
The IG-DMR and the -DMR at Human Chromosome 14q32.2: Hierarchical Interaction and Distinct Functional Properties as Imprinting Control Centers
Human chromosome 14q32.2 harbors the germline-derived primary DLK1-MEG3 intergenic differentially methylated region (IG-DMR) and the postfertilization-derived secondary MEG3-DMR, together with multiple imprinted genes. Although previous studies in cases with microdeletions and epimutations affecting both DMRs and paternal/maternal uniparental disomy 14-like phenotypes argue for a critical regulatory function of the two DMRs for the 14q32.2 imprinted region, the precise role of the individual DMR remains to be clarified. We studied an infant with upd(14)pat body and placental phenotypes and a heterozygous microdeletion involving the IG-DMR alone (patient 1) and a neonate with upd(14)pat body, but no placental phenotype and a heterozygous microdeletion involving the MEG3-DMR alone (patient 2). The results generated from the analysis of these two patients imply that the IG-DMR and the MEG3-DMR function as imprinting control centers in the placenta and the body, respectively, with a hierarchical interaction for the methylation pattern in the body governed by the IG-DMR. To our knowledge, this is the first study demonstrating an essential long-range imprinting regulatory function for the secondary DMR.
Published in the journal:
. PLoS Genet 6(6): e32767. doi:10.1371/journal.pgen.1000992
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000992
Summary
Human chromosome 14q32.2 harbors the germline-derived primary DLK1-MEG3 intergenic differentially methylated region (IG-DMR) and the postfertilization-derived secondary MEG3-DMR, together with multiple imprinted genes. Although previous studies in cases with microdeletions and epimutations affecting both DMRs and paternal/maternal uniparental disomy 14-like phenotypes argue for a critical regulatory function of the two DMRs for the 14q32.2 imprinted region, the precise role of the individual DMR remains to be clarified. We studied an infant with upd(14)pat body and placental phenotypes and a heterozygous microdeletion involving the IG-DMR alone (patient 1) and a neonate with upd(14)pat body, but no placental phenotype and a heterozygous microdeletion involving the MEG3-DMR alone (patient 2). The results generated from the analysis of these two patients imply that the IG-DMR and the MEG3-DMR function as imprinting control centers in the placenta and the body, respectively, with a hierarchical interaction for the methylation pattern in the body governed by the IG-DMR. To our knowledge, this is the first study demonstrating an essential long-range imprinting regulatory function for the secondary DMR.
Introduction
Human chromosome 14q32.2 carries a cluster of protein-coding paternally expressed genes (PEGs) such as DLK1 and RTL1 and non-coding maternally expressed genes (MEGs) such as MEG3 (alias, GTL2), RTL1as (RTL1 antisense), MEG8, snoRNAs, and microRNAs [1], [2]. Consistent with this, paternal uniparental disomy 14 (upd(14)pat) results in a unique phenotype characterized by facial abnormality, small bell-shaped thorax, abdominal wall defects, placentomegaly, and polyhydramnios [2], [3], and maternal uniparental disomy 14 (upd(14)mat) leads to less-characteristic but clinically discernible features including growth failure [2], [4].
The 14q32.2 imprinted region also harbors two differentially methylated regions (DMRs), i.e., the germline-derived primary DLK1-MEG3 intergenic DMR (IG-DMR) and the postfertilization-derived secondary MEG3-DMR [1], [2]. Both DMRs are hypermethylated after paternal transmission and hypomethylated after maternal transmission in the body, whereas in the placenta the IG-DMR alone remains as a DMR and the MEG3-DMR is rather hypomethylated [1], [2]. Furthermore, previous studies in cases with upd(14)pat/mat-like phenotypes have revealed that epimutations (hypermethylation) and microdeletions affecting both DMRs of maternal origin cause paternalization of the 14q32.2 imprinted region, and that epimutations (hypomethylation) affecting both DMRs of paternal origin cause maternalization of the 14q32.2 imprinted region, while microdeletions involving the DMRs of paternal origin have no effect on the imprinting status [2], [5]–[8]. These findings, together with the notion that parent-of-origin specific expression patterns of imprinted genes are primarily dependent on the methylation status of the DMRs [9], argue for a critical regulatory function of the two DMRs for the 14q32.2 imprinted region, with possible different effects between the body and the placenta.
However, the precise role of individual DMR remains to be clarified. Here, we report that the IG-DMR and the MEG3-DMR show a hierarchical interaction for the methylation pattern in the body, and function as imprinting control centers in the placenta and the body, respectively. To our knowledge, this is the first study demonstrating not only different roles between the primary and secondary DMRs at a single imprinted region, but also an essential regulatory function for the secondary DMR.
Results
Clinical reports
We studied an infant with upd(14)pat body and placental phenotypes (patient 1) and a neonate with upd(14)pat body, but no placental, phenotype (patient 2) (Figure 1). Detailed clinical features of patients 1 and 2 are shown in Table 1. In brief, patient 1 was delivered by a caesarean section at 33 weeks of gestation due to progressive polyhydramnios despite amnioreduction at 28 and 30 weeks of gestation, whereas patient 2 was born at 28 weeks of gestation by a vaginal delivery due to progressive labor without discernible polyhydramnios. Placentomegaly was observed in patient 1 but not in patient 2. Patients 1 and 2 were found to have characteristic face, small bell-shaped thorax with coat hanger appearance of the ribs, and omphalocele. Patient 1 received surgical treatment for omphalocele immediately after birth and mechanical ventilation for several months. At present, she is 5.5 months of age, and still requires intensive care including oxygen administration and tube feeding. Patient 2 died at four days of age due to massive intracranial hemorrhage, while receiving intensive care including mechanical ventilation. The mother of patient 1 had several non-specific clinical features such as short stature and obesity. The father of patient 1 and the parents of patient 2 were clinically normal.


Sample preparation
We isolated genomic DNA (gDNA) and transcripts (mRNAs, snoRNAs, and microRNAs) from fresh leukocytes of patients 1 and the parents of patients 1 and 2, from fresh skin fibroblasts of patient 2, and from formalin-fixed and paraffin-embedded placental samples of patient 1 and similarly treated pituitary and adrenal samples of patient 2 (although multiple body tissues were available in patient 2, useful gDNA and transcript samples were not obtained from other tissues probably due to drastic post-mortem degradation). We also made metaphase spreads from leukocytes and skin fibroblasts. For comparison, we obtained control samples from fresh normal adult leukocytes, neonatal skin fibroblasts, and placenta at 38 weeks of gestation, and from fresh leukocytes of upd(14)pat/mat patients and formalin-fixed and paraffin-embedded placenta of a upd(14)pat patient [2], [3].
Structural analysis of the imprinted region
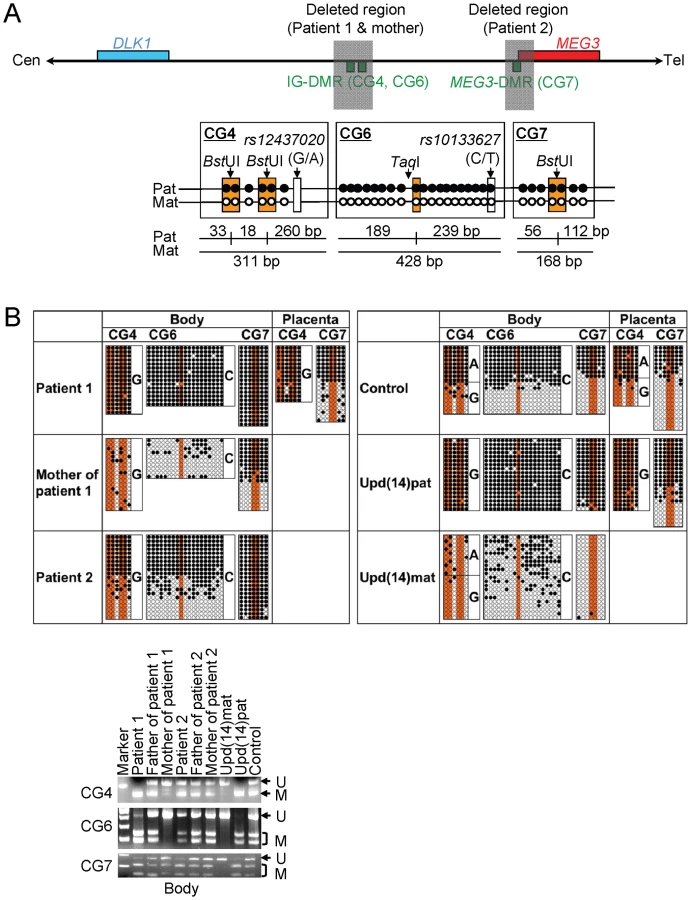
We first examined the structure of the 14q32.2 imprinted region (Figure 2). Upd(14) was excluded in patients 1 and 2 as well as in the mother of patient 1 by microsatellite analysis (Table S1), and FISH analysis for the two DMRs identified a familial heterozygous deletion encompassing the IG-DMR alone in patient 1 and her mother and a de novo heterozygous deletion encompassing the MEG3-DMR alone in patient 2 (Figure 2). The microdeletions were further localized by SNP genotyping for 70 loci (Table S1) and quantitative real-time PCR (q-PCR) analysis for four regions around the DMRs (Figure S1A), and serial direct sequencing for the long PCR products harboring the deletion junctions successfully identified the fusion points of the microdeletions in patient 1 and her mother and in patient 2 (Figure 2). According to the NT_026437 sequence data at the NCBI Database (Genome Build 36.3) (http://preview.ncbi.nlm.nih.gov/guide/), the deletion size was 8,558 bp (82,270,449–82,279,006 bp) for the microdeletion in patient 1 and her mother, and 4,303 bp (82,290,978–82,295,280 bp) for the microdeletion in patient 2. The microdeletion in patient 2 also involved the 5′ part of MEG3 and five of the seven putative CTCF binding sites A–G [10], and was accompanied by insertion of a 66 bp sequence duplicated from MEG3 intron 5 (82,299,727–82,299,792 bp on NT_026437). Direct sequencing of the exonic or transcribed regions detected no mutation in DLK1, MEG3, and RTL1, although several cDNA polymorphisms (cSNPs) were identified (Table S1). Oligoarray comparative genomic hybridization identified no other discernible structural abnormality (Figure S1B).

Methylation analysis of the two DMRs and the seven putative CTCF binding sites
We next studied methylation patterns of the previously reported IG-DMR (CG4 and CG6) and MEG3-DMR (CG7) (Figure 3A) [2], using bisulfite treated gDNA samples. Bisulfite sequencing and combined bisulfite restriction analysis using body samples revealed a hypermethylated IG-DMR and MEG3-DMR in patient 1, a hypomethylated IG-DMR and differentially methylated MEG3-DMR in the mother of patient 1, and a differentially methylated IG-DMR and hypermethylated MEG3-DMR in patient 2, and bisulfite sequencing using placental samples showed a hypermethylated IG-DMR and rather hypomethylated MEG3-DMR in patient 1 (Figure 3B).
We also examined methylation patterns of the seven putative CTCF binding sites by bisulfite sequencing (Figure 4A). The sites C and D alone exhibited DMRs in the body and were rather hypomethylated in the placenta (Figure 4B), as observed in CG7. Furthermore, to identify an informative SNP(s) pattern for allele-specific bisulfite sequencing, we examined a 349 bp region encompassing the site C and a 356 bp region encompassing the site D as well as a 300 bp region spanning the previously reported three SNPs near the site D, in 120 control subjects, the cases with upd(14)pat/mat, and patients 1 and 2 and their parents. Consequently, an informative polymorphism was identified for a novel G/A SNP near the site D in only a single control subject, and the parent-of-origin specific methylation pattern was confirmed (Figure 4C). No informative SNP was found in the examined region around the site C, and no other informative SNP was identified in the two examined regions around the site D, with the previously known three SNPs being present in a homozygous condition in all the subjects analyzed.

Expression analysis of the imprinted genes
Finally, we performed expression analyses, using standard reverse transcriptase (RT)-PCR and/or q-PCR analysis for multiple imprinted genes in this region (Figure 5A–5C). For leukocytes, weak expression was detected for MEG3 and SNORD114-29 in a control subject and the mother of patient 1 but not in patient 1. For skin fibroblasts, although all MEGs but no PEGs were expressed in control subjects, neither MEGs nor PEGs were expressed in patient 2. For placentas, although all imprinted genes were expressed in control subjects, PEGs only were expressed in patient 1. For the pituitary and adrenal of patient 2, DLK1 expression alone was identified.

Expression pattern analyses using informative cSNPs revealed monoallelic MEG3 expression in the leukocytes of the mother of patient 1 (Figure 5D), and biparental RTL1 expression in the placenta of patient 1 (no informative cSNP was detected for DLK1) and biparental DLK1 expression in the pituitary and adrenal of patient 2 (RTL1 was not expressed in the pituitary and adrenal) (Figure 5E), as well as maternal MEG3 expression in the control leukocytes and paternal RTL1 expression in the control placentas (Figure S2). Although we also attempted q-PCR analysis, precise assessment was impossible for MEG3 in the mother of patient 1 because of faint expression level in leukocytes and for RTL1 in patient 1 and DLK1 in patient 2 because of poor quality of mRNAs obtained from formalin-fixed and paraffin-embedded tissues.
Discussion
The data of the present study are summarized in Figure 6. Parental origin of the microdeletion positive chromosomes is based on the methylation patterns of the preserved DMRs in patients 1 and 2 and the mother of patient 1 as well as maternal transmission in patient 1. Loss of the hypomethylated IG-DMR of maternal origin in patient 1 was associated with epimutation (hypermethylation) of the MEG3-DMR in the body and caused paternalization of the imprinted region and typical upd(14)pat body and placental phenotypes, whereas loss of the hypomethylated MEG3-DMR of maternal origin in patient 2 permitted normal methylation pattern of the IG-DMR in the body and resulted in maternal to paternal epigenotypic alteration and typical upd(14)pat body, but no placental, phenotype. In this regard, while a 66 bp segment was inserted in patient 2, this segment contains no known regulatory sequence [11] or evolutionarily conserved element [12] (also examined with a VISTA program, http://genome.lbl.gov/vista/index.shtml). Similarly, while no control samples were available for pituitary and adrenal, the previous study in human subjects has shown paternal DLK1 expression in adrenal as well as monoallelic DLK1 and MEG3 expressions in various tissues [11]. Furthermore, the present and the previous studies [2] indicate that this region is imprinted in the placenta as well as in the body. Thus, these results, in conjunction with the finding that the IG-DMR remains as a DMR and the MEG3-DMR exhibits a non-DMR in the placenta [2], imply the following: (1) the IG-DMR functions hierarchically as an upstream regulator for the methylation pattern of the MEG3-DMR on the maternally inherited chromosome in the body, but not in the placenta; (2) the hypomethylated MEG3-DMR functions as an essential imprinting regulator for both PEGs and MEGs in the body; and (3) in the placenta, the hypomethylated IG-DMR directly controls the imprinting pattern of both PEGs and MEGs. These notions also explain the epigenotypic alteration in the previous cases with epimutations or microdeletions affecting both DMRs (Figure S3).

It remains to be clarified how the IG-DMR and the MEG3-DMR interact hierarchically in the body. However, the present data, together with the previous findings in cases with epimutations [2], [5]–[8], imply that MEG3-DMR can remain hypomethylated only in the presence of a hypomethylated IG-DMR and is methylated when the IG-DMR is deleted or methylated irrespective of the parental origin. Furthermore, mouse studies have suggested that the methylation pattern of the postfertilization-derived Gtl2-DMR (the mouse homolog for the MEG3-DMR) is dependent on that of the germline-derive IG-DMR [13]. Thus, a preferential binding of some factor(s) to the unmethylated IG-DMR may cause a conformational alteration of the genomic structure, thereby protecting the methylation of the MEG3-DMR.
It also remains to be elucidated how the IG-DMR and the MEG3-DMR regulate the expression of both PEGs and MEGs in the placenta and the body, respectively. For the MEG3-DMR, however, the CTCF binding sites C and D may play a pivotal role in the imprinting regulation. The methylation analysis indicates that the two sites reside within the MEG3-DMR, and it is known that the CTCF protein with versatile functions preferentially binds to unmethylated target sequences including the sites C and D [10], [14]–[16]. In this regard, all the MEGs in this imprinted region can be transcribed together in the same orientation and show a strikingly similar tissue expressions pattern [1], [12], whereas PEGs are transcribed in different directions and are co-expressed with MEGs only in limited cell-types [1], [17]. It is possible, therefore, that preferential CTCF binding to the grossly unmethylated sites C and D activates all the MEGs as a large transcription unit and represses all the PEGs perhaps by influencing chromatin structure and histone modification independently of the effects of expressed MEGs. In support of this, CTCF protein acts as a transcriptional activator for Gtl2 (the mouse homolog for MEG3) in the mouse [18].
Such an imprinting control model has not been proposed previously. It is different from the CTCF protein-mediated insulator model indicated for the H19-DMR and from the non-coding RNA-mediated model implicated for several imprinted regions including the KvDMR1 [19]. However, the KvDMR1 harbors two putative CTCF binding sites that may mediate non-coding RNA independent imprinting regulation [20], and the imprinting control center for Prader-Willi syndrome [21] also carries three CTCF binding sites (examined with a Search for CTCF DNA Binding Sites program, http://www.essex.ac.uk/bs/molonc/spa.html). Thus, while each imprinted region would be regulated by a different mechanism, a CTCF protein may be involved in the imprinting control of multiple regions, in various manners.
This imprinted region has also been studied in the mouse. Clinical and molecular findings in wildtype mice [1], [22], [23], mice with PatDi(12) (paternal disomy for chromosome 12 harboring this imprinted region) [13], [24], [25], and mice with targeted deletions for the IG-DMR (ΔIG-DMR) [22], [26] and for the Gtl2-DMR (ΔGtl2-DMR) [27] are summarized in Table 2. These data, together with human data, provide several informative findings. First, in both the human and the mouse, the IG-DMR is differentially methylated in both the body and the placenta, whereas the MEG3/Gtl2-DMR is differentially methylated in the body and exhibits non-DMR in the placenta. Second, the IG-DMR and the MEG3/Gtl2-DMR show a hierarchical interaction on the maternally derived chromosome in both the human and the mouse bodies. Indeed, the MEG3/Gtl2-DMR is epimutated in patient 1 and mice with maternally inherited ΔIG-DMR, and the IG-DMR is normally methylated in patient 2 and mice with maternally inherited ΔGtl2-DMR. Third, the function of the IG-DMR is comparable between human and mouse bodies and different between human and mouse placentas. Indeed, patient 1 has upd(14)pat body and placental phenotypes, whereas mice with the ΔIG-DMR of maternal origin have PatDi(12)-compatible body phenotype and apparently normal placental phenotype. It is likely that imprinting regulation in the mouse placenta is contributed by some mechanism(s) other than the methylation pattern of the IG-DMR, such as chromatin conformation [22], [28], [29].

Unfortunately, however, the data of ΔGtl2-DMR mice appears to be drastically complicated by the retained neomycin cassette in the upstream region of Gtl2. Indeed, it has been shown that the insertion of a lacZ gene or a neomycin gene in the similar upstream region of Gtl2 causes severely dysregulated expression patterns and abnormal phenotypes after both paternal and maternal transmissions [30], [31], and that deletion of the inserted neomycin gene results in apparently normal expression patterns and phenotypes after both paternal and maternal transmissions [31]. (In this regard, although a possible influence of the inserted 66 bp segment can not be excluded formally in patient 2, phenotype and expression data in patient 2 are compatible with simple paternalization of the imprinted region.) In addition, since the apparently normal phenotype in mice homozygous for ΔGtl2-DMR is reminiscent of that in sheep homozygous for the callipyge mutation [32], a complicated mechanism(s) such as the polar overdominance may be operating in the ΔGtl2-DMR mice [33]. Thus, it remains to be clarified whether the MEG3/Gtl2-DMR has a similar or different function between the human and the mouse.
Two points should be made in reference to the present study. First, the proposed functions of the two DMRs are based on the results of single patients. This must be kept in mind, because there might be a hidden patient-specific abnormality or event that might explain the results. For example, the abnormal placental phenotype in patient 1 might be caused by some co-incidental aberration, and the apparently normal placenta in patient 2 might be due to mosaicism with grossly preserved MEG3-DMR in the placenta and grossly deleted MEG3-DMR in the body. Second, the clinical features in the mother of patient 1 such as short stature and obesity are often observed in cases with upd(14)mat (Table S2). However, the clinical features are non-specific and appear to be irrelevant to the microdeletion involving the IG-DMR, because loss of the paternally derived IG-DMR does not affect the imprinted status [2], [26]. Indeed, MEG3 in the mother of patient 1 showed normal monoallelic expression in the presence of the differentially methylated MEG3-DMR. Nevertheless, since the upd(14)mat phenotype is primarily ascribed to loss of functional DLK1 (Figure S3B) [2], [34], it might be possible that the microdeletion involving the IG-DMR has affected a cis-acting regulatory element for DLK1 expression (for details, see Note in the legend for Table S2). Further studies in cases with similar microdeletions will permit clarification of these two points.
In summary, the results show a hierarchical interaction and distinct functional properties of the IG-DMR and the MEG3-DMR in imprinting control. Thus, this study provides significant advance in the clarification of mechanisms involved in the imprinting regulation at the 14q32.2 imprinted region and the development of upd(14) phenotype.
Materials and Methods
Ethics statement
This study was approved by the Institutional Review Board Committees at National Center for Child health and Development, University College Dublin, and Dokkyo University School of Medicine, and performed after obtaining written informed consent.
Primers
All the primers utilized in this study are summarized in Table S3.
Sample preparation
For leukocytes and skin fibroblasts, genomic DNA (gDNA) samples were extracted with FlexiGene DNA Kit (Qiagen), and RNA samples were prepared with RNeasy Plus Mini (Qiagen) for DLK1, MEG3, RTL1, MEG8 and snoRNAs, and with mirVana miRNA Isolation Kit (Ambion) for microRNAs. For paraffin-embedded tissues including the placenta, brain, lung, heart, liver, spleen, kidney, bladder, and small intestine, gDNA and RNA samples were extracted with RecoverAll Total Nucleic Acids Isolation Kit (Ambion) using slices of 40 µm thick. For fresh control placental samples, gDNA and RNA were extracted using ISOGEN (Nippon Gene). After treating total RNA samples with DNase, cDNA samples for DLK1, MEG3, MEG8, and snoRNAs were prepared with oligo(dT) primers from 1 µg of RNA using Superscript III Reverse Transcriptase (Invitrogen), and those for microRNAs were synthesized from 300 ng of RNA using TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems). For RTL1, cDNA samples were synthesized with RTL1-specific primers that do not amplify RTL1as. Control gDNA and cDNA samples were extracted from adult leukocytes and neonatal skin fibroblasts purchased from Takara Bio Inc. Japan, and from a fresh placenta of 38 weeks of gestation. Metaphase spreads were prepared from leukocytes and skin fibroblasts using colcemide (Invitrogen).
Structural analysis
Microsatellite analysis and SNP genotyping were performed as described previously [2]. For FISH analysis, metaphase spreads were hybridized with a 5,104 bp FISH-1 probe and a 5,182 bp FISH-2 probe produced by long PCR, together with an RP11-566I2 probe for 14q12 used as an internal control [2]. The FISH-1 and FISH-2 probes were labeled with digoxigenin and detected by rhodamine anti-digoxigenin, and the RP11-566I2 probe was labeled with biotin and detected by avidin conjugated to fluorescein isothiocyanate. For quantitative real-time PCR analysis, the relative copy number to RNaseP (catalog No: 4316831, Applied Biosystems) was determined by the Taqman real-time PCR method using the probe-primer mix on an ABI PRISM 7000 (Applied Biosystems). To determine the breakpoints of microdeletions, sequence analysis was performed for long PCR products harboring the fusion points, using serial forward primers on the CEQ 8000 autosequencer (Beckman Coulter). Direct sequencing was also performed on the CEQ 8000 autosequencer. Oligoarray comparative genomic hybridization was performed with 1×244K Human Genome Array (catalog No: G4411B) (Agilent Technologies), according to the manufacturer's protocol.
Methylation analysis
Methylation analysis was performed for gDNA treated with bisulfite using the EZ DNA Methylation Kit (Zymo Research). After PCR amplification using primer sets that hybridize both methylated and unmethylated clones because of lack of CpG dinucleotides within the primer sequences, the PCR products were digested with appropriate restriction enzymes for combined bisulfite restriction analysis. For bisulfite sequencing, the PCR products were subcloned with TOPO TA Cloning Kit (Invitrogen) and subjected to direct sequencing on the CEQ 8000 autosequencer.
Expression analysis
Standard RT-PCR was performed for DLK1, RTL1, MEG3, MEG8, and snoRNAs using primers hybridizing to exonic or transcribed sequences, and one µl of PCR reaction solutions was loaded onto Gel-Dye Mix (Agilent). Taqman real-time PCR was carried out using the probe-primer mixtures (assay No: Hs00292028 for MEG3 and Hs00419701 for MEG8; assay ID: 001028 for miR433, 000452 for miR127, 000568 for miR379, and 000477 for miR154) on the ABI PRISM 7000. Data were normalized against GAPDH (catalog No: 4326317E) for MEG3 and MEG8 and against RNU48 (assay ID: 0010006) for the remaining miRs. The expression studies were performed three times for each sample.
To examine the imprinting status of MEG3 in the leukocytes of the mother of patient 1, direct sequence data for informative cSNPs were compared between gDNA and cDNA. To analyze the imprinting status of RTL1 in the placental sample of patient 1 and that of DLK1 in the pituitary and adrenal samples of patient 2, RT-PCR products containing exonic cSNPs informative for the parental origin were subcloned with TOPO TA Cloning Kit, and multiple clones were subjected to direct sequencing on the CEQ 8000 autosequencer. Furthermore, MEG3 expression pattern was examined using leukocyte gDNA and cDNA samples from multiple normal subjects and leukocyte gDNA samples from their mothers, and RTL1 expression pattern was analyzed using gDNA and cDNA samples from multiple fresh normal placentas and leukocyte gDNA from the mothers.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. da RochaST
EdwardsCA
ItoM
OgataT
Ferguson-SmithAC
2008 Genomic imprinting at the mammalian Dlk1-Dio3 domain. Trends Genet 24 306 316
2. KagamiM
SekitaY
NishimuraG
IrieM
KatoF
2008 Deletions and epimutations affecting the human 14q32.2 imprinted region in individuals with paternal and maternal upd(14)-like phenotypes. Nat Genet 40 237 242
3. KagamiM
YamazawaK
MatsubaraK
MatsuoN
OgataT
2008 Placentomegaly in paternal uniparental disomy for human chromosome 14. Placenta 29 760 761
4. KotzotD
2004 Maternal uniparental disomy 14 dissection of the phenotype with respect to rare autosomal recessively inherited traits, trisomy mosaicism, and genomic imprinting. Ann Genet 47 251 260
5. TempleIK
ShrubbV
LeverM
BullmanH
MackayDJ
2007 Isolated imprinting mutation of the DLK1/GTL2 locus associated with a clinical presentation of maternal uniparental disomy of chromosome 14. J Med Genet 44 637 640
6. BuitingK
KanberD
Martín-SuberoJI
LiebW
TerhalP
2008 Clinical features of maternal uniparental disomy 14 in patients with an epimutation and a deletion of the imprinted DLK1/GTL2 gene cluster. Hum Mutat 29 1141 1146
7. HosokiK
OgataT
KagamiM
TanakaT
SaitohS
2008 Epimutation (hypomethylation) affecting the chromosome 14q32.2 imprinted region in a girl with upd(14)mat-like phenotype. Eur J Hum Genet 16 1019 1023
8. ZechnerU
KohlschmidtN
RittnerG
DamatovaN
BeyerV
2009 Epimutation at human chromosome 14q32.2 in a boy with a upd(14)mat-like clinical phenotype. Clin Genet 75 251 258
9. LiE
BeardC
JaenischR
1993 Role for DNA methylation in genomic imprinting. Nature 366 362 365
10. RosaAL
WuYQ
Kwabi-AddoB
CovelerKJ
Reid SuttonV
2005 Allele-specific methylation of a functional CTCF binding site upstream of MEG3 in the human imprinted domain of 14q32. Chromosome Res 13 809 818
11. WylieAA
MurphySK
OrtonTC
JirtleRL
2000 Novel imprinted DLK1/GTL2 domain on human chromosome 14 contains motifs that mimic those implicated in IGF2/H19 regulation. Genome Res 10 1711 1718
12. TierlingS
DalbertS
SchoppenhorstS
TsaiCE
OligerS
2007 High-resolution map and imprinting analysis of the Gtl2-Dnchc1 domain on mouse chromosome 12. Genomics 87 225 235
13. TakadaS
PaulsenM
TevendaleM
TsaiCE
KelseyG
2002 Epigenetic analysis of the Dlk1-Gtl2 imprinted domain on mouse chromosome 12: implications for imprinting control from comparison with Igf2-H19. Hum Mol Genet 11 77 86
14. OhlssonR
RenkawitzR
LobanenkovV
2001 CTCF is a uniquely versatile transcription regulator linked to epigenetics and disease. Trends Genet 17 520 527
15. HarkAT
SchoenherrCJ
KatzDJ
IngramRS
LevorseJM
2000 CTCF mediates methylation-sensitive enhancer-blocking activity at the H19/Igf2 locus. Nature 405 486 489
16. KanduriC
PantV
LoukinovD
PugachevaE
QiCF
2000 Functional association of CTCF with the insulator upstream of the H19 gene is parent of origin-specific and methylation-sensitive. Curr Biol 10 853 856
17. da RochaST
TevendaleM
KnowlesE
TakadaS
WatkinsM
2007 Restricted co-expression of Dlk1 and the reciprocally imprinted non-coding RNA, Gtl2: implications for cis-acting control. Dev Biol 306 810 823
18. WanLB
PanH
HannenhalliS
ChengY
MaJ
2008 Maternal depletion of CTCF reveals multiple functions during oocyte and preimplantation embryo development. Development 135 2729 2738
19. IderaabdullahFY
VigneauS
BartolomeiMS
2008 Genomic imprinting mechanisms in mammals. Mutat Res 647 77 85
20. FitzpatrickGV
PugachevaEM
ShinJY
AbdullaevZ
YangY
2007 Allele-specific binding of CTCF to the multipartite imprinting control region KvDMR1. Mol Cell Biol 27 2636 2647
21. HorsthemkeB
WagstaffJ
2008 Mechanisms of imprinting of the Prader-Willi/Angelman region. Am J Med Genet A 146A 2041 2052
22. LinSP
CoanP
da RochaST
SeitzH
CavailleJ
2007 Differential regulation of imprinting in the murine embryo and placenta by the Dlk1-Dio3 imprinting control region. Development 134 417 426
23. CoanPM
BurtonGJ
Ferguson-SmithAC
2005 Imprinted genes in the placenta–a review. Placenta 26 Suppl A S10 20
24. GeorgiadesP
WatkinsM
SuraniMA
Ferguson-SmithAC
2000 Parental origin-specific developmental defects in mice with uniparental disomy for chromosome 12. Development 127 4719 4728
25. TakadaS
TevendaleM
BakerJ
GeorgiadesP
CampbellE
2000 Delta-like and gtl2 are reciprocally expressed, differentially methylated linked imprinted genes on mouse chromosome 12. Curr Biol 10 1135 1138
26. LinSP
YoungsonN
TakadaS
SeitzH
ReikW
2003 Asymmetric regulation of imprinting on the maternal and paternal chromosomes at the Dlk1-Gtl2 imprinted cluster on mouse chromosome 12. Nat Genet 35 97 102
27. TakahashiN
OkamotoA
KobayashiR
ShiraiM
ObataY
2009 Deletion of Gtl2, imprinted non-coding RNA, with its differentially methylated region induces lethal parent-origin-dependent defects in mice. Hum Mol Genet 18 1879 1888
28. LewisA
MitsuyaK
UmlaufD
SmithP
DeanW
2004 Imprinting on distal chromosome 7 in the placenta involves repressive histone methylation independent of DNA methylation. Nat Genet 36 1291 1295
29. UmlaufD
GotoY
CaoR
CerqueiraF
WagschalA
2004 Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat Genet 36 1296 1300
30. SekitaY
WagatsumaH
IrieM
KobayashiS
KohdaT
2006 Aberrant regulation of imprinted gene expression in Gtl2lacZ mice. Cytogenet. Genome Res 113 223 229
31. SteshinaEY
CarrMS
GlickEA
YevtodiyenkoA
AppelbeOK
2006 Loss of imprinting at the Dlk1-Gtl2 locus caused by insertional mutagenesis in the Gtl2 5′ region. BMC Genet 7 44
32. CharlierC
SegersK
KarimL
ShayT
GyapayG
2001 The callipyge mutation enhances the expression of coregulated imprinted genes in cis without affecting their imprinting status. Nat Genet 27 367 369
33. GeorgesM
CharlierC
CockettN
2003 The callipyge locus: evidence for the trans interaction of reciprocally imprinted genes. Trends Genet 19 248 252
34. MoonYS
SmasCM
LeeK
VillenaJA
KimKH
2002 Mice lacking paternally expressed Pref-1/Dlk1 display growth retardation and accelerated adiposity. Mol Cell Biol 22 5585 5592
35. TsaiCE
LinSP
ItoM
TakagiN
TakadaS
2002 Genomic imprinting contributes to thyroid hormone metabolism in the mouse embryo. Curr Biol 12 1221 1226
36. SekitaY
WagatsumaH
NakamuraK
OnoR
KagamiM
2008 Role of retrotransposon-derived imprinted gene, Rtl1, in the feto-maternal interface of mouse placenta. Nat Genet 40 243 248
37. SeitzH
YoungsonN
LinSP
DalbertS
PaulsenM
2003 Imprinted microRNA genes transcribed antisense to a reciprocally imprinted retrotransposon-like gene. Nat Genet 34 261 262
38. DavisE
CaimentF
TordoirX
CavailléJ
Ferguson-SmithA
2005 RNAi-mediated allelic trans-interaction at the imprinted Rtl1/Peg11 locus. Curr Biol 15 743 749
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Web-Based, Participant-Driven Studies Yield Novel Genetic Associations for Common Traits
- The IG-DMR and the -DMR at Human Chromosome 14q32.2: Hierarchical Interaction and Distinct Functional Properties as Imprinting Control Centers
- Cushing's Syndrome and Fetal Features Resurgence in Adrenal Cortex–Specific Knockout Mice
- Amplification of a Cytochrome P450 Gene Is Associated with Resistance to Neonicotinoid Insecticides in the Aphid
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy