
Mice with Alopecia, Osteoporosis, and Systemic Amyloidosis Due to Mutation in , a Gene Coding for Palmitoyl Acyltransferase
Protein palmitoylation has emerged as an important mechanism for regulating protein trafficking, stability, and protein–protein interactions; however, its relevance to disease processes is not clear. Using a genome-wide, phenotype driven N-ethyl-N-nitrosourea–mediated mutagenesis screen, we identified mice with failure to thrive, shortened life span, skin and hair abnormalities including alopecia, severe osteoporosis, and systemic amyloidosis (both AA and AL amyloids depositions). Whole-genome homozygosity mapping with 295 SNP markers and fine mapping with an additional 50 SNPs localized the disease gene to chromosome 7 between 53.9 and 56.3 Mb. A nonsense mutation (c.1273A>T) was located in exon 12 of the Zdhhc13 gene (Zinc finger, DHHC domain containing 13), a gene coding for palmitoyl transferase. The mutation predicted a truncated protein (R425X), and real-time PCR showed markedly reduced Zdhhc13 mRNA. A second gene trap allele of Zdhhc13 has the same phenotypes, suggesting that this is a loss of function allele. This is the first report that palmitoyl transferase deficiency causes a severe phenotype, and it establishes a direct link between protein palmitoylation and regulation of diverse physiologic functions where its absence can result in profound disease pathology. This mouse model can be used to investigate mechanisms where improper palmitoylation leads to disease processes and to understand molecular mechanisms underlying human alopecia, osteoporosis, and amyloidosis and many other neurodegenerative diseases caused by protein misfolding and amyloidosis.
Published in the journal:
. PLoS Genet 6(6): e32767. doi:10.1371/journal.pgen.1000985
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000985
Summary
Protein palmitoylation has emerged as an important mechanism for regulating protein trafficking, stability, and protein–protein interactions; however, its relevance to disease processes is not clear. Using a genome-wide, phenotype driven N-ethyl-N-nitrosourea–mediated mutagenesis screen, we identified mice with failure to thrive, shortened life span, skin and hair abnormalities including alopecia, severe osteoporosis, and systemic amyloidosis (both AA and AL amyloids depositions). Whole-genome homozygosity mapping with 295 SNP markers and fine mapping with an additional 50 SNPs localized the disease gene to chromosome 7 between 53.9 and 56.3 Mb. A nonsense mutation (c.1273A>T) was located in exon 12 of the Zdhhc13 gene (Zinc finger, DHHC domain containing 13), a gene coding for palmitoyl transferase. The mutation predicted a truncated protein (R425X), and real-time PCR showed markedly reduced Zdhhc13 mRNA. A second gene trap allele of Zdhhc13 has the same phenotypes, suggesting that this is a loss of function allele. This is the first report that palmitoyl transferase deficiency causes a severe phenotype, and it establishes a direct link between protein palmitoylation and regulation of diverse physiologic functions where its absence can result in profound disease pathology. This mouse model can be used to investigate mechanisms where improper palmitoylation leads to disease processes and to understand molecular mechanisms underlying human alopecia, osteoporosis, and amyloidosis and many other neurodegenerative diseases caused by protein misfolding and amyloidosis.
Introduction
Proteins can be modified by a variety of lipids, including myristate (C14), farnesyl (C15), palmitate (C16), geranylgeranyl (C20) and glycosylphosphatidylinositol (GPI). Palmitoylation is one of the most common post-translational lipid modifications that involve the addition of palmitate to specific cysteine residues of proteins via a thioester linkage [1]–[3]. Although most of the lipid modifications are irreversible, protein S-palmitoylation can be either permanent or transient, which allows it to dynamically regulate protein function [1],[4]. Numerous soluble and integral membrane proteins have been shown to be palmitoylated including signaling proteins, enzymes, scaffolding proteins, ion channels, cell adhesion molecules and neuronal proteins. Specific examples are oncogenic Ras proteins, trimeric G protein α subunit, Rap2b, RhoB, eNOS, SNAP-25, PSD-95 postsynaptic scaffolding protein, huntingtin and anthrax toxin receptor [1], [2], [5]–[8].
Palmitoyl post-translational modification has recently emerged as an important mechanism for modulating protein targeting, trafficking, stability and protein-protein interactions, and plays roles in numerous cellular processes, including signaling, apoptosis and neuronal transmission [1], [3].
Although palmitoylation was first described over 30 years ago, the genes coding for enzymes involved in protein palmitoylation, the palmitoyl acyltransferase (PATs), have only recently been discovered [7], [9]. To date, at least 23 members of PATs have been identified in the mammalian genome [9], [10]. This family of proteins contains a cysteine-rich domain (CRD) with a core Asp-His-His-Cys (DHHC) motif that is essential for PAT activity [7], [9], [11]. The presence of so many PATs in a single organism could be due to differences in substrate specificities, intracellular localizations or tissue distributions. For example, DHHC2 and DHHC15 are more specific to PSD-95 and GAP-43, DHHC9 and DHHC18 are specific to H-Ras and N-Ras, while DHHC3 and the closely related DHHC7 have broad substrate specificities [2], [8]. In neuronal tissue, DHHC13 and 17 modulate huntingtin palmitoylation and DHHC8 modulates paralemmin-1 [11]. The substrate specificity appears to be determined by the regulatory domains outside the DHHC domains of the enzymes [8], [11].
Despite the functional importance of protein palmitoylation at the cellular and biochemical levels, its physiological role and its relevance to disease processes is not clear. Oncogenic Ras proteins and huntingtin are direct targets for palmitoylation thus, they may be involved in the disease process. Disregulation of DHHC2 may be involved in cancer metastasis [12]. The sole mouse model of DHHC deficiency is the Zdhhc8 knockout; these mice have a mild behavior phenotype with a decrease in exploratory activity and a deficiency in prepulse inhibition. These behavioral changes are only observed in female mice. The phenotypes together with genetic evidence may support the hypothesis that DHHC8 is a risk factor for schizophrenia [13]. Systematic knockdown of the Zdhhc genes has not been done, which could provide some unexpected physiological roles for DHHC proteins. Here we report on mice with a mutation in the Zdhhc13, a gene coding for palmitoyl acyltransferase, which catalyzes the reaction of protein palmitoylation [11]. Mutant mice exhibit a severe phenotype and profound pathology involving multi-organ/systems. These mice, developed cachexia, alopecia, osteoporosis, systemic amyloidosis, failed to thrive and succumbed to early death.
Results
The mutant mice were analyzed either on a C57BL/6×129S6/SvEv or C57BL/6×129S6/SvEv × C3H mixed genetic background. The phenotypes described herein are all penetrant in both genetic backgrounds.
Clinical Phenotypes
General appearance
Affected mice appeared normal at birth, but by postnatal day 7 were small in size and developed hypotrichosis; these features differentiated affected from normal siblings. The affected male mice had poor weight gain and weighed 50% less than the unaffected siblings (Figure 1A). Affected females mice also showed similar poor weight gain (data not shown). In addition, these mice, regardless of sex, had a shortened life span; about 50% died before 7.5 months of age and only 20% survived beyond one year of age (Figure 1B). Furthermore, the affected mice showed generalized hypotrichosis and hair loss particularly certain body parts, some hairs remained over the head and back, although, they were thin, short and had decreased luster (Figure 2). Skin was loose with wrinkling and folding (Figure 2C). Kyphosis was evident beginning at day 28 (Figure 2, also shown in Figure 3A). When the gene responsible for these phenotypes was identified as Zhddc13 (see below under Identification of the mutated gene), it was clear that only homozygous Zdhhc13 -/- exihibited abnormal phenotypes, while heterozygous Zhddc13 +/ − displayed normalcy like the wild-type (+/+).



Hematology and blood chemistry
Complete blood counts obtained from affected mice at 4 weeks and 30 weeks of age were comparable to the wild-type, except that adult mutant mice showed neutrophilia (mutant = 58.8±2.24 and unaffected = 38.5±4.13, P<0.05) and lymphocytopenia (mutant = 33.1±1.86 and 53.8±4.88 unaffected) despite normal WBC counts (Table S1). Blood biochemistry revealed elevations in AST and ALT enzymes in the adult (ALT in mutant = 100.6±15.3 and in unaffected = 52.8±5.07, AST in mutant = 44.5±7.18 and in unaffected = 24.8±2.72, P<0.05). However, at 4 weeks of age only AST was elevated (mutant = 101.6±15.6 and unaffected = 56.4±3.09, P<0.05) (Table S2). About 10% of adult mice also showed elevations in BUN (up to 180 mg/dl), CPK and total bilirubin. Serum calcium, magnesium and C-reactive protein were all within normal limits in the affected mice at both 4 weeks and 30 weeks of age.
Bone studies
Radiographic examinations showed severe kyphosis with marked increased spinal angle in the affected mice (Figure 3A). Osteoporosis was also profound, as evidenced by a decrease in the trabecular number of femur (Figure 3B and Table 1) and by other trabecular bone parameters, including a decrease in bone volume density (BV/TV) and bone mineral density (BMD), along with an increase in the structure model index (SMI), which indicated an abundance of rod-like trabeculae (Table 1). These features of osteoporosis could be seen as early as 4 weeks of age (data not shown).

Histopathological analyses
Post mortem examinations revealed hepatosplenomegaly (2-3 times normal), severe muscle wasting and reduced white and brown adipose tissue. Skin histopathology of the affected mice showed hyperkeratosis and epidermis hyperplasia with thin dermis and scanty adipose tissue (Figure 4A). Hair follicles in different stages, such as anagen, catagen, and telogen, could be observed in wild-type mice (Figure 4D), while mutant mice had significantly fewer active hair follicles with most remaining in the late telogen phase. There were no hair shafts in the affected hair follicles and the upper portion was dilated (Figure 4C). Amyloid deposition was observed in the entire dermis in the affected mice as homogenous eosinophilic substances by H&E stain (Figure 5A). This was confirmed by Congo red stain as pink-red deposits (Figure 5B) and under a polarizing microscope as yellow-green birefringence (Figure 5C).


Amyloid depositions were also found in most of the other major organs examined, except for muscle. Amyloids were seen in liver, spleen, kidney, adrenal gland, pancreas, salivary glands, heart, lung, intestine and brain. The amyloids deposited in organs and their severity in young (4 weeks) and older animals (20–32 weeks) are summarized in Table 2. In general, there was a progressive increase of amyloid with age.

In the liver, amyloid deposits were observed mainly in sinusoids around perivascular areas (Portal and central veins) (Figure 6A). Enlarged Kupffer cells containing amyloid substance were also seen and in severe cases, massive amyloid deposits disrupted the hepatic architecture (data not shown). Mild amyloid deposition in the space of Disse or sinusoid was also found in young mice as early as 4 weeks of age (Table 2).

In the spleen, accumulations of amyloid appeared in peri-white pulp, connective tissue frameworks of red pulp and central arterial walls of white pulp (Figure 6B). Mild amyloid deposition was also observed in young animals; however, the arterial wall findings were not detectable in young mutants (Table 2).
In the kidney, amyloid accumulated in the glomerulus (Figure 6C), renal tubules (Figure 6D) and perivascular areas. Thickenings of the basement membranes of glomerular capillaries and mesangial matrix were common lesions in mutants. In the advanced stage, the glomerular capillaries were obliterated, and the glomerular structures were completely destroyed. The renal tubules became dilated with large amounts of filtrated substances (as an eosinophilic substance, presumably albumin that is observed as a result of a glomerular filtration defect) and some tubular epithelial cells showed atrophy. In young animals, renal amyloidosis was milder, but could be seen in both glomerulus and tubules (Table 2).
Massive amyloid deposits were also observed in the adrenal glands in adult mice. Amyloid accumulated in the zona fasciculate, zona reticularis, and in part of the zona glomerulosa and medulla (Figure 6E).
In the pancreas, amyloid accumulated in both exocrine pancreatic tissues and islet of Langerhans (Figure 6F) and, in the advanced stage, acinar cells were degenerated and completely replaced by amyloid. Acinar cells in salivary glands were also full of amyloid (Figure 6G).
In the heart, amyloid deposition could be seen in the blood vessel walls, and only in severe cases, amyloid was observed in the myocardium (Figure 6H). Small amounts of amyloid deposits could also been found in other organs such as lung, intestine and brain. No amyloid deposition was found in skeletal muscle.
Immunohistochemistry Staining
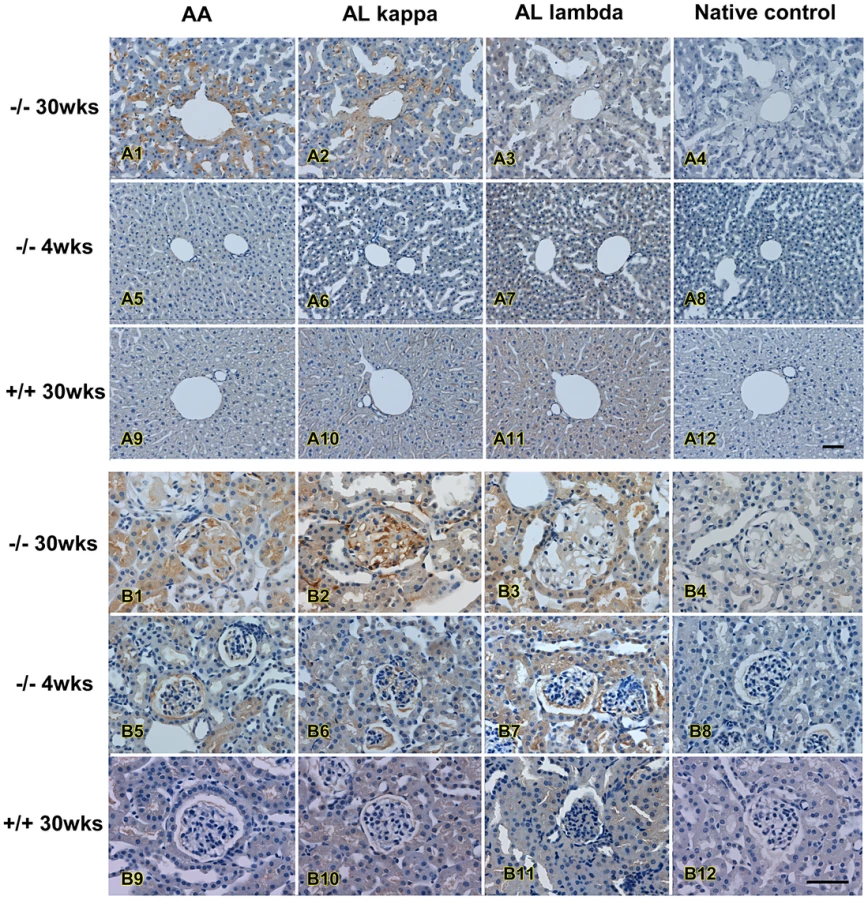
Immunohistochemistry staining was performed using anti-amyloid A, anti-kappa light chain and anti-lambda light chain to confirm the amyloidosis and to differentiate AA and AL type of amyloidosis. In liver, amyloid AA and AL κ were the major amyloid detected, primarily in sinusoids and around the portal vein, while the amount of AL λ amyloid was less (Figure 7, upper panel). A similar pattern was also observed in kidney glomerulus and tubular cells in which AA and AL κ were the predominant amyloids (Figure 7, lower panel). Both AA and AL κ could also be detected in the kidneys of the young animals (Table 2).
Identification of the Mutant Gene
To map the gene responsible for these abnormal phenotypes, affected mice in the B6×129 mix background were out-crossed to C3H/HeJ mice to generate N1 (B6 and C3H hybrid) affected offspring, then were intercrossed. The offspring of this intercross (N1F1) were used in the genomic analysis. Whole genome SNP homozygosity mapping revealed one region located between 46.4 and 64.7 Mb (18.3 Mb) of chromosome seven with 90% B6 homozygosity in consecutive SNPs (Figure 8A). Fine mapping narrowed down the candidate region to within 2.4 Mb (between 53.9 and 56.3 Mb) on chromosome 7 (Figure 8B). This region contained 64 genes. Because our affected mice showed generalized amyloidosis, we concentrated on the amyloid related genes located in this region which included Saa1l (serum amyloid A-like 1), Saa3 (serum amyloid A 3), Saa4 (serum amyloid A 4), Saa1 (serum amyloid A 1), Saa2 (serum amyloid A 2) and Zdhhc13 (zinc finger, DHHC domain containing 13). Direct DNA sequencing of genes in affected mice revealed a homozygous A to T substitution in exon 12 of Zdhhc13 (c.1273A>T) (Figure 8C). The parents were heterozygous for this mutation. Further study showed that the homozygous c.1273A>T mutation (−/−) completely segregated with the abnormal phenotypes. Siblings, as well as parents, that were heterozygous for this mutation (+/−) were phenotypically normal. This A to T substitution resulted in a stop codon (AGA>TGA) (arg-425-stop codon) and predicted a truncated protein. No other mutations were found in the remaining exons of gene Zdhhc13 or in its promoter region. All other amyloid-related genes in the candidate region were also normal without detectable mutations.

Real time RT-PCR showed that the tissue expression of Zdhhc13 mRNA was significantly reduced to 26.23% in the liver and 15.59% in the kidney of the affected mice compared to the wild type, (Table 3). The decreased mRNA indicated nonsense RNA decay due to the premature stop codon in our mutant Zdhhc13 mice.

To confirm that we had identified the correct gene, we obtained a gene trap allele in Zdhhc13 from the SIGTR. This allele produced the same phenotype as the ENU-induced allele, with the exception of pink-eyes and a dilute coat color, which are associated with the closely linked Oca2 locus carried in the 129/Ola gene trap ES cells. The skin which showed abnormal hair follicles, lack of hair and thickened epidermis (Figure 9E) which are similar to the mutant mice identified by the ENU mutation.

Expression of Zdhhc13
We carried out Northern analysis of a variety of tissues to determine where Zdhhc13 is expressed. We found that Zdhhc13 is expressed in most adult tissues, but at low levels in the liver, skin, and lung (Figure 9A). We hypothesized that Zdhhc13 may be needed most during development of these tissues, so we analyzed expression of Zdhhc13 in liver, skin, lung and brain at three different time points: postnatal day (P) 2, P8 and P30. Zdhhc13 was expressed most highly in the liver, lung, and brain at P2, showing that transcripts are developmentally regulated (Figure 9B). In contrast, Zdhhc13 is expressed most highly in skin at P8, when hair follicles are maturing. We examined the gene trap allele, which contains a lacZ reporter gene, for expression of Zdhhc13 in the skin, and found that it is expressed in the epithelium surrounding the hair follicles, consistent with a role in hair growth (Figure 9).
Mutation in Zdhhc13 Affects Protein Palmitoylation
Huntingtin is a known substrate of Zdhhc13 [11]. To demonstrate the palmitoylation defect caused by the mutation, HEK 293 T cells were co-transfected with huntingtin-myc and Zdhhc13-flag (WT and mutant). We examined the palmitoylation levels using acyl-biotin exchange assay after immunoprecipitation of huntingtin with anti-myc antibody and found that the huntingtin was palmitoylated by the wild-type Zdhhc13. The ability of mutant Zdhhc13 to palmitoylate huntingtin was greatly reduced by the mutant Zdhhc13, to a level indistinguishable from the endogenous palmitoyl activity present in the control (Figure 10A).

Moreover, when we examined the IgG light chain purified from serum of wild and mutant mice, we observed much less palmitoylated signals in the mutant mice as compared to the wild type mice (Figure 10C).
Discussion
Using ENU-mutagenesis, we have identified mice with severe phenotypes manifested by failure to thrive, alopecia, osteoporosis, systemic amyloidosis and early death. We found that a nonsense mutation (R425X) in Zdhhc13 was the cause of these abnormal phenotypes. To ensure that other mutations induced by ENU would not confound the observed phenotypes, mice examined after 6 generations of outcross breeding continued to show 100% phenotype and genotype correlations [14]. Further, a gene trap allele of Zdhhc13, which contains a vector inserted into the first intron, exhibits the similar phenotypes (data not shown).
Zdhhc13, also named huntingtin-interacting protein-like (HIP14L), shares 51% identity and 69% similarity with huntingtin-interacting protein-14 (HIP14) or Zdhhc17 between #45 and #611 of the 622 residues [15]. Both Zdhhc13 and 17 belong to a family of enzymes that are involved in attaching lipids to proteins, the palmitoyl acyltransferases (PATs). Zdhhc13, in addition to being a PAT, is also a mediator of Mg2+ transport. Inhibition of palmitoylation by 2-bromopalmitate (2BP) diminished Mg2+ transport by about 50% [16]. The causal mutation (R425X) in Zdhhc13 mice would predict the synthesis of a truncated protein lacking the zinc-finger DHHC-CRD domain (#426-476) and the active site (C456) for the formation of an S-palmitoyl cysteine intermediate (Figure S1). Thus, it is unlikely that this truncated protein can perform any palmitoylation function, consistent with the similarity of phenotypes with the gene trap allele. Because our mutant mice had normal serum magnesium and calcium levels and demonstrated no clinical evidence of magnesium deficiency, we propose that the observed phenotypes originated from the loss of the enzymatic function of Zdhhc13 as a PAT. Indeed, we have shown that mutation in the Zdhhc13 affects the protein palmitoylation.
The exact mechanism by which the mutation of Zdhhc13 resulted in such diverse pathologies is not clear. Amyloidosis is a devastating group of disorders in which normally soluble proteins are misfolded and aggregate to form insoluble amyloid fibrils with a β-sheet structure and presumably trigger an unfolded protein response (UPR) and its downstream pathways, including autophagy and cell death by apoptosis [17]-[19]. There are two major types of systemic amyloidosis. AA-amyloidosis, also called secondary or reactive amyloidosis, is a consequence of prolonged high level expression, mainly in the liver, of the acute-phase protein Serum Amyloid A precursor protein (SAA). AA-amyloidosis is usually associated with chronic inflammatory conditions but it can also be caused by mutations in a constitutively expressed protein, resulting in its greater tendency to aggregate [20], [21]. The second type of amyloidosis is called AL-amyloidosis. It is either a primary or a multiple myeloma-associated amyloidosis; its fibrils are derived from fragments of monoclonal immunoglobulin light chain (λ and κ) condensed into β-pleated sheet structures as a result of incomplete breakdown in the autophagolysosomes [22]. It is rare to find both AA and AL types in the same patient, and such cases only account for ∼2–3% of all amyloidosis patients [23].
Our Zdhhc13 mutant mice manifested both AA and AL amyloids (Figure 5, Figure 6, Figure 7). Liver, spleen, kidneys, skin, adrenal and salivary glands are the most affected organs resulting in hepatosplenomegaly, nephromegaly, sialadenosis (non-inflammatory swelling of the salivary glands) and skin involvement. We cannot attribute this observed amyloidosis to inflammation, as CRP levels were normal (Table S2). Nor can we attribute it to multiple myeloma, as blood and bone marrow contained no excess plasma cells (data not shown). Furthermore, sequences of all amyloid-related genes in the candidate regions were normal except for the single mutation (R425X) in Zdhhc13. The coexistence of AL and AA systemic amyloidosis could be attributed to the fact that the presence of AL type amyloid fibrils acting as an amyloid-enhancing factor (AEF) and enhance the AA amyloid deposition [23]–[26]. Alternatively, a deficiency of Zdhhc13 may play a role in the amyloidogenesis. Palmitoylation is known to affect protein stability by influencing a protein's access to an ubiquitinating enzyme [27]. Palmitoylation is also known to protect huntingtin from aggregation [28], and prevent oligomerization of certain proteins [29]. Moreover, defective palmitoylation results in aggregation of amyloid β-sheet, which leads to the formation of fibril [30].
Therefore, we propose that a deficiency of Zdhhc13 PAT activity may have caused amyloidosis in our mice. The lack of Zdhhc13 PAT activity affected palmitoylation of a set of unspecified protein targets and compromised their conformational stability and subcellular localization, eventually causing systemic amyloidosis of both the AA and AL types [26]. Coincidentally, a computer algorithm predicted the presence of palmitoylation site(s) on both SAA and the light chains of IgG [31]. Consistent with this notion was our demonstration of reduced level of palmitoylation of IgG light chain in the mutant mice.
Deficiency of protein palmitoylation in the Zdhhc13 mutant mice could also explain the apparent osteoporosis because palmitoylation regulates osteoblast differentiation through bone morphogenesis protein (BMP)-induced Osterix expression [6]. Deletion of Osterix leads to a loss of mature osteoblasts and a lack of calcified bones (Osteoporosis), other signaling pathways such as NF-κB may be also involved in the development of the severe osteoporosis phenotype observed as early as weaning [32].
The Zdhhc13 mutant mice also showed significant skin pathology with hypotrichosis, alopecia and loose skin with wrinkling and folding. Histopathology revealed epidermal hyperplasia with a thin dermis, inactive hair follicles and amyloid deposition. Expression analysis shows that expression of Zdhhc13 is upregulated at the time of follicle maturation (P8), consistent with a direct role of Zdhhc13 in hair formation. Although the exact molecular mechanisms are not clear, these phenotypes are consistent with defects in the NF-κB signaling pathways [33]. Another possible mechanism responsible for the skin pathology is the BMP-induced MAPK pathway as protein palmitoylation plays an important role in BMP-induced MAPK pathway activation [6]. BMP is involved not only in osteoblast differentiation but also in epidermal proliferation and differentiation, hair follicle cycling and innervations [34]. Since Zdhhc13 normally upregulates both NF-κB and MAPK signaling pathways [35] the Zdhhc13 mutation may significantly affect these pathways, leading to the disease phenotypes.
In summary, we report that deficiency of a single palmitoyl acyltransferase (Zdhhc13) can cause severe systemic phenotypes, including failure to thrive, cachexia, osteoporosis, alopecia, multi-organs/systems dysfunction secondary to systemic amyloidosis and early death. Our results established a direct link between protein palmitoylation and regulation of important diverse physiological functions and indicated that its absence can result in profound disease pathology. This mouse model will be useful for further investigation of the mechanisms by which improper palmitoylation leads to disease processes. The identification of target proteins of ZDHHC13 would be an important first step for understanding the molecular mechanisms underlying human alopecia, osteoporosis and many neurodegenerative diseases caused by protein misfolding and amyloidosis.
Materials and Methods
Mouse Lines
The first recessive mutant allele was generated by a conventional ENU mutagenesis regimen. [36]. Briefly, multiple doses of N-ethyl-N-nitrosourea (ENU) (100 mg per kg body weight) were injected to mutagenize spermatogonia of C57BL/6J males (G0 generation). Recessive mutations were isolated in the third generation of breeding to females that carried either the balancer chromosome 129S6.Inv(11)8Brd or 129.Rex mutations, both of which had been made congenic on a 129S6/SvEvTac genetic background (N = 10). The mutant mice reported here were identified by their small size and hypotrichosis as early as postnatal day 7 and were designated as skin and coat mutation 4 (skcm04Jus). The mutant line was inherited as a recessive trait that segregated independently of the chromosome 11 balancer and the phenotype was completely penetrant in the 129S6/SvEv genetic background, and, later on, in the C3H background. The experimental protocols in this study were reviewed and approved by the Institutional Animal Care and Utilization Committee of Academia Sinica.
The gene trap allele was produced from embryonic stem (ES) cells AC0492 obtained from the Sanger Institute Gene Trap Resource (SIGTR). ES cells were injected into C57BL/6J blastocysts by the Darwin Genetics Core at Baylor College of Medicine. Chimeras were obtained, mated to C57BL/6J mice, and the allele was transmitted through the germline to generate Zdhhc13SIGTR mice. Subsequent experiments were performed on mice from this mixed 129/B6 genetic background. After initial genotyping of ES cells for the gene trap allele per protocols available from the resource, the phenotype was used to follow transmission of the allele. Zdhhc13 is located at 56 Mb on Chromosome 7, which is only 7 Mb from Oculocutaneous albinism 2 (Oca2; pink-eyed dilution), which is located at 63 Mb. Therefore, mice carrying the gene trap allele were also pink-eyed and had dilute coat colors because of the Oca2 mutation carried in the 129/Ola ES cells used to generate the gene trap.
Blood Chemistry
Blood samples were obtained through an incision of the tail artery or by cardiac puncture at the time of sacrifice and collected in a heparinized tube (MICROTAINER, BD Diagnostics, Franklin Lakes, NJ). Complete hemogram was carried out using Abbott Cell-DYN 3700 Veterinary Haematology Analyzer (Abbott Laboratory, Illinois, USA). Thin blood smears were taken directly from the tail artery, fixed with absolute methanol for 5 minutes and stained by modified Wright's Giemsa stain for Plasma cells (Plasma B cells) identification. Plasma was analyzed using the FUJI DRI-CHEM SYSTEM 3500s (Fuji Photo Film Co. Ltd.) for measurement of aspartate aminotransferase (AST; U/l), alanine aminotransferase (ALT; U/l), creatinine phosphokinase (CPK; U/l), total cholesterol (TCHO; mg/dl), total protein (TP; g/dl), albumin (ALB; g/dl), globulin (GLO; g/dl), total bilirubin (TBIL; mg/d), blood urea nitrogen (BUN; mg/dl), C-reactive protein (CRP; mg/dl), calcium (Ca; g/dl) and magnesium (Mg; g/dl).
Micro-Computed Tomography (Micro–CT) Analysis of Bone
For trabecular bone analysis and 3D images, a micro-CT scanner (Skyscan-1076, Skyscan, Belgium) was operated at 50 kV, 200 uA, 0.4° of rotation step, 0.5 mm Al filter and 9 um/pixel of scan resolution. For bone mineral density (BMD) analysis, it was operated at 50KV, 200 uA, 1° of rotation step, 0.5 mm Al filter and 35 um/pixel of scan resolution. Cross-sections were reconstructed using a cone-beam algorithm (software Cone_rec; Skyscan, Belgium). Files were then imported into CTAn software (Skyscan) for three-dimensional analysis and three-dimensional image generation. BMD for each femur was measured by CTAn, which was calibrated using of phantoms with known BMD (0.25∼0.75 g/cm3).
Histopathology
Mice were sacrificed with overdoses of sodium pentobarbital for the histop-athological examinations. After flushing with normal saline, mice were perfused through the heart with 4% paraformaldehyde in 0.1M PBS, pH7.4, the perfusion flow rate, (4 ml/min) was controlled by an infusion pump (Bio-Rad, Econo Pump). A total of 37 organs and tissues, including heart, lung, liver, kidney, spleen, pancreas, adrenal gland, salivary gland, brain, skin, adipose tissues, skeletal muscles and bone, were removed, embedded in paraffin, cut into 5 µm sections and stained with hematoxylin-eosin (H&E) for general pathological examinations. Other serial sections were also processed for Congo Red staining to detect amyloid; and immunohistochemistry staining for amyloid classification. Bone marrow was aspirated from both femora for bone marrow smears immediately after sacrificing by cervical vertebral dislocation. Smears were fixed with absolute ethanol for 5 minutes and stained by modified Wright's Giemsa stain for Plasma cells identification.
Immunohistochemistry
Immunostaining used the following antibodies: rabbit anti-human λ light chains polyclonal antibody, rabbit anti-human κ light chains polyclonal antibody, mouse anti-human amyloid A monoclonal antibody (DakoCytomation). Tissue sections were pretreated with concentrated formic acid for 1 min, washed in tris-buffered saline (TBS) for 10 min, then incubated in Rodent Block M (BioCare) or tris-buffered saline Tween (TBST) containing 2% bovine serum albumin and 3% normal goat serum at 37°C for 30 min. Then, tissue sections were incubated with primary antibody, washed in TBST, incubated in 3% H2O2 for 15 min at room temperature and then washed again in TBST followed by incubation with horseradish peroxidase-conjugated secondary antibody (anti-rabbit IgG and goat anti-mouse IgG (Jackson ImmunoResearch. West Grove, USA). Color was developed with 0.1% 3,3′-diaminobenzidine.
Mapping of Gene Responsible for Abnormal Phenotypes
For the purpose of rough mapping, the affected skcm04Jus mice, which were in a mixed 129S6/SvEv and C57BL/6 mixed genetic background at the N = 4 generation on 129S6/SvEvTac (obtained from Baylor College of Medicine) were outcrossed to the C3He/HeJ strain in Academia Sinica to generate N1 offspring, and N1 mice were then intercrossed with generate N1F1 offspring. DNA was collected from 32 affected N1F1 mice. A panel of 295 single nucleotide polymorphism (SNP) markers located on all 19 mouse autosomes and the X-chromosome for mouse strains C3H/HeJ, C57BL/6, DBA/2J or BALB/cByJ was selected from a SNP dataset containing 10,915 SNPs from 48 mouse strains (provided by Tim Wiltshire, Genomics Institute of the Novartis Research Foundation, San Diego, California). SNPs were chosen based on the criterion that the genotype of these strains at the 295 loci was different from that of C57BL/6J. Since point mutations were introduced into C57BL/6J genome by ENU, the recessive mutant phenotype will always associated with a homozygous B6 SNP genotype at the mutant locus. SNP genotyping using genomic DNA isolated from mouse tails (Puregene DNA purification kit, Gentra Systems, Minneapolis, MN, USA) was performed using high-throughput MALDI-TOF mass spectrometry [37], [38]. Primers and probes flanking the SNPs were designed in multiplex format using SpectroDESIGNER software (Sequenom, San Diego, CA, USA). PCRs were performed in a volume of 5 µl containing 0.15 U of Taq polymerase (HotStarTaq, Qiagen, Valencia, CA, and USA), 5.0 ng of genomic DNA, 1.0 pmol of each PCR primer and 2.5 nmol of dNTP. Thermocycling conditions were one cycle at 94°C for 15 min, 45 cycles of 94°C for 20 s, 56°C for 30 s, 72°C for 30 s and one final cycle of extension at 72°C for 3 min. Unincorporated dNTPs were dephosphorylated using 0.3 U of Shrimp Alkaline Phosphatase (Hoffman-LaRoche, Basel, Switzerland) followed by primer extension using 9 pmol of each primer extension probe, 4.5 nmole of the appropriate dNTP/ddNTP combination, and 1.28 U of Thermosequenase (Amersham Pharmacia, Piscataway, NJ, USA). Reactions were cycled at 94°C for 2 min, followed by 55 cycles of 94°C for 5 s, 52°C for 5 s and 72°C for 5 s. Following the addition of a cation exchange resin (SpectroCLEAN, Sequenom) to remove residual salt from the reactions, 15 nl of the purified primer extension reaction was spotted onto a 384-element silicon chip preloaded with 3-hydroxypicoloinic acid matrix (SpectroCHIP, Sequenom), using the SpectroPOINT (Sequenom). SpectroCHIPs were analyzed using a Bruker Biflex III MALDI-TOF SpectroREADER mass spectrometer (Sequenom) and spectra processed with SpectroTYPER (Sequenom).
For fine mapping, 52 SNPs covering the candidate region from 46468726 bp (SNP rs30814649) to 64723695 bp (SNP rs32491610) on chromosome 7 (Mouse Genomic Informatics (MGI) http://www.informatics.jax.org/javawi2/servlet/WIFetch?page=snpQF were selected. Strain C57BL/6 was used as a selected strain and C3He/HeJ and 129/SvEv as reference strains. DNA samples from 84 affected N1F1 mice and 10 parental heterozygous mice in a 96 well plate (MicroAmp Optical 96-Well Reaction Plate, Applied Biosystems) were used for SNP genotyping using high-throughput MALDI-TOF mass spectrometry.
Identification of the Mutant Gene
All exons, exon-intron junctions and 2.5 kb promoter regions of candidate genes, Saa11, Saa3, Saa4, Saa1, Saa2 and Zdhhc13, were amplified and sequenced. Primers were designed using the Primer3 program http://biotools.umassmed.edu/bioapps/primer3_www.cgi. The primers used for the detection of an exon 12 mutation in the Zdhhc13 gene were F, 5′-CTGGGTTGAGAGTATTCCACA-3′ and R, 5′-GAGATTAGCCACAGAGCTTCG-3′. PCR reactions were performed in a final volume of 25 µl, containing 50 pmol of each primer (0.5 ul), 10× Taq Buffer (10 mM Tris–HCl (pH 8.3), 50 mM KCl) with 1.5 mM MgCl2 (2.5 ul), 2.5 mM dNTPs (2.5 ul) and Taq DNA polymerase (5 U/ul) MDBio, Inc. (0.25 ul). Amplification conditions were an initial denaturation of 4 min. at 94°C, followed by 20 cycles of touchdown PCR in 30 s at 94°C, 30 s at 65°C (decrease 0.5°C per cycle), 40 s at 72°C; and a final 20 cycles in 30 s at 94°C, 30 s at 55°C, followed by 40 s at 72°C and then a final extension at 72°C for 5 min. All amplified PCR fragments were digested with shrimp alkaline phosphatase and ExoI to remove unincorporated primers and sequenced using the BigDye Terminator Cycle Sequencing Kit v1.1/3.1 (Applied Biosystems, Foster City, CA, USA) following the manufacturer's instructions. Sequencing products were separated on either ABI PRISM 3100 Genetic Analyzer or ABI PRISM 3700 DNA Analyzer (Applied Biosystems). Raw sequencing data were analyzed with the DNA Sequencing Analysis Software v3.7 (Applied Biosystems).
Expression Analysis
To examine the differences in the tissue expression of Zdhhc13, total RNA samples were extracted from liver and kidney of three 6-month old mutants as well as three aged-matched wild type C3He/HeJ mice using Trizol following the manufacturer's protocol. First-strand cDNA was synthesized using 1 µl oligo-dT 15 primer and 1 µl SuperScript III RT (200 U/1 µl) in 20 µl volume of 2 µg of total RNA, 1 µl of 10 mM dNTPs, 1 µl of reaction buffer (10 mM Tris-HCl pH 8.3, 2.5 mM KCl, 0.6 mM MgCl2), µl of RNase inhibitor40 U/µl, and 1 µl of 0.1 M DTT. Real-time quantitative RT-PCR analysis used the ABI PRISM 7700 Sequence Detection System (Applied Biosystems). RT-PCR amplification of Zdhhc13 was carried out using the following primer set: 5′ - GACTGGACGCTGCATAGGTT, forward strand in Zdhhc13 exon13, and 5′ - TGGCACAATGATTTGACCAG, reverse strand in Zdhhc13 exon 15. The primers were designed using Primer Express (Applied Biosystems).
The cDNA corresponding to 75 ng of reversed transcribed total RNA was amplified in a final volume of 20µl using Power SYBER green PCR Master mix in 20µl total reaction volume in duplicate assays for Zdhhc13 and endogenous B-actin as an internal control.
An analysis of the results was based on the Ct calculation, where Ct represents the cycle number at which fluorescence of the PCR samples crossed a given threshold. The expression level of β-actin was taken as the first “calibrator” to normalize the total Zdhhc13 mRNA in each tissue (ΔCt). Expression of, Zdhhc13 in each of the control mouse tissue was then taken as the second “calibrator” to normalize the expression of Zdhhc13 in the affected tissue accordingly (ΔΔCt). Final results were given as the relative amounts of Zdhhc13 mRNA in the affected mouse tissues as compared to the control (2ΔΔCt).
Northern analysis was carried out as previously described by Lorenzetti et al. [39], using 10 ug of total RNA isolated using RNA STAT-60 reagent (TEL-TEST, Inc., Friendswood, TX) according to the manufacturer's protocol, and transferred to a nylon membrane. The blot was hybridized using Ultrahyb (Ambion) with a probe for Zdhhc13 is the N-terminal 571 base pairs, which was PCR amplified with two primers (F-5′-ATGGAGGGCCCGGGCCT-3′, R-5′-TAAGCCGATAGCATGAGCG-3′). The probe for GAPDH is the N-terminal 509 base pairs, which was PCR amplified with two primers (F-5′-GGTCGGTGTGAACGGATTTGG-3′, R-5′-CATGAGCCCTTCCACAATGCC-3′). [39] B-galactosidase staining was carried out using X-gal staining, the skins of p1, p6 and p10 mice were embedded in Tissue Tek and frozen after fixation with 4% paraformaldehyde and sucrose protection. The 10 mm vertical cryosections were fixed again in 2% paraformaldehyde followed by serial washing (three times washing with 2 mM MgCl2 containing PBS, 0.02% NP40 and 0.01% deoxycholate). The sections were pre-incubated with the staining buffer (PBS supplemented with 5 mM K3Fe(CN)6, 5 mM K3Fe(CN)6, 2 mM MgCl2) for 2 min, and further incubated with the staining buffer supplemented with 1 mg/ml Xgal for 3 hrs. After washing with PBS, the sections were counterstained with Nuclear Fast Red (Vector) and then mounted.
Evaluation of Enzyme Activity of Zdhhc13
We used huntingtin, a known substrate for Zdhhc13, and acyl-biotin exchange assay [11] to measure the palmitoyl acyl transferase activity. Plasmid construction: The cDNA of wild type and mutant Zdhhc13 were subcloned into C-terminal p3xFLAG-CMV vector (Sigma). Huntingtin (1–548 aa) cDNA was subscloned into pcDNA4/myc-His (Invitrogen). Cell culture: HEK293T cell were used for transiently transfected with Lipofectamine 2000 (Invitrogen). At 24–48 hours posttransfection, cells were harvested with PBS and proteins were extracted with lysis buffer (LB, 150 mM NaCl, 50 mM Tris-HCl, 5 mM EDTA, 1 mM PMSF, 1X protease inhibitor (Roche), 0.2% Triton X-100, pH 7.4) containing 50 mM N-ethylmaleimide (NEM) (Sigma). Immunoprecipitation (IP) and immunobloting (IB) of huntingtin protein and acyl-biotin exchange assay to label S-palmitoylated protein were performed using Myc antibody (Invitrogen), 1∶250 for IP; 1∶5000 for IB; and FLAG, mouse, antibody (Sigma) 1∶2000 for IB; as described previously [11].
IgG Light Chains Purification and the Evaluation of S-Palmitoylation
Serum from 5 month old mice was used to purify the IgG light chain. Serum was incubated with Protein A/G agarose beads (Santa Cruze Biotechnology) for 1 hour at room temperature. After washing the beads with PBS for 3 times, IgG light chain were eluted with 0.2 M Glycine (pH 2.5) and neutralized with 1M Tris-HCl (pH 8.5). The eluted fractions were precipitated using the chloroform/methanol (C/M) precipitation method. Protein pellets were redissolved with 4% SDS buffer (50 mM Tris-HCl, 4% SDS, 5 mM EDTA, pH 7.4) containing 50 mM N-ethylmaleimide (NEM) (Sigma) for 10 minutes at 37°C. Following dilution with 3 vol of lysis buffer, the fraction was rotated end-over-end overnight at 4°C. Excess NEM was removed with 3 sequential C/M precipitations. Protein pellets were redissolved with 4% SDS buffer and divided to 2 portions. First portion was added to 3 vol of lysis buffer containing 1 M hydroxylamine, pH 7.2 and incubate for 1 hour at room temperature to remove palmitate group from the protein. Control portion was in lysis buffer without hydroxylamine. After incubation, 3 sequential C/M precipitations were performed on samples to remove hydroxylamine. Protein pellets were redissolved with 4% SDS buffer and were added with 3 vol of lysis buffer containing 0.5 µM biotin-BMCC (Pierce), pH 6.2 and incubate for 1 hour at 4°C to label the protein, followed by SDS-PAGE and Western blotting. Streptavidin protein - HRP (abcam) was used against biotinylated protein. Rabbit anti-human κ light chain polyclonal antibody (DakoCytomation) was used against IgG light chain.
Supporting Information
{kind=link}
Zdroje
1. LinderME
DeschenesRJ
2007 Palmitoylation: policing protein stability and traffic. Nat Rev Mol Cell Biol 8 74 84
2. IwanagaT
TsutsumiR
NoritakeJ
FukataY
FukataM
2009 Dynamic protein palmitoylation in cellular signaling. Prog Lipid Res 48 117 127
3. CharollaisJ
Van Der GootFG
2009 Palmitoylation of membrane proteins (Review). Mol Membr Biol 26 55 66
4. NadolskiMJ
LinderME
2007 Protein lipidation. Febs J 274 5202 5210
5. ReshMD
1999 Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim Biophys Acta 1451 1 16
6. LeongWF
ZhouT
LimGL
LiB
2009 Protein palmitoylation regulates osteoblast differentiation through BMP-induced osterix expression. PLoS ONE 4 e4135 doi:10.1371/journal.pone.0004135
7. LoboS
GreentreeWK
LinderME
DeschenesRJ
2002 Identification of a Ras palmitoyltransferase in Saccharomyces cerevisiae. J Biol Chem 277 41268 41273
8. TsutsumiR
FukataY
FukataM
2008 Discovery of protein-palmitoylating enzymes. Pflugers Arch 456 1199 1206
9. RothAF
FengY
ChenL
DavisNG
2002 The yeast DHHC cysteine-rich domain protein Akr1p is a palmitoyl transferase. J Cell Biol 159 23 28
10. LinderME
DeschenesRJ
2004 Model organisms lead the way to protein palmitoyltransferases. J Cell Sci 117 521 526
11. HuangK
SandersS
SingarajaR
OrbanP
CijsouwT
2009 Neuronal palmitoyl acyl transferases exhibit distinct substrate specificity. Faseb J 19 19
12. OyamaT
MiyoshiY
KoyamaK
NakagawaH
YamoriT
2000 Isolation of a novel gene on 8p21.3-22 whose expression is reduced significantly in human colorectal cancers with liver metastasis. Genes Chromosomes Cancer 29 9 15
13. MukaiJ
LiuH
BurtRA
SworDE
LaiWS
2004 Evidence that the gene encoding ZDHHC8 contributes to the risk of schizophrenia. Nat Genet 36 725 731
14. NoveroskeJK
WeberJS
JusticeMJ
2000 The mutagenic action of N-ethyl-N-nitrosourea in the mouse. Mamm Genome 11 478 483
15. SingarajaRR
HadanoS
MetzlerM
GivanS
WellingtonCL
2002 HIP14, a novel ankyrin domain-containing protein, links huntingtin to intracellular trafficking and endocytosis. Hum Mol Genet 11 2815 2828
16. GoytainA
HinesRM
QuammeGA
2008 Huntingtin-interacting proteins, HIP14 and HIP14L, mediate dual functions, palmitoyl acyltransferase and Mg2+ transport. J Biol Chem 283 33365 33374
17. RonD
WalterP
2007 Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8 519 529
18. HazenbergBP
vanGII
BijzetJ
JagerPL
van RijswijkMH
2004 Diagnostic and therapeutic approach of systemic amyloidosis. Neth J Med 62 121 128
19. ChienP
WeissmanJS
DePaceAH
2004 Emerging principles of conformation-based prion inheritance. Annu Rev Biochem 73 617 656
20. WestermarkP
BensonMD
BuxbaumJN
CohenAS
FrangioneB
2007 A primer of amyloid nomenclature. Amyloid 14 179 183
21. SelkoeDJ
2003 Folding proteins in fatal ways. Nature 426 900 904
22. MerliniG
BellottiV
2003 Molecular mechanisms of amyloidosis. N Engl J Med 349 583 596
23. RekhtmanN
HashKS
MoresiJM
2006 Mucocutaneous bullous amyloidosis with an unusual mixed protein composition of amyloid deposits. Br J Dermatol 154 751 754
24. van der HilstJC
van der MeerJW
DrenthJP
SimonA
2007 AL amyloidosis enhances development of amyloid A amyloidosis. Br J Dermatol 156 748 749
25. FuX
KorenagaT
FuL
XingY
GuoZ
2004 Induction of AApoAII amyloidosis by various heterogeneous amyloid fibrils. FEBS Lett 563 179 184
26. SolomonA
MacySD
WooliverC
WeissDT
WestermarkP
2009 Splenic plasma cells can serve as a source of amyloidogenic light chains. Blood 113 1501 1503
27. Valdez-TaubasJ
PelhamH
2005 Swf1-dependent palmitoylation of the SNARE Tlg1 prevents its ubiquitination and degradation. Embo J 24 2524 2532
28. YanaiA
HuangK
KangR
SingarajaRR
ArstikaitisP
2006 Palmitoylation of huntingtin by HIP14 is essential for its trafficking and function. Nat Neurosci 9 824 831
29. GustafssonM
ThybergJ
NaslundJ
EliassonE
JohanssonJ
1999 Amyloid fibril formation by pulmonary surfactant protein C. FEBS Lett 464 138 142
30. JohanssonJ
2001 Membrane properties and amyloid fibril formation of lung surfactant protein C. Biochem Soc Trans 29 601 606
31. RenJ
WenL
GaoX
JinC
XueY
2008 CSS-Palm 2.0: an updated software for palmitoylation sites prediction. Protein Eng Des Sel 21 639 644
32. ChangJ
WangZ
TangE
FanZ
McCauleyL
2009 Inhibition of osteoblastic bone formation by nuclear factor-[kappa]B. Nat Med 15 682 689
33. BellS
DegitzK
QuirlingM
JilgN
PageS
2003 Involvement of NF-kappaB signalling in skin physiology and disease. Cell Signal 15 1 7
34. CazeneuveC
AjrapetyanH
PapinS
Roudot-ThoravalF
GenevieveD
2000 Identification of MEFV-independent modifying genetic factors for familial Mediterranean fever. Am J Hum Genet 67 1136 1143
35. MatsudaA
SuzukiY
HondaG
MuramatsuS
MatsuzakiO
2003 Large-scale identification and characterization of human genes that activate NF-kappaB and MAPK signaling pathways. Oncogene 22 3307 3318
36. KileBT
HentgesKE
ClarkAT
NakamuraH
SalingerAP
2003 Functional genetic analysis of mouse chromosome 11. Nature 425 81 86
37. KaoHJ
ChengCF
ChenYH
HungSI
HuangCC
2006 ENU mutagenesis identifies mice with cardiac fibrosis and hepatic steatosis caused by a mutation in the mitochondrial trifunctional protein beta-subunit. Hum Mol Genet 15 3569 3577
38. WuJY
KaoHJ
LiSC
StevensR
HillmanS
2004 ENU mutagenesis identifies mice with mitochondrial branched-chain aminotransferase deficiency resembling human maple syrup urine disease. J Clin Invest 113 434 440
39. LorenzettiD
BishopCE
JusticeMJ
2004 Deletion of the Parkin coregulated gene causes male sterility in the quaking(viable) mouse mutant. Proc Natl Acad Sci U S A 101 8402 8407
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Web-Based, Participant-Driven Studies Yield Novel Genetic Associations for Common Traits
- The IG-DMR and the -DMR at Human Chromosome 14q32.2: Hierarchical Interaction and Distinct Functional Properties as Imprinting Control Centers
- Cushing's Syndrome and Fetal Features Resurgence in Adrenal Cortex–Specific Knockout Mice
- Amplification of a Cytochrome P450 Gene Is Associated with Resistance to Neonicotinoid Insecticides in the Aphid
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy