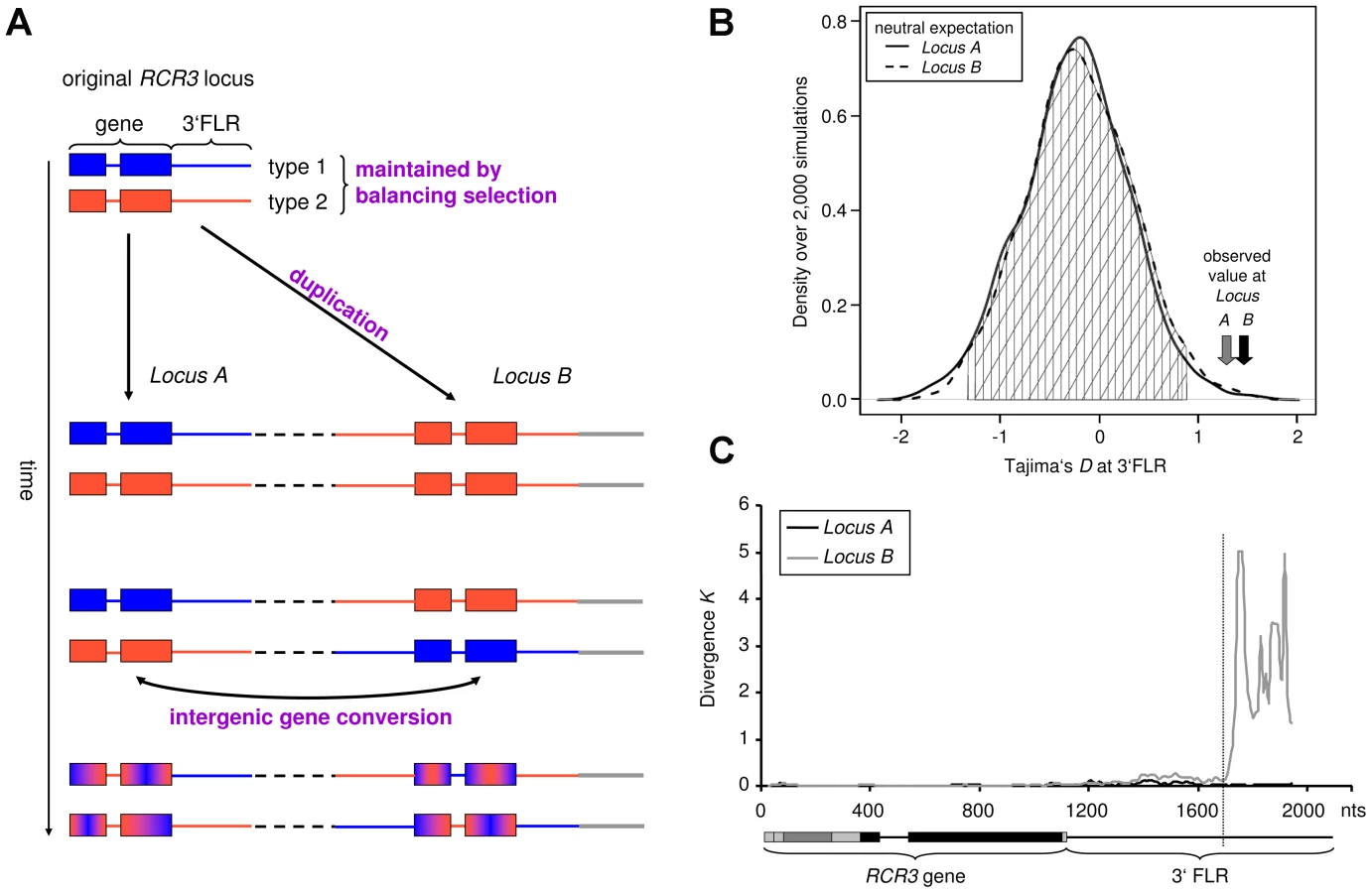
Our approach allowed us to unambiguously match 27 RCR3 sequences with their corresponding 3′FLR and therefore assign 27 of 43 RCR3 sequences to the different loci: 14 alleles to Locus A, nine alleles to Locus B and four to Locus C (Table S2). The copy number of the gene varies between individuals of S. peruvianum and no individual seemed to carry all three RCR3 copies. However, all but two tested individuals carried alleles that were assigned to two different RCR3 loci and, in most cases, at least one allele originated from Locus A (Figure S1). For population genetic analyses, only alleles that could be unambiguously assigned to their corresponding locus were used. Due to small sample size (n = 4), alleles originating from Locus C were excluded from the analysis. The genomic origin of each assigned allele is indicated by the respective letter (A, B or C) in the nomenclature used in this study.
The two RCR3 loci undergo frequent gene conversion
Gene duplication and subsequent (functional) divergence of duplicates are typical mechanisms generating diversity at genes involved in host-pathogen coevolution [42]–[44]. However, young duplicates that have not had time to diverge from one another can be homogenized by frequent intergenic gene conversion [42]. The high sequence similarity between the RCR3 ORFs and the presence of copy number variants within populations are consistent with the recent origin of the RCR3 gene family in S. peruvianum. We therefore developed an Approximate Bayesian Computation (ABC) method [32], [45] to evaluate whether gene conversion occurs and, if so, at what rate [46]. A model of evolution with gene conversion was largely favored over a model without gene conversion (Bayes Factor>1,000). The population gene conversion rate C between the RCR3 ORFs was consistently estimated to be significantly greater than zero (C = 1.08, credibility interval CI = [0.19–7.7], Figure S2, Table S3) and more than 100 times larger than the population mutation rate estimated at 14 reference loci in S. peruvianum (0.014; Table S4) or at the RCR3 gene (0.0085; Table S4).
A survey of the site frequency spectrum (SFS) of shared and private polymorphisms [47] also confirms this high rate of gene conversion (Text S1, Figure S3). We therefore suggest that functional divergence between the two copies on the protein level is unlikely at this stage of evolution because adaptive mutations appearing at one locus can be transferred to the other locus by gene conversion [46]. In contrast, signatures of gene conversion could not be detected at the 3′FLRs based on ABC analysis and the shape of the SFS (fewer shared polymorphisms, excess of fixed differences between loci, Text S2, Figure S3). This suggests that gene conversion does not happen as frequently in the 3′FLRs of the RCR3 gene as compared to the RCR3 ORFs.
Two differentiated sequence types are maintained in the RCR3 gene family by balancing selection
We analyzed sequence variation within the population to evaluate which selective forces act on the RCR3 gene. Phylogenetic analyses of the coding sequence of all assigned RCR3 alleles revealed two differentiated sequence types (Figure 2 and Figure S4), which segregate within all three loci. The haplotypic structure of the sequence types is mainly due to two different intronic sequence types and variation in linkage disequilibrium with this intron. The two sequence types are highly differentiated from one another: The index of fixation at RCR3 (FST = 0.311) is higher than the average FST between populations of S. peruvianum at eight reference genes (FST = 0.198, minimum 0.081, maximum 0.352, [48]). However, polymorphism within each sequence type is low (πsequence type 1 = 0.007, πsequence type 2 = 0.005) consistent with the maintenance of the two sequence types via long-term balancing selection.

To evaluate whether natural selection contributed to the maintenance of the distinct sequence types at the RCR3 locus, several population genetic statistics were calculated for the alleles of RCR3 Locus A and Locus B. Putative pseudogenes (see below and Text S3) were excluded from these analyses. To rule out demographic effects, which could interfere with the detection of the signature of natural selection acting at the RCR3 locus, all statistics were compared to a set of 14 reference loci that had previously been sequenced in the same individuals of S. peruvianum [49], [50].
We computed Tajima's D (DT), which summarizes the SFS of mutations in a given dataset [51]. Positive DT values indicate an excess of polymorphism at intermediate frequency, a pattern indicative of balancing selection. A sliding window analysis depicting DT across the entire RCR3 ORFs revealed regions with highly positive values. To test whether DT at the RCR3 ORF would globally deviate from neutrality, we derived the expected distributions of DT for the studied population under neutrality, taking demography and the respective gene conversion rate into account (Texts S1 and S2). The observed values at the RCR3 ORFs do not deviate significantly from neutrality (Figure S5, Table S4). However, the 3′FLRs of both loci exhibit significantly positive DT values compared to the expected neutral distribution for this population (Figure 1B, Text S2, Figure S6). Taken together, our findings suggest the following evolutionary scenario for the RCR3 loci in S. peruvianum (Figure 1A). Since both sequence types segregate at each locus and the FLRs show positive Tajima's D values, the two sequence types most likely pre-date the formation of the gene family and have been maintained by balancing selection. At the initial time of duplication, only a single sequence type would have been transferred to the new genomic region (8.2 kb downstream of Locus A in the S. lycopersicum genome), for example sequence type 1 from Locus A to Locus B. Then, following the origin of Locus B, the second sequence type (e.g. type 2) was also introduced at Locus B by recombination events such as gene conversion. High levels of recombination within the coding region (perhaps via gene conversion as described above) have subsequently intermixed the two sequence types and likely obscured the signature of balancing selection in the coding region by whittling down the region targeted by natural selection. In contrast, the signature of balancing selection is apparent in the 3′FLRs, where gene conversion does not occur as frequently.
The balanced polymorphism underlies differences in the strength of HR
The presence of two distinct sequence types differentiated especially in their intron sequences suggests three potential targets of selection: 1) selection on different regulatory motifs in the intron, 2) selection for different splicing variants or 3) selection on one or more amino acid polymorphism(s) in linkage with the intron. In silico analysis did not reveal different regulatory motifs between the two intronic sequence types, although we cannot rule out the possibility that novel regulatory motifs have been overlooked. Nucleotide sequencing of mRNA from the two sequence types did not indicate the existence of different splicing variants at the RCR3 locus. Therefore, we reason that balancing selection is most likely acting on amino acid polymorphism(s) linked to the intron.
To evaluate functional differences between sequence types at the protein level, we took a four-pronged approach. Using an over-expression vector in planta, we first evaluated whether protein accumulated for all sequence types in apoplastic fluids (AFs) of Nicotiana benthamiana. In total, 54 different allelic protein variants were chosen for these assays as follows. Eleven of these protein variants were chosen from the set of 27 alleles of S. peruvianum that could be assigned to Locus A, B or C. These eleven variants represented the protein diversity found in this set of S. peruvianum alleles. These alleles originated from all three loci and included both sequence types. The remaining 43 variants were chosen from the set of S. peruvianum alleles that could not be assigned to their corresponding locus and from closely related tomato species to maximize the amount of amino acid variation assayed. Of the total number of tested alleles, 47 were detected in AFs by Western blotting (Figure S7, Table S5). The remaining seven RCR3 proteins did not accumulate in independent expression assays, although the accumulation of mRNA was confirmed by RT-PCR (class I alleles in Figure 2, Figure S8). One of these alleles originated from S. corneliomulleri and six alleles originated from S. peruvianum (one from Locus A, one from Locus B, and four could not be assigned to their corresponding locus). In all cases in which no protein accumulated, the causative mutations (frame shifts leading to premature stop codons in five of these alleles and point mutations in the remaining two) could be identified (Text S3, Figure S9, Table S5). Since these seven alleles appear to be pseudogenes, they were excluded from population genetic analyses described above.
The second assay was a protease enzymatic assay. The activity of the RCR3 proteins in AFs was detected by Activity-based Protein Profiling (ABPP) using fluorescent DCG-04. DCG-04 is an inhibitor of papain-like cysteine proteases and reacts irreversibly and covalently to the active site cysteine of proteases in an activity-dependent manner [52]. This assay has been applied frequently to detect the activity of plant proteases and their inhibition by pathogenic protease inhibitors [24], [25], [53]–[55]. All 47 expressed RCR3 proteins could be labeled by DCG-04 to similar levels, confirming that they all are active proteases (Figure S10, Table S5).
Our third and fourth functional assays were designed to detect differences among alleles in their sensitivity to AVR2 and in their ability to elicit HR upon co-infiltration with and without AVR2 into rcr3-mutant tomato plants (cv. Money Maker Cf-2/rcr3-3, [56]). Inhibition assays based on competitive ABPP were performed to determine which RCR3 can be inhibited by the fungal protease inhibitor AVR2. Of the 47 tested RCR3 proteins, 41 (including all tested alleles from Locus A, B and C) were inhibited by AVR2 resulting in the activation of the Cf-2 dependent defense response in planta (alleles in classes III and IV in Figure 2, Figure S11, Table S5). The six alleles that failed to be inhibited by AVR2 were isolated from individuals of S. peruvianum and S. chilense (alleles in class II in Figure 2). A single nonsynonymous substitution at position 692, resulting in a change from asparagine (N) to aspartic acid (D) at position 194 in the protein (N194D), is significantly associated with this phenotypic difference (at 1% after Bonferroni correction; R2 = 0.842, P = 1.05×10−26, Figure S12). This supports previous results using site directed mutagenesis by Shabab et al. (2008) [24], which found that RCR3 alleles carrying the N194D substitution are insensitive to inhibition by AVR2. Additionally we show here that alleles that carry the N194D mutation fail to activate the defense response in planta, in the presence of AVR2 (Figures S11 and S12). Due to the large sample size used in this study (54 alleles), we had power to detect epistatic interactions between amino acid variants, such as the substitution R151Q in an allele carrying the N194D mutation (peru1954_1). This allele with the Q variant at site 151 was inhibited by AVR2, contrary to other alleles with the D variant at site 194, implicating potential epistatic interactions between these two polymorphisms (Figures S9 and S13, Table S5). Among all tested alleles that do not carry the N194D substitution only a single allele, peru7233_2, did not induce HR despite being sensitive to inhibition by AVR2 (Table S5). This allele has one amino acid difference (R138I) compared to other alleles that induced HR upon co-infiltration with AVR2 (Figure S9).
In addition to the N194D polymorphism, nucleotide polymorphisms at positions 717 (synonymous mutation) and 750 (causes amino acid difference R213S) were associated with insensitivity to inhibition by AVR2 (Figure S12, R2 = 0.254, P = 2.9×10−6; and R2 = 0.336, P = 9.8×10−8). However, since alleles that have the polymorphism at bp 750 (R213S), but not N194D, can be inhibited by AVR2 and elicit HR in planta, it is likely that the association between phenotype and sequence variation for this polymorphism is due to linkage disequilibrium. In our data set encompassing nine Solanum species, the amino acid substitution N194D was found exclusively in six individuals of S. peruvianum and S. chilense and was only represented by two alleles in the dataset used for the population genetic study. Therefore, it is unlikely that this polymorphism alone can account for the signature of balancing selection at this locus.
According to the ‘Guard-Hypothesis’ the defense response relies upon two distinct events: modification of the guardee by the effector and activation of the defense signaling through the guard molecule [17], [18]. Our approach enabled us to investigate both events. The inhibition assays did not show differential sensitivity for modification by AVR2 among most of the tested RCR3 alleles besides the alleles carrying the N194D mutation. However, our functional assay for HR activation revealed differences in the strength of activation of the defense response. Despite similar levels of sensitivity to AVR2, the tested RCR3 variants differ substantially in the strength of the defense response they elicit, with 13 protein variants (two of which originating from Locus A and two originating from Locus B) showing weaker HR (slower HR, smaller extent of cell death) compared to the others (alleles in class III in Figure 2, Figure 3, and Figure S11, Table S5). Five SNPs are correlated with phenotypic variation in the strength of the HR; one (at nucleotide position 102) remaining statistically significant at the 1% level after Bonferroni correction (Figure 3, Figures S9 and S13). All five mutations associated with this phenotype are linked with one another and with the intron despite frequent gene conversion at the locus. The most likely polymorphisms targeted by natural selection are at nucleotide positions 728 (R2 = 0.1, P = 0.015), 775 (R2 = 0.132, P = 0.0044) and 1099 (R2 = 0.146, P = 0.0026) since all three polymorphisms result in amino acid changes (I206K, Q222E and S330A). One of these polymorphisms (Q222E) is identical to an amino acid substitution that has previously been suggested to be involved in auto-necrosis due to incompatibility between RCR3 and Cf-2 ([56], Figure S13). In a putative structural model of the RCR3 protease domain the remaining two amino acid polymorphisms are located on the same protein surface as four additional positions that may be involved in the incompatibility between RCR3 and Cf-2 ([56], Figure S13). All three amino acid changes result in dissimilar amino acid substitutions and could have an impact on the protein conformation and function, while the two polymorphisms at synonymous sites (Figure 3B) may affect RCR3 transcript stability and could also be targets of selection [57]. Together with the intron, all five mutations are located in the regions of positive Tajima's D values in the sliding window analysis and likely underlie the signature of balancing selection at the RCR3 locus (Figure 3).
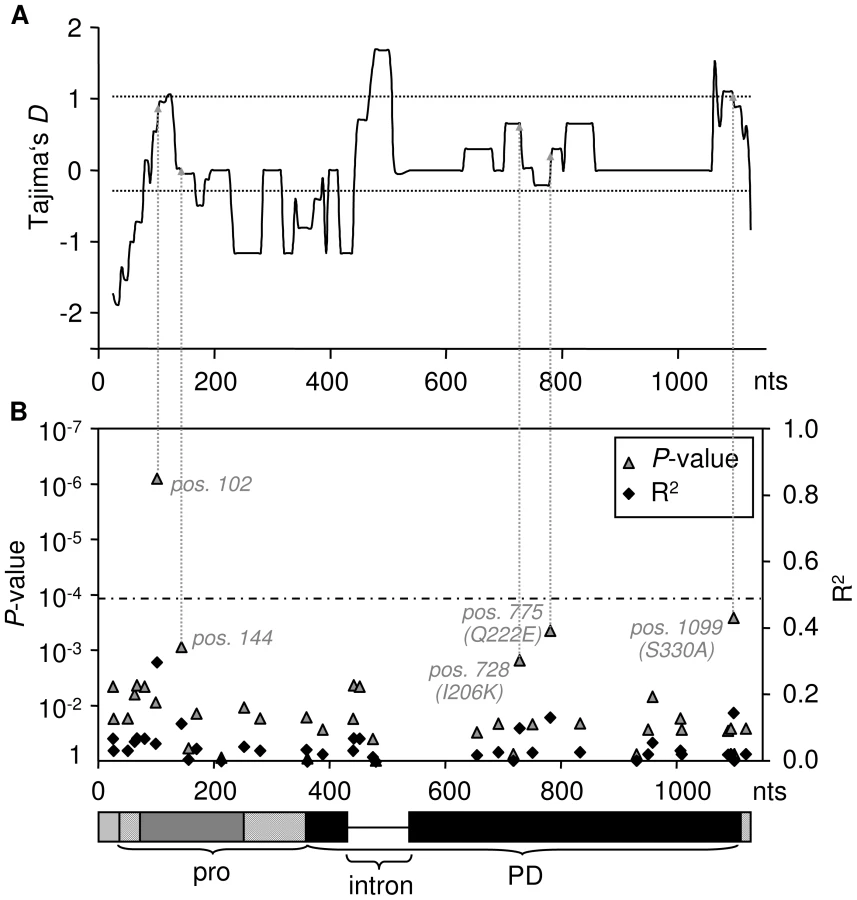
A balanced polymorphism underlying coevolution between guard and guardee?
Previous studies on R gene evolution demonstrated the maintenance of variation in pathogen recognition via balancing selection [4], [5], [34]. Our combination of functional assays, population genetic tools, computational and statistical methods allowed us to pinpoint specific amino acid polymorphisms affecting guardee function. We find that a balanced polymorphism is present at each copy of the young guardee gene family in S. peruvianum because (or in spite) of the homogenizing force of gene conversion. Balancing selection, gene duplication and gene conversion are mechanisms known to play a major role in R gene evolution [4], [42] and appear to be important in the evolution of the guardee RCR3, at least in S. peruvianum. The signature of balancing selection persists in this species and while there is no evidence that gene duplication and gene conversion are involved in evolution of RCR3 in other species, it is possible that balancing selection could play a role in the evolution of RCR3 also in other species. However, contrary to what is reported at R genes, variation in pathogen recognition does not seem to underlie the balanced polymorphism at RCR3. Instead, our results suggest that variation in the activation of the defense response, rather than effector recognition per se, underlies the balanced polymorphism. Two alternative scenarios of the evolution of RCR3 could explain these observations:
1) The diversity detected at the RCR3 locus could be due to coevolution with allelic types of AVR2 or with other pathogen effectors not included in this study. In our assay, the phenotypic response to a single allelic type of AVR2 was similar in all but six tested RCR3 alleles. Therefore, functional differences between the different RCR3 alleles regarding interaction with other allelic variants of AVR2 are improbable. Furthermore, the polymorphisms underlying the signature of balancing selection were not associated with phenotypic variation in AVR2 sensitivity. Therefore, balancing selection for differential recognition of AVR2 alone cannot account for the maintenance of the two functional types at RCR3.
RCR3 can be targeted by other pathogen effectors, including EPIC1/2B which are secreted by the oomycete Phytophthora infestans [58]. These effectors are protease inhibitors and are thought to target cysteine proteases similar to RCR3 close to the substrate binding groove [59]. Unlike the N194D mutation, which has been shown to have indeed an effect on the interaction between RCR3 and AVR2, the polymorphisms underlying the signature of balancing selection are not located close to the putative site of interaction between RCR3 and these effectors. It is therefore unlikely that the observed signature of balancing selection is due to coevolution between RCR3 and other protease inhibitors. Note however, that the molecular and structural details of the interaction between RCR3 and these other effectors and its role in disease resistance are not yet well-understood. It will be of great interest to study the coevolution between RCR3 and its full effector repertoire once their roles in disease resistance have been resolved.
2) Alternatively, variation could be maintained at the RCR3 locus through coevolution at the interface between guardee and interacting host molecules and involve balancing selection for resistance/susceptibility at the level of defense signal activation. Balanced polymorphisms for resistance and susceptibility due to a potential cost of resistance and/or ecological/epidemiological factors have been studied theoretically [22] and empirically [4], [60] at R genes. To our knowledge, this study provides first evidence that this mechanism can also drive guardee evolution. In the case of the RCR3 gene, a potential cost of resistance could be the result of coevolution with its guard Cf-2, which also exists as a gene family in wild tomatoes (S. pimpinellifolium; [61], [62]). The interaction between RCR3 and Cf-2 requires a precise matching between allelic variants. A mismatch between allelic variants from the closely related species S. lycopersicum (RCR3esc) and S. pimpinellifolium (RCR3pim) results in an auto-necrotic response [56] and may be an example of Dobzhansky-Muller incompatibility between tomato species [63]. One of the amino acid changes differing between RCR3pim and RCR3esc and potentially contributing to the reported incompatibility between RCR3 and Cf-2 (Q222E, [56]) is associated with the attenuated HR phenotype observed in this study. The remaining amino acid changes associated with this phenotype are not identical to, but are located on the same protein surface as the potential causative mutations of the RCR3-Cf-2 incompatibility (Figure S13). The amino acid changes underlying the balanced polymorphism at the RCR3 locus and causing differences in the strength of HR therefore likely play a role in the interaction between RCR3 and Cf-2. Different combinations of RCR3-Cf-2 allelic variants might thus result in differential activation of the defense response.
For practical reasons, we tested the RCR3 proteins in standard S. lycopersicum backgrounds. Since we did not conduct our assays in S. peruvianum, we cannot exclude the possibility that RCR3 and Cf-2 function differently in this species. However, the fact that RCR3 alleles from S. peruvianum do activate resistance in the presence of Cf-2 alleles in the S. lycopersicum background as expected from previous studies in S. lycopersicum and S. pimpinellifolium suggests that this interaction is conserved throughout the tomato clade.
Furthermore, even if the RCR3-Cf-2 interaction is not conserved in S. peruvianum, RCR3 may be coevolving with other host endogenous molecules which could explain the pattern of variation observed at RCR3.
Since in our study all RCR3 alleles were tested in an identical genetic background, some alleles may not be matched with their optimal Cf-2 partner, explaining the observed attenuated response for some pairings of RCR3s with Cf-2. However, we are confident that the different observed HR phenotypes are not an artifact of using a particular Cf-2 protein, because both RCR3 types are maintained by balancing selection. In nature, while an attenuated response due to weaker interaction between guard and guardee may result in reduced resistance in the presence of the pathogen, these alleles may be selectively advantageous in the absence of the pathogen, because they carry a lower cost and/or risk for activation of auto-immunity [23]. Therefore, the optimal RCR3 allele will depend upon this delicate balance between sufficient activation in the presence of the pathogen, but limited risk for auto-activation in the absence of the pathogen, explaining why both RCR3 types segregate within a single population. The optimization of defense activation may be a very important component of guard-guardee coevolution, especially when the selection pressure by the pathogen is variable in time and space.
Materials and Methods
Plant material and DNA sequencing
For population genetic and functional analyses, the ORF of the RCR3 gene was cloned and amplified from genomic DNA from eleven heterozygous individuals of S. peruvianum (accession LA2744 from Tarapaca, Chile), collected by Charles M. Rick (Table S2). Seeds from different field collected plants were grown under standard greenhouse conditions in Davis, CA. DNA was isolated using the CTAB method (Doyle and Doyle, 1987). Alleles from single individuals from eight additional species of Solanum were cloned and sequenced and their RCR3 alleles were functionally tested (Table S2). These species included: S. peruvianum (accessions LA1954 and LA0446), S. chilense (accessions LA2748, LA1930 and LA1958), S. corneliomulleri (accessions LA1274 and LA1973), S. pimpinellifolium (accession LA0400), S. lycopersicum (cv. VFNT Cherry and cv. Rio Grande), S. chmielewskii (accession LA3653), S. habrochaites (accession LA1777), S. pennellii (accessions LA0716 and LA3791). For outgroup comparisons, the RCR3 gene from S. lycopersicoides (accession LA2951) was sequenced and tested. All accessions were obtained from the Tomato Genetics Resource Center (TGRC) in Davis, CA. Plant growth conditions and DNA extraction for these additional accessions (with exception of LA1954, LA0446, LA2748, LA1930, LA1958, LA1274 and LA1973) were identical as for S. peruvianum from Tarapaca (LA2744). DNA from these other accessions was extracted using the Dneasy DNA Extraction Kit (Qiagen).
The RCR3 gene was identified using the RCR3 reference sequence from S. lycopersicum cv. ‘Mogeor’ (GenBank, accession number AF493234). Restriction sites for cloning were introduced into the primer sequences, which were designed to cover the start and stop codon (Table S1). The gene was PCR amplified using the Phusion proofreading polymerase (Finnzymes, Espoo, Finland) and cloned into Zero Blunt TOPO vectors (Invitrogen, Carlsbad, CA). Direct sequencing of PCR products and sequencing of miniprepped plasmid DNA from clones were conducted in parallel (Big Dye Terminator v 1.1, Applied Biosystems). Sequencing was performed according to the Sanger sequencing protocol using the DNA analyzer ABI 3730 (Applied Biosystems & Hitachi). Multiple clones per gene per individual were sequenced and ambiguous positions were compared to the direct sequences from the original PCR products. When necessary, independent rounds of PCRs, cloning and sequencing were conducted to resolve ambiguities. Raw sequences were edited and aligned in Sequencher 4.8 (1991–2007 Gene Codes Corporation) and alignments were refined by hand with MacClade (Version 4.0, Maddison and Maddison 2000, Sinauer Associates).
Analysis of RCR3 flanking regions
Due to low sequence divergence at the RCR3 ORF, it was not possible to distinguish allelic and paralogous sequences using the ORFs exclusively. Paralogs and orthologs may be distinguished by their flanking sequences since allelic sequences originate from the same locus in a genome and should possess the same (or very similar) flanking sequences. Paralogs, which are located at different positions in the genome, should have different flanking sequences. To distinguish between paralogs and orthologs, fragments of 400–2000 bp of RCR3 flanking DNA (with a minimum of 200 bp overlap with the gene to identify the matching allele) were amplified, cloned and sequenced from individuals from the Tarapaca population of S. peruvianum (individuals 7232–7241). A three-step Tail-PCR protocol with a set of random and nested RCR3 specific primers was used (Table S1, [64]). The location of the amplified RCR3 flanking regions in the tomato genome was assessed using BLASTn searches (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and phylogenetic reconstruction (PAUP v. 4.0b10, Swofford 1999, Sinauer Associates). FLRs were assigned stringently to the different allele sequences of the RCR3 gene, and only unambiguous pairs of alleles and FLRs were retained. Subsequently, the genomic origin of alleles with matching FLR was defined according to the BLAST search. Only RCR3 alleles matched unambiguously to a given FLR were used for population genetic analysis.
Population genetic analyses
The standard summary statistics including π, divergence, Tajima's D (DT) and Fu and Li's D test statistics were calculated using DnaSP v. 5.10 [65]. Sliding window analyses were also conducted using DnaSP. Phylogenetic analyses were performed using PAUP v. 4.0b10 (Swofford 1999, Sinauer Associates). The phylogenetic relationships between the sequences were determined using maximum parsimony (MP) and neighbor-joining (NJ) and these methods yielded similar topologies. To test whether gene conversion occurred between RCR3 copies, simulations were performed assuming a recent gene duplication event with copy number variation using the coalescent simulator by Thornton (2007) [66]. We then developed an ABC method using ABCest [67] to perform the model choice procedure (between models with and without gene conversion based on the code by Beaumont et al. (2002) [68]) and estimate the gene conversion rate (Text S1 and S2). Additionally, we surveyed the SFS of private and shared polymorphisms for the duplicated loci [47].
To investigate whether demographic effects could explain the pattern of sequence variation at the RCR3 locus, our results were compared to values obtained from 14 single-copy reference loci (CT066, CT093, CT099, CT114, CT143, CT148, CT166, CT179, CT189, CT198, CT208, CT251, CT268 and sucr), previously amplified from the same individuals of S. peruvianum [31], [49], [50]. A summary of their predicted gene products is found in Table 1 of [50]. Additionally, these loci were used to simulate expected neutral distributions of DT for comparison with the observed values at the RCR3 locus (Text S2).
Cloning procedure and Agrobacterium-mediated transient expression
A total of 54 RCR3 variants, which had been cloned into TOPO Zero Blunt for sequence analyses, were selected for functional testing. Cloning procedures of these variants were conducted according to the protocol described in [24]. Each RCR3 variant to be functionally tested was excised from the Zero Blunt TOPO vector using the restriction enzymes XhoI and NcoI, for which restriction sites resided in the PCR primers. Excised fragments were cloned into the pFK26 vector carrying the 35S overexpression promoter. 35S::RCR3 cassettes were shuttled into the binary vector pTP05 using the restriction enzymes XbaI and SalI [24]. All clones were verified by sequencing and electroporated into Agrobacterium tumefaciens strain GV3101. Agroinfiltration into leaves of N. benthamiana plants was performed as described previously [24]. After protein expression, RCR3 is secreted into the intercellular space outside the cytoplasm membrane. To recover expressed RCR3, infiltrated N. benthamiana leaves were harvested 72 h post inoculation, and the apoplastic intercellular fluids (AFs) of all infiltrated leaves were isolated according to [24]. Volumes of AFs containing equal concentrations of active RCR3 were used for all further experiments. Western Blot analysis was used to confirm the expression of RCR3 using RCR3 antibodies described previously [25].
Activity-based protein profiling and inhibition assays
Activity-based protein profiling (ABPP) using fluorescent DCG-04 [52] was used to detect RCR3 activity in the isolated AFs. 45 µl of AF were labeled with 2 µM fluorescent Bodipy-DCG-04 at pH 5.5 in the presence of 1 mM DTT for 5 h as described previously [24]. Concentrations of active RCR3 were adjusted based on the fluorescence signal. Accuracy of these adjustments was confirmed by independent ABPPs. Inhibition studies were performed by pre-incubation with 100 nM affinity-purified AVR2 [24], followed by ABPP. Proteins were separated via sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) and fluorescently-labeled proteins were detected by in-gel fluorescence scanning using a Typhoon 8600 scanner (GE Healthcare Life Sciences, http://www.gelifesciences.com) at ex/em 580 nm.
HR assays
We investigated whether different RCR3 constructs could activate the hypersensitive response upon exposure to AVR2 in cultivated tomato plants (S. lycopersicum cv. Money Maker). For this purpose, 100 µl of AF containing equal concentrations of expressed, active RCR3 with or without 100 nM AVR2 were infiltrated into Cf-2/rcr3-3 and Cf0/RCR3 tomato leaves. Tissue collapse was monitored daily until six days post inoculation (dpi) and recorded photographically.
Association of genotypic and observed phenotypic data
A general linear model algorithm implemented in TASSEL v. 3.0 (http://www.maizegenetics.net/) was used to evaluate correlations between phenotypic variation and sequence polymorphism at the RCR3 locus. The genotypic data was filtered such that only mutations that occurred in frequencies greater than 25% were included. Resulting P-values were Bonferroni-corrected for multiple testing. A structural model of the RCR3 protease domain was created as previously described in [24] using papain (PDB code 9papA) as a template.
RT–PCR
Seven RCR3 constructs failed to be expressed in N. benthamiana leaves. To confirm that the construct was designed correctly and that the agroinfiltration was successful, RNA of infiltrated leaves was isolated and RT-PCR with RCR3-specific primers was performed. The extraction of RNA was conducted using the Rneasy Plus Mini Kit (Qiagen) starting with 40–80 mg of plant material. cDNA-banks were created by reverse transcription using SuperScript Reverse Transcriptase (Invitrogen). RT-PCR was conducted for the RCR3 gene and a portion of the Ribulose-bisphosphate-carboxylase-oxigenase as a RNA-extraction control (Table S1).
All sequences have been deposited on GenBank under accession numbers JQ927229–JQ927299.
Supporting Information
Zdroje
1. DawkinsRKrebsJR 1979 Arms races between and within species. Proc R Soc Lond Ser B-Biol Sci 205 489 511
2. ChisholmSTCoakerGDayBStaskawiczBJ 2006 Host-microbe interactions: Shaping the evolution of the plant immune response. Cell 124 803 814
3. Schmid-HempelP 2008 Parasite immune evasion: a momentous molecular war. Trends Ecol Evol 23 318 326
4. StahlEADwyerGMauricioRKreitmanMBergelsonJ 1999 Dynamics of disease resistance polymorphism at the Rpm1 locus of Arabidopsis. Nature 400 667 671
5. BergelsonJKreitmanMStahlEATianDC 2001 Evolutionary dynamics of plant R-genes. Science 292 2281 2285
6. HolubEB 2001 The arms race is ancient history in Arabidopsis, the wildflower. Nat Rev Genet 2 516 527
7. BakkerEGToomajianCKreitmanMBergelsonJ 2006 A genome-wide survey of R gene polymorphisms in Arabidopsis. Plant Cell 18 1803 1818
8. NishimuraMTDanglJL 2010 Arabidopsis and the plant immune system. Plant J 61 1053 1066
9. JonesJDGDanglJL 2006 The plant immune system. Nature 444 323 329
10. Hammond-KosackKEJonesJDG 1997 Plant disease resistance genes. Annu Rev Plant Physiol Plant Molec Biol 48 575 607
11. ZipfelCFelixG 2005 Plants and animals: a different taste for microbes? Curr Opin Plant Biol 8 353 360
12. FlorHH 1956 The complementary genic systems in flax and flax rust. Adv Genet 8 29 54
13. ThompsonJNBurdonJJ 1992 Gene-for-gene coevolution between plants and parasites. Nature 360 121 125
14. NurnbergerTBrunnerFKemmerlingBPiaterL 2004 Innate immunity in plants and animals: striking similarities and obvious differences. Immunol Rev 198 249 266
15. JiaYMcAdamsSABryanGTHersheyHPValentB 2000 Direct interaction of resistance gene and avirulence gene products confers rice blast resistance. Embo J 19 4004 4014
16. DoddsPNLawrenceGJCatanzaritiAMTehTWangCIA 2006 Direct protein interaction underlies gene-for-gene specificity and coevolution of the flax resistance genes and flax rust avirulence genes. Proc Natl Acad Sci USA 103 8888 8893
17. van der BiezenEAJonesJDG 1998 Plant disease-resistance proteins and the gene-for-gene concept. Trends Biochem Sci 23 454 456
18. DanglJLJonesJDG 2001 Plant pathogens and integrated defence responses to infection. Nature 411 826 833
19. van der HoornRALKamounS 2008 From Guard to Decoy: A new model for perception of plant pathogen effectors. Plant Cell 20 2009 2017
20. CaldwellKSMichelmoreRW 2009 Arabidopsis thaliana genes encoding defense signaling and recognition proteins exhibit contrasting evolutionary dynamics. Genetics 181 671 684
21. BombliesKWeigelD 2007 Hybrid necrosis: autoimmunity as a potential gene-flow barrier in plant species. Nature Rev Genet 8 382 393
22. BrownJKMTellierA 2011 Plant-parasite coevolution: Bridging the gap between genetics and ecology. Annu Rev Phytopathol 49 345 367
23. IspolatovIDoebeliM 2009 Speciation due to hybrid necrosis in plant-pathogen models. Evolution 63 3076 3084
24. ShababMShindoTGuCKaschaniFPansuriyaT 2008 Fungal effector protein AVR2 targets diversifying defense-related Cys proteases of tomato. Plant Cell 20 1169 1183
25. RooneyHCEvan 't KloosterJWvan der HoornRALJoostenMJonesJDG 2005 Cladosporium Avr2 inhibits tomato Rcr3 protease required for Cf-2-dependent disease resistance. Science 308 1783 1786
26. WulffBBHChakrabartiAJonesDA 2009 Recognitional Specificity and Evolution in the Tomato-Cladosporium fulvum Pathosystem. Mol Plant-Microbe Interact 22 1191 1202
27. BondTET 1938 Infection experiments with Cladosporium fulvum Cooke and related species. Ann Appl Biol 25 277 307
28. PeraltaIESpoonerDMKnappS 2008 The taxonomy of tomatoes: a revision of wild tomatoes (Solanum section Lycopersicon) and their outgroup relatives in sections Juglandifolium and Lycopersicoides. Syst Bot Monogr 84 1 186
29. YoungKRUlloaCULuteynJLKnappS 2002 Plant evolution and endemism in Andean South America: An introduction. Bot Rev 68 4 21
30. NakazatoTWarrenDLMoyleLC 2010 Ecological and geographic modes of species divergence in wild tomatoes. Am J Bot 97 680 693
31. StädlerTRoseliusKStephanW 2005 Genealogical footprints of speciation processes in wild tomatoes: Demography and evidence for historical gene flow. Evolution 59 1268 1279
32. TellierALaurentSYLainerHPavlidisPStephanW 2011 Inference of seed bank parameters in two wild tomato species using ecological and genetic data. Proc Natl Acad Sci USA 108 17052 17057
33. RoseLELangleyCHBernalAJMichelmoreRW 2005 Natural variation in the Pto pathogen resistance gene within species of wild tomato (Lycopersicon). I. Functional analysis of Pto alleles. Genetics 171 345 357
34. RoseLEMichelmoreRWLangleyCH 2007 Natural variation in the Pto pathogen resistance gene within species of wild tomato (Lycopersicon). II. Population genetics of Pto. Genetics 175 1307 1319
35. RoseLEGrzeskowiakLHörgerACGrothMStephanW 2011 Targets of selection in a disease resistance network in wild tomatoes. Mol Plant Pathol 12 921 927
36. KerrEAPatrickZABaileyDL 1971 Resistance in tomato species to new races of leaf mold (Cladosporium fulvum Cke.). Hort Res 11 84 92
37. KruijtMKipDJJoostenMHAJBrandwagtBFde WitPJGM 2005 The Cf-4 and Cf-9 resistance genes against Cladosporium fulvum are conserved in wild tomato species. Mol Plant-Microbe Interact 18 1011 1021
38. KerrEABaileyDL 1964 Resistance to Cladosporium fulvum Cke. obtained from wild species of tomato. Can J Bot 42 1541 1554
39. RoseLEBittner-EddyPDLangleyCHHolubEBMichelmoreRW 2004 The maintenance of extreme amino acid diversity at the disease resistance gene, RPP13, in Arabidopsis thaliana. Genetics 166 1517 1527
40. TianMYWinJSongJvan der HoornRvan der KnaapE 2007 A Phytophthora infestans cystatin-like protein targets a novel tomato papain-like apoplastic protease. Plant Physiol 143 364 377
41. LabateJARobertsonLDBaldoAM 2009 Multilocus sequence data reveal extensive departures from equilibrium in domesticated tomato (Solanum lycopersicum L.). Heredity 103 257 267
42. MichelmoreRWMeyersBC 1998 Clusters of resistance genes in plants evolve by divergent selection and a birth-and-death process. Genome Res 8 1113 1130
43. RaffaeleSFarrerRACanoLMStudholmeDJMacLeanD 2010 Genome evolution following host jumps in the Irish Potato Famine pathogen lineage. Science 330 1540 1543
44. SpanuPDAbbottJCAmselemJBurgisTASoanesDM 2010 Genome expansion and gene loss in powdery mildew fungi reveal functional tradeoffs in parasitism. Science 330 1543 1546
45. BeaumontMA 2010 Approximate Bayesian computation in evolution and ecology. Annu Rev Ecol Evol Syst 41 379 405
46. InnanH 2003 A two-locus gene conversion model with selection and its application to the human RHCE and RHD genes. Proc Natl Acad Sci USA 100 8793 8798
47. InnanH 2003 The coalescent and infinite-site model of a small multigene family. Genetics 163 803 810
48. ArunyawatUStephanWStädlerT 2007 Using multilocus sequence data to assess population structure, natural selection, and linkage disequilibrium in wild tomatoes. Mol Biol Evol 24 2310 2322
49. BaudryEKerdelhueCInnanHStephanW 2001 Species and recombination effects on DNA variability in the tomato genus. Genetics 158 1725 1735
50. RoseliusKStephanWStädlerT 2005 The relationship of nucleotide polymorphism, recombination rate and selection in wild tomato species. Genetics 171 753 763
51. TajimaF 1989 Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123 585 595
52. GreenbaumDMedzihradszkyKFBurlingameABogyoM 2000 Epoxide electrophiles as activity-dependent cysteine protease profiling and discovery tools. Chem Biol 7 569 581
53. van der HoornRALLeeuwenburghMABogyoMJoostenMHPeckSC 2004 Activity profiling of papain-like cysteine proteases in plants. Plant Physiol 135 1170 1178
54. van EsseHPvan't KloosterJWBoltonMDYadetaKAvan BaarlenP 2008 The Cladosporium fulvum virulence protein Avr2 inhibits host proteases required for basal defense. Plant Cell 20 1948 1963
55. KaschaniFShababMBozkurtTShindoTSchornackS 2010 An effector-targeted protease contributes to defense against Phytophthora infestans and is under diversifying selection in natural hosts. Plant Physiol 154 1794 1804
56. KrügerJThomasCMGolsteinCDixonMSSmokerM 2002 A tomato cysteine protease required for Cf-2-dependent disease resistance and suppression of autonecrosis. Science 296 744 747
57. ChamaryJVParmleyJLHurstLD 2006 Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet 7 98 108
58. SongJWinJTianMYSchornackSKaschaniF 2009 Apoplastic effectors secreted by two unrelated eukaryotic plant pathogens target the tomato defense protease Rcr3. Proc Natl Acad Sci USA 106 1654 1659
59. KaschaniFvan der HoornRAL 2011 A model of the C14-EPIC complex indicates hotspots for a protease-inhibitor arms race in the oomycete-potato interaction. Plant Signal Behav 6 109 112
60. TianDTrawMBChenJQKreitmanMBergelsonJ 2003 Fitness costs of R-gene-mediated resistance in Arabidopsis thaliana. Nature 423 74 77
61. DixonMSJonesDAKeddieJSThomasCMHarrisonK 1996 The tomato Cf-2 disease resistance locus comprises two functional genes encoding leucine-rich repeat proteins. Cell 84 451 459
62. CaicedoALSchaalBA 2004 Heterogeneous evolutionary processes affect R gene diversity in natural populations of Solanum pimpinellifolium. Proc Natl Acad Sci USA 101 17444 17449
63. BombliesKLempeJEpplePWarthmannNLanzC 2007 Autoimmune response as a mechanism for a Bateson-Dobzhansky-Muller-type incompatibility syndrome in plants. PLoS Biol 5 e236
64. LiuYGMitsukawaNOosumiTWhittierRF 1995 Efficient isolation and mapping of Arabidopsis thaliana T-DNA insertjunctions by thermal assymmetric interlaced PCR. Plant J 8 457 463
65. LibradoPRozasJ 2009 DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25 1451 1452
66. ThorntonKR 2007 The neutral coalescent process for recent gene duplications and copy-number variants. Genetics 177 987 1000
67. ExcoffierLEstoupACornuetJM 2005 Bayesian analysis of an admixture model with mutations and arbitrarily linked markers. Genetics 169 1727 1738
68. BeaumontMAZhangWYBaldingDJ 2002 Approximate Bayesian computation in population genetics. Genetics 162 2025 2035
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2012 Číslo 7
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Guidelines for Genome-Wide Association Studies
- The Role of Rice HEI10 in the Formation of Meiotic Crossovers
- Identification of Chromatin-Associated Regulators of MSL Complex Targeting in Dosage Compensation
- GWAS Identifies Novel Susceptibility Loci on 6p21.32 and 21q21.3 for Hepatocellular Carcinoma in Chronic Hepatitis B Virus Carriers
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy