
Dynamic Switch of Negative Feedback Regulation in Akt–TOR Signaling
Akt represents a nodal point between the Insulin receptor and TOR signaling, and its activation by phosphorylation controls cell proliferation, cell size, and metabolism. The activity of Akt must be carefully balanced, as increased Akt signaling is frequently associated with cancer and as insufficient Akt signaling is linked to metabolic disease and diabetes mellitus. Using a genome-wide RNAi screen in Drosophila cells in culture, and in vivo analyses in the third instar wing imaginal disc, we studied the regulatory circuitries that define dAkt activation. We provide evidence that negative feedback regulation of dAkt occurs during normal Drosophila development in vivo. Whereas in cell culture dAkt is regulated by S6 Kinase (S6K)–dependent negative feedback, this feedback inhibition only plays a minor role in vivo. In contrast, dAkt activation under wild-type conditions is defined by feedback inhibition that depends on TOR Complex 1 (TORC1), but is S6K–independent. This feedback inhibition is switched from TORC1 to S6K only in the context of enhanced TORC1 activity, as triggered by mutations in tsc2. These results illustrate how the Akt–TOR pathway dynamically adapts the routing of negative feedback in response to the activity load of its signaling circuit in vivo.
Published in the journal:
. PLoS Genet 6(6): e32767. doi:10.1371/journal.pgen.1000990
Category:
Research Article
doi:
https://doi.org/10.1371/journal.pgen.1000990
Summary
Akt represents a nodal point between the Insulin receptor and TOR signaling, and its activation by phosphorylation controls cell proliferation, cell size, and metabolism. The activity of Akt must be carefully balanced, as increased Akt signaling is frequently associated with cancer and as insufficient Akt signaling is linked to metabolic disease and diabetes mellitus. Using a genome-wide RNAi screen in Drosophila cells in culture, and in vivo analyses in the third instar wing imaginal disc, we studied the regulatory circuitries that define dAkt activation. We provide evidence that negative feedback regulation of dAkt occurs during normal Drosophila development in vivo. Whereas in cell culture dAkt is regulated by S6 Kinase (S6K)–dependent negative feedback, this feedback inhibition only plays a minor role in vivo. In contrast, dAkt activation under wild-type conditions is defined by feedback inhibition that depends on TOR Complex 1 (TORC1), but is S6K–independent. This feedback inhibition is switched from TORC1 to S6K only in the context of enhanced TORC1 activity, as triggered by mutations in tsc2. These results illustrate how the Akt–TOR pathway dynamically adapts the routing of negative feedback in response to the activity load of its signaling circuit in vivo.
Introduction
The development of multi-cellular organisms depends on the precise choreography of a diverse array of signal transduction pathways. Besides the requirement of some signaling events to occur in a spatial or temporal on-off manner, other pathways need to stay homeostatically active within physiological boundaries. This requires balanced regulation by activating as well as repressing signals.
Mechanistically, three basic concepts of downregulating signaling pathway have emerged: (1) control via specific inhibitory ligands or receptors [1], [2], (2) negative cross-regulation by distinct signaling pathways [3], and (3) auto-regulation by negative feedback mechanisms [4], [5]. In most cases, the molecular component that executes the feedback-mediated inhibition is transcriptionally targeted by the very pathway that it regulates. This mechanism ensures an interdependence of signaling activity and feedback regulation and is often viewed as an inherent means to downregulate signaling pathways after stimulation.
Loss of negative feedback regulation has been correlated with the initiation, growth and progression of tumors. For example, loss of negative feedback in Hedgehog (Hh) signaling by impeding patched function results in ectopic Hh signaling, basal cell carcinoma and medulloblastomas [6]. The expression of axin2 or dickkopf-1, which encode feedback inhibitors of Wnt signaling, is silenced in colon and breast carcinomas and early lung adenocarcinoma [7], [8]. Negative feedback regulators of Ras signaling, such as Sprouty proteins and MAPK phosphatases, are downregulated in liver, prostate and breast cancers [9], [10], [11], [12]. Similarly, inhibition of negative feedback regulation has been reported for JAK/STAT, TGF-beta and NF-kappaB signaling pathways [13], [14], [15]. These observations indicate that some cancers arise by “breaching” auto-regulatory control mechanisms of signaling pathways via mutational inactivation or epigenetic silencing of negative feedback regulators.
The Akt-TOR pathway has emerged as a central signaling nexus that integrates responses to growth factors, nutrients, metabolites and stress. Most prominently, activation of Akt is initiated by the insulin receptor (InR), relayed via an intracellular signaling cascade comprising insulin receptor substrate (IRS), class IA PI3 Kinase (PI3K), PDK1 and TOR complex 2 (TORC2), consisting of TOR, Rictor, Sin1, Lst8 and PRR5L [16], [17], [18], [19], [20]. Among other substrates, Akt inhibits the activities of the transcription factor FoxO [21] and the Rheb-specific GTPase activating protein (GAP) Tsc2. In turn, Rheb regulates the TOR complex 1 (TORC1), containing TOR, Raptor and Lst8 [17], [22], [23]. TORC1 targets several well characterized substrates, most notably S6 Kinase (S6K) [24], [25]. Hence, the two distinct TOR complexes TORC1 and TORC2 both participate in Akt-TOR signaling, but act at different levels in the Akt-TOR signaling pathway and integrate distinct stimuli. TORC2 responds to growth factors and might determine the substrate specificity of Akt [26], [27], [28], [29], while TORC1 mediates signaling by amino acids and cellular energy stress [30], [31], [32], [33]. Ectopic activation of the core Akt-TOR signaling pathway by a variety of mechanisms is a frequent event in cancer biology 18,34. Moreover, chronic diseases such as obesity and type II diabetes show pathological alteration of Akt-TOR activity [35].
Negative feedback mechanisms regulate the signaling input into the Akt-TOR pathway. Indeed, FoxO transcription factors inhibit the activity of the phosphatases PP2A and calcineurin by driving the expression of Atrogin-1, causing elevated levels of Akt phosphorylation and activity [36], [37]. Furthermore, Akt-dependent inhibition of the FoxO transcription factor results in reduced transcription of the inR gene. Conversely, low Akt-TOR signaling selectively increases InR mRNA translation relative to the total mRNA pool. In conjunction, both mechanisms reduce the relative levels of InR expression when Akt-TOR activity is high, thereby desensitizing against a stimulating ligand [38], [39], [40]. In addition, an S6K-dependent negative feedback mechanism leads to IRS1 destabilization, thus decreasing Akt activity [41], [42], [43], [44]. While these negative feedback mechanisms have been defined in cell culture, it is currently unknown whether and how feedback regulation within the Akt-TOR signaling pathway is exerted during development in vivo.
In Drosophila, the dAkt-TOR signaling pathway is conserved and regulates cell proliferation, and developmental timing and sizing of cells, organs and the whole fly [45], [46], [47]. As with the mammalian counterparts, Drosophila Akt receives regulatory inputs from TORC2 as well as PDK1. The phosphorylation site in the C-terminal hydrophobic motif of Drosophila Akt is conserved, and, while dispensable for normal Drosophila development, is required for relaying high PI3K signaling levels [29], [48], [49], [50]. Similarly, prostate-specific ablation of C-terminal Akt phosphorylation in mice conditionally mutant for Rictor delays lethality of Pten+/- induced prostate cancer [51]. In general, the C-terminal phosphorylation of Akt has emerged as a valuable and reliable tool to detect Akt activity in vivo and in vitro [52], [53]. In contrast to the three Akt genes in mammalian genomes, Drosophila contains only a single dAkt gene. In addition, the InR and IRS families are represented solely as single genes, and the insulin/InR-related IGF-1/IGFR system is absent in flies. This simplicity underscores the suitability of the fly as a model organism for studying complex processes like the in vivo analysis of feedback mechanisms.
To date, the analysis of feedback-mediated Akt-TOR pathway adaptation has been pursued under genetic or metabolic conditions that trigger high, possibly supra-physiological activity of TORC1 and S6K, and mostly in cell culture systems [18], [54]. In this study, we present evidence that regulation by negative feedback is an integral part of the dAkt-TOR pathway in vivo. Importantly, we demonstrate that the pathway utilizes two distinct modes of negative feedback to downregulate its activity in vivo, independently of FoxO. Conditions of wild-type TORC1 activity favor a dampening feedback signal emanating from TORC1 itself, independent of S6K. In contrast, conditions that induce high TORC1 activity trigger an S6K-dependent feedback mechanism to dampen dAkt-TOR pathway signaling. Our observations suggest that S6K acts as a load-sensitive regulator of Akt-TOR signaling. We propose the presence of a novel dual “overload protection” circuit that emphasizes the importance of tight control over Akt-TOR pathway signal levels.
Results
An assay for Drosophila phospho-Akt
We established a cell-based assay for regulators of insulin signaling in Drosophila that could be used in a genome-wide RNAi screen. Testing of more than 64 commercially available phospho-antibodies against components of this signaling cascade revealed that none of them recognized an insulin-induced antigen using immunohistochemistry (data not shown). Thus, we generated a phospho-Akt antiserum recognizing the phosphorylation of the C-terminal hydrophobic motif of Drosophila Akt. The single dakt gene encodes two splice forms of 513 and 611 amino acids in length. The antibody (hereafter referred to as anti P-dAkt) recognizes two bands in a western blot assay, likely corresponding to the phosphorylated forms of the short and long splice form, respectively.
In order to test if the phosphorylation of this hydrophobic motif correlates well with activity of Akt [52], [53], we stimulated Drosophila Kc167 cells with insulin for 10 min and induced a robust P-dAkt signal. The hydrophobic motif phosphorylation was strongly suppressed when known components of the insulin signaling cascade, including InR, Chico, the catalytic subunit of PI3K, PI3K92E and dAkt itself were silenced by RNAi (Figure 1A–1E and 1A'–1E'). We next asked whether the anti P-dAkt antibody detected differences in dAkt phosphorylation in the third instar imaginal disc, an established system to study cell and tissue size alterations dependent InR signaling in vivo [55], [56], [57]. To validate our assay, we expressed dominant negative insulin receptor (InRDN) or a constitutively active catalytic subunit of PI3K (PI3KCAAX), utilizing the UAS-Gal4 expression system [58]. Using apterous-Gal4 (ap-Gal4) to drive expression of InRDN and PI3KCAAX concomitant with membrane-tagged GFP in the dorsal compartment of the wing disc, we compared the levels of P-dAkt immunoreactivity in the dorsal compartment cells to non-expressing ventral cells as controls (Figure 1K–1L and 1K'–1L'). Expression of InRDN resulted in a reduction of P-dAkt levels (Figure 1F and 1F'), whereas PI3KCAAX expression drastically increased the P-dAkt intensity when compared to ventral control cells (Figure 1G and 1G'). Staining of wild-type imaginal wing discs did not reveal a pattern of P-dAkt immunoreactivity associated with compartments or their boundaries (not shown).
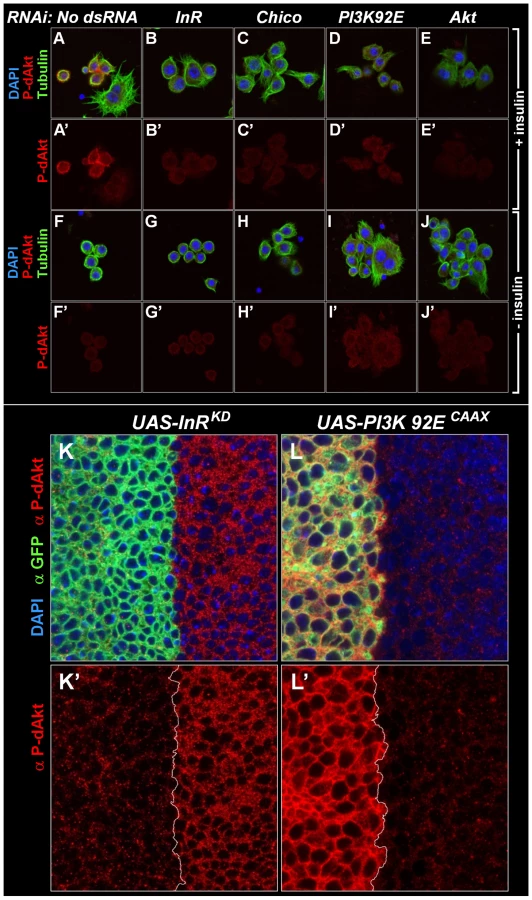
Western blotting of extracts of Kc167 cells treated with various dsRNAs against components of the insulin signaling pathway confirmed the specificity found by immunostaining of cells and Drosophila tissue (Figure S1). RNAi-mediated knockdown of InR, PI3K92E or dAkt abolished the anti-P-dAkt reactivity. Together, our data show that anti P-dAkt faithfully detects dAkt phosphorylation, and that the hydrophobic phosphorylation motif correlates with InR-PI3K regulated dAkt activity in cell culture and in vivo.
A genome-wide RNAi screen for regulators of P-dAkt reveals negative feedback regulation by Tsc1/Tsc2-TOR-S6K signaling
To identify novel regulatory inputs in the insulin signal transduction pathway, we used the Cytoblotting/In Cell Western method in combination with the newly generated anti P-dAkt antibody (Figure S2A) as a fast and quantitative cell-based high throughput assay. Cells were grown in 384-well plates and, after three days in the presence of gene-specific dsRNAs of the genome-wide dsRNA library [59], were fixed and immunostained with anti P-dAkt antiserum. Bound primary antibody was quantified and normalized to cell number. Using this approach, we carried out genome-wide RNAi screens in duplicates without stimulation and after 10 min. of insulin stimulation. We identified 79 dsRNAs that conferred suppression of dAkt phosphorylation, and 56 dsRNAs that enhanced P-dAkt immunoreactivity (Table S1). Importantly, five out of eight known components functioning upstream of dAkt were identified, validating the reliability of this method (Figure S2B, S2C, and Table S1). dsRNAs against Chico, PHLLP and Pten, the remaining 3 regulators of dAkt, scored below the cutoff threshold.
In our screens, we found that dsRNAs against the small GTPase Rheb, the TORC1 component Raptor and S6K, all downstream mediators required for insulin signal transduction, induced enhanced phosphorylation of dAkt in the absence of insulin. Conversely, dsRNAs against the negative regulators Tsc1 and Tsc2 suppressed the P-dAkt signal when the pathway was activated by insulin. In total, we identified ten out of eleven components known to participate in the Tsc1/Tsc1-TOR signaling branch, with Tctp [60] as the single component not identified by any of our screens (Figure S2 and Table S1). Interestingly, the function of Tctp as a regulator of Rheb is controversial [61], [62]. The results of the genome-wide RNAi screen were validated using independent dsRNAs against known insulin pathway components (Figure 2A and 2B). dsRNAs against CSK, MEKK1 and Thread were used as negative controls, and Pten dsRNA as positive control. As observed in the genome-wide screen, removal of the negative regulators Tsc1 and Tsc2 resulted in suppression of P-dAkt in the presence of insulin, while knock down of S6K elevated P-dAkt at baseline conditions. Thus, dAkt phosphorylation is sensitive to interference by Tsc1/Tsc2-TOR-S6K signaling, classically viewed as signaling downstream of dAkt [23], [63], [64], [65], [66], [67]. These results are consistent with the existence of an inhibitory feedback signal by the components downstream of dAkt, namely Rheb, Raptor, Tsc1/2 and S6K [50], [65].

To test the feedback by different means than RNAi, two different strategies were used to inhibit the activator of S6K, TORC1 (Figure 2C). In a chemical approach, we exposed cultured cells to rapamycin, an effective, small molecule inhibitor of TORC1 [68], [69], [70]. In a metabolic approach, we starved cultured cells in amino acid-free media, thereby potently inhibiting TORC1 activity [65], [68], [69], [70], [71]. Rapamycin-induced TORC1 inhibition and amino acid starvation both led to a highly significant increase in P-dAkt compared to control cells treated with solvent control or amino acid-containing medium, respectively. These results confirm the RNAi data and validate the existence of a negative feedback loop that regulates the activation of the pathway by insulin [65].
Activation of S6K correlates with an inhibition of P-dAkt
Since dsRNA-mediated knockdown of S6K enhanced dAkt phosphorylation, we asked whether the inhibitory effect of S6K on dAkt phosphorylation was related to its activity. The activation of Drosophila S6K can be scored using phosphorylation of Thr 398 (orthologous to Thr 389 in mammalian S6K1) as readout (Figure 3A) [50], [65]. We analyzed lysates of Drosophila Kc167 cells pretreated with dsRNAs against Tsc2, Raptor, S6K and Rheb, for both S6K and dAkt phosphorylation. Cells treated with dsRNA against luciferase and non-RNAi treated cells served as negative controls (Figure 3A, lanes 4 and 6). Enhanced P-dAkt reactivity correlated with suppression of S6K phosphorylation, with a clear elevation of dAkt phosphorylation when Rheb, Raptor or S6K expression was knocked down.

To address how S6K mediates its feedback inhibition of dAkt phosphorylation, we induced dAkt phosphorylation by exclusively removing the negative feedback inhibition in Kc167 cells using RNAi against S6K in the absence of insulin stimulation. The robust enhancement of P-dAkt due to the knockdown of S6K expression was not affected by further RNAi-mediated knockdown of control genes such as GFP, CSK or MEKK1/4. We then knocked down the individual components of the insulin signaling pathway to assess whether they were required for the enhanced dAkt phosphorylation caused by S6K silencing (Figure 3B). In the S6KRNAi background, RNAi-mediated silencing of Pten (positive control) further enhanced the P-dAkt levels, while dsRNA to dAkt (negative control) reduced P-dAkt to baseline levels. Importantly, RNAi against the signaling effectors InR or PI3K suppressed the enhanced dAkt phosphorylation conferred by S6K RNAi, indicating that the S6K-dependent feedback inhibition requires the functions of these two upstream signaling effectors.
Phosphorylation of dAkt in vivo is regulated by negative feedback
Cell autonomous regulation of dAkt phosphorylation by direct negative feedback has to date not been shown to occur in vivo. To test whether the negative feedback on dAkt phosphorylation also occurs in vivo, i.e. during Drosophila development, we used the wing imaginal disc of the third instar larva. We took advantage of the unique features of the aktq allele [72], a loss of function allele that encodes a kinase-inactive dAkt protein due to mutation of the DF327G motif in kinase subdomain VII into DI327G. This dAkt mutant protein is unable to phosphorylate downstream components, but is readily expressed and can be phosphorylated by upstream signaling components [72]. We generated homozygous mutant aktq clones in the wing imaginal discs using FLP-FRT-mediated mitotic recombination using the MARCM technique [73], [74]. This confers GFP expression to the cells expressing the mutant allele only. dAkt protein expression in aktq mutant clones and in the neighboring cells expressing wild-type dAkt were at similar levels (Figure S3). We then visualized dAkt phosphorylation of aktq mutant and wild-type cells by immunofluorescence using the anti P-dAkt antibody. The presence of an inhibitory mechanism that depends on the activity of the dAkt kinase and negatively feeds back on dAkt phosphorylation predicts enhanced P-dAkt levels in aktq mutant cells when compared to cells expressing wild-type dAkt. Consistently, we observed drastically enhanced phosphorylation of dAkt in clones homozygous for the aktq mutation (Figure 4A). The increased dAkt phosphorylation in aktq mutant cells thus indicates that inactivation of the dAkt kinase function removes repression on dAkt phosphorylation by negative feedback. It further demonstrates the cell autonomous presence of this regulatory loop in imaginal wing discs of third instar larvae.

Regulation of dAkt phosphorylation by the Tsc1/Tsc2 tumor suppressor complex
Having established that feedback inhibition leads to repression of dAkt phosphorylation in vivo, we asked whether changes in Tsc1/Tsc2 function would affect the feedback activity on dAkt phosphorylation (Figure 4). dAkt-mediated phosphorylation of Tsc2 inhibits the function of the Tsc1/Tsc2 tumor suppressor complex [63], [75]. First, we co-expressed Tsc1 and Tsc2 in the dorsal compartment of the third instar imaginal wing disc under the control of ap-Gal4. If the Tsc1/Tsc2 complex defines the feedback inhibition of dAkt phosphorylation in vivo, overexpression of Tsc1/Tsc2 is expected to result in increased dAkt phosphorylation. Indeed, compared to ventral control cells, dAkt phosphorylation is clearly elevated in dorsal cells (Figure 4B). This result indicates that forced Tsc1/Tsc2 expression represses the feedback inhibition. Conversely, we induced homozygous mutant clones of either tsc1Q87X or tsc2192, resulting in the lack of functional Tsc1/Tsc2 tumor suppressor complex [76], [77], [78]. Complementary to the Tsc1/Tsc2 overexpression experiment, we expected a decrease in dAkt phosphorylation. Indeed, we found reduced dAkt phosphorylation levels in tsc1Q87X homozygous mutant cells, when compared to wild-type control cells (Figure 4C).
Next, we addressed whether the dAkt feedback signaling is routed from dAkt to the Tsc1/Tsc2 complex by evaluating the P-dAkt immunoreactivity in aktq, tsc1Q87X double mutant clones. If Tsc1/Tsc2 transduces the feedback signal originating from dAkt, we expect that the additional elimination of tsc1 function in an aktq clone reverses the increased dAkt phosphorylation found in a clone of aktq single mutant cells. Consistent with this prediction, the level of dAkt phosphorylation in aktq, tsc1Q87X double mutant clones was not elevated when compared to cells with wild-type expression of Tsc1/Tsc2 and dAkt. To the contrary, P-dAkt levels were decreased, more like cells singly deficient in tsc1 function (Figure 4C). We therefore conclude that the Tsc1/Tsc2 tumor suppressor complex controls dAkt phosphorylation in vivo by defining the feedback inhibition.
Negative feedback regulation of dAkt phosphorylation is independent of FoxO
Transcription factors of the FoxO family have emerged as central mediators of the PI3K-dAkt signal transduction pathway [21], [79]. In Drosophila cell culture and the adult fly, the InR transcript is selectively transcribed and translated under conditions of low dAkt signaling levels [38], [39], [40]. Since these data suggests an alternative route of feedback mediated regulation of dAkt phosphorylation that is independent of TORC1 and S6K, we wanted to test the role of dFOXO in the negative feedback regulation of dAkt phosphorylation in the third instar imaginal wing disc. To do so, we used an activated form of dFOXO (dFOXO-TM), in which the dAkt phosphorylation sites have been replaced by alanines (Figure S4) [80]. These mutations result in constitutively nuclear localization of dFOXO, and strong transactivation of target genes both in vitro and in vivo [38], [39], [81]. Expression of dFOXO-TM by means of ap-Gal4 did not reveal any discernable differences in dAkt phosphorylation between dFOXO-TM expressing versus non-expressing cells (Figure S4A). Furthermore, mitotic clones homozygous for the dfoxo25 loss of function allele, which is predicted to encode a truncated protein [81], retain a similar amount of P-dAkt as wild-type control cells (Figure S4B). Finally, aktq, dfoxo25 double mutant clones show increased dAkt phosphorylation, similar to homozygous clones of aktq alone (Figure S4C). These data indicate that dFOXO is not involved in the negative feedback regulation of dAkt phosphorylation in the developing wing disc.
Tsc1/Tsc2 regulates dAkt phosphorylation via TORC1
To further delineate the feedback inhibition pathway mediated by the Tsc1/Tsc2 complex, we analyzed the requirement of TORC1 in the downregulation of dAkt phosphorylation. The protein kinase TOR has been found in close physical proximity of Tsc1 and Tsc2, and biochemical and genetic evidence have established that TOR is a central mediator of Tsc1/Tsc2 signaling [76], [82], [83], [84], [85]. However, TOR is part of TORC1 as well as TORC2, and the former is required for S6K activation, which, in cell culture, brings forth negative feedback on dAkt phosphorylation, while the latter is required for hydrophobic motif phosphorylation of dAkt [16]. To interfere specifically with TORC1 function, we expressed an RNAi hairpin construct against Raptor (raptorRNAi), a component present only in TORC1 and not in TORC2 [49], [86]. Using ap-Gal4, we compared the dAkt phosphorylation levels in UAS-RaptorRNAi expressing, GFP-positive dorsal cells to those in wild-type, GFP-negative control cells in the ventral compartment (Figure 5A). If TORC1 is required to drive feedback inhibition of dAkt phosphorylation, its inactivation should augment the level of P-dAkt immunoreactivity. Accordingly, we observed increased P-dAkt staining in RaptorRNAi cells.

We further tested whether TORC1 is required for the decrease in dAkt phosphorylation observed in tsc1 mutant wing disc cells. We expressed raptorRNAi in mitotic clones homozygously mutant for tsc1W243X. Loss of tsc1 results in derepression of Rheb and TORC1 activity [23], [64], which, accordingly, resulted in excessive feedback inhibition of dAkt phosphorylation in tsc1 mutant cells (Figure 5B). Feedback inhibition by Tsc1/Tsc2 mediated through TORC1 predicts that loss of tsc1 concomitant with a reduction of raptor function by RNAi confers the same P-dAkt phenotype as that of raptorRNAi alone. Indeed, raptorRNAi expression in tsc1W243X mutant cells displayed an increase in P-dAkt immunostaining, as seen in cells expressing raptorRNAi alone (Figure 5C). This is consistent with the loss of negative feedback inhibition of dAkt phosphorylation in raptorRNAi, tsc1W243X cells, indicating an epistatic relationship of TORC1 to Tsc1 in the negative feedback circuit.
Two distinct modes of inhibitory feedback signaling to dAkt
S6K is a central player downstream of TORC1, and TORC1 activity is directly required for the activation of S6K [25], [35]. To test the role of S6K in the feedback inhibition of dAkt phosphorylation, we generated homozygous clones of an s6K null allele (s6Kl-1) [24], and investigated the level of dAkt phosphorylation in reference to wild-type tissue. If S6K mediates the feedback inhibition emanating from TORC1, dAkt phosphorylation should be increased in s6Kl-1 cells, since the inhibitory feedback on dAkt would be released. To our surprise, no change in P-dAkt level was apparent (Figure 6A), suggesting that S6K does not regulate the negative feedback signaling mediated by Tsc1/Tsc2 and TORC1 at this stage of wing imaginal disc development. Because this in vivo finding differed strikingly from the results in Drosophila cell culture, we verified that the s6Kl-1 chromosome did not carry additional mutations. The lethality associated with our s6Kl-1 stock [24] was rescued by expressing a S6KWT cDNA from an act-Gal4 driver. To further assess if the phosphorylation status of dAkt varies in the s6K l-1 mutant background in a tissue dependent fashion, we performed a western blot analysis of extracts from whole third instar wild-type and s6Kl-1 mutant larvae (Figure S5). Consistent with our result in wing imaginal disc clones of s6K l-1, we did not observe an increase of P-dAkt. However, we detected a downregulation of total Akt protein expression in extracts from s6Kl-1 mutant larvae when compared to wt, thus suggesting additional regulatory mechanisms controlling dAkt in different tissues.

Most studies on feedback regulation of dAkt by S6K are carried out in the context of either tsc1 or tsc2 mutants or other experimental settings of putatively high TORC1 activity [18], [87]. This led us to probe the dependence of the feedback inhibition on S6K in a high TORC1 signaling background induced by a tsc2 mutant context. First, we confirmed that, similar to tsc1Q87X (Figure 4C), P-dAkt is downregulated in cells homozygously mutant for tsc2192 (Figure 6B). Subsequently, we generated s6kl-1, tsc2192 double mutant clones and stained the cells with anti P-dAkt antiserum (Figure 6C). Surprisingly, and in contrast to s6kl-1 single mutant clones and tsc2192 single mutant clones, tsc2192, s6kl-1 double mutant tissue of the wing imaginal disc displayed elevated P-dAkt levels compared to wild-type cells (Figure 6). We observed a similar result using a different allelic combination, s6Kl-1, tsc2* (data not shown). This result suggests that the feedback inhibition of dAkt phosphorylation depends on S6K only when TORC1 signaling is elevated above its wild-type activity.
S6K as sensor and regulator of dAkt–TOR signaling activity
The observation that ablation of S6K function only affects feedback inhibition of dAkt phosphorylation when TORC1 signaling is elevated suggests that activation of S6K by TORC1 in a wild-type context is insufficient to affect dAkt phosphorylation. The activation of S6K by TORC1 involves phosphorylation of several sites in the auto-inhibitory domain and the linker region of S6K [88], [89]. We used ap-Gal4 to express either wild-type S6K (S6KWT), or mutant S6K forms (S6KTE, S6KSTDE or S6KSTDETE [90], which are intrinsically activated due to substitution of several serine and threonine residues by acidic amino acids in the linker (S6KTE), the autoinhibitory domain (S6KSTDE), or both (S6KSTDETE). Overexpression of S6KWT did not visibly change the level of dAkt phosphorylation when compared to ventral, non-S6K-expressing control cells (Figure S6). However, expression of the activated alleles S6KTE, S6KSTDETE and, to a limited extent, S6KSTDE, resulted in decreased dAkt phosphorylation, when compared to ventral non-expressing cells, reminiscent of the effect of high TORC1 signaling. We further addressed whether activated S6K is also sufficient to elicit inhibition of P-dAkt when TORC1 activity is low. To this end, we co-expressed Tsc1 and Tsc2, to dominantly inhibit TORC1, and assessed P-dAkt levels in the absence or presence of S6KTE co-expression (Figure S7). As observed above (Figure 4), Tsc1/Tsc2 expression caused a pronounced increase in P-dAkt (Figure S7A and S7A'). Strikingly, simultaneous expression of dominant active S6K (S6KTE, Figure S7B and S7B') reversed the elevated P-dAkt down to a near wild-type level. Altogether, these results suggest that activation of S6K is sufficient to elicit feedback inhibition of dAkt phosphorylation under normal or inhibited TORC1 activity levels. However, in wild-type wing imaginal disc cells, endogenous S6K is not sufficiently activated to regulate the feedback inhibition of dAkt phosphorylation. These results suggest that S6K serves as a sensor and homeostatic regulator of dAkt-TOR signaling intensity in vivo.
Discussion
Akt signaling provides a critical nexus between PI3K and TORC1 signaling. Excessive activation of dAkt and its effector pathways drives unrestricted cell growth and proliferation, as observed in cancers. Conversely, an impaired response of Akt to stimulating factors like insulin in peripheral tissues contributes to metabolic syndromes and diabetes mellitus. Hence, the activity of Akt-TOR signaling needs to be maintained within well-defined physiological boundaries within a critical upper as well as lower limit. We now provide the first evidence that, during development, negative feedback monitors and autoregulates the activity of the dAkt-TOR signaling pathway in wing imaginal disc tissue in vivo.
We studied the control of dAkt activation in Drosophila Kc167 cells and in vivo using the wing disc of the Drosophila third instar larva. Our results highlight that dAkt centers itself in a circular pathway with two modes of negative feedback regulation (Figure 7). Consequently, the levels of dAkt phosphorylation in vivo are controlled not only by the input by extracellular factors into the signaling pathway, but also by the amplitude of the dAkt-TORC1 signal itself. In the wild-type context of the third instar wing disc, the negative feedback on dAkt phosphorylation is mediated by TORC1 and is S6K-independent. Interestingly, increased dAkt-TOR signaling activity switches the mechanism of negative feedback from an S6K-independent - to an S6K-dependent feedback mode (Figure 6). We interpret this finding as a rewiring of the dAkt-TOR signaling network that illustrates a dynamic response of a signaling circuit to signaling loads.

TORC1–dependent feedback control of dAkt phosphorylation in vivo
Our initial experiments on dAkt phosphorylation in wing imaginal discs demonstrated that expression of activated PI3K (PI3KCAAX) or dominant negative InR (InRK1409A) elevated or repressed, respectively, the level of dAkt phosphorylation. This observation indicates that in these cells, dAkt phosphorylation can be enhanced or repressed, depending on the signaling input, and thus that dAkt has “room” for quantitative regulation of activation. Accordingly, high levels of negative feedback, caused by mutational inactivation of tsc1 or tsc2, suppress dAkt phosphorylation, while low levels of negative feedback signaling, as in RaptorRNAi-expressing cells, enhance dAkt phosphorylation. These results lead to the conclusion that the wild-type level of dAkt phosphorylation in these cells is set by negative feedback regulation that is executed by the Tsc1/Tsc2-Rheb-TORC1 arm of the dAkt-TOR pathway. Interestingly, in s6Kl-1 mutant clones, levels of P-dAkt are unchanged, indicating the independence of the negative feedback circuit from S6K activity under these conditions. These results differ from the reported elevated Akt kinase activity in extracts from whole s6Kl-1 second instar larvae [65], [91]. Studies of whole larval extracts may reflect the regulation in endoreplicating tissues, which dominate the body mass at that stage of development, whereas our results of s6Kl-1 mutant clones in the wing disc examine Akt phosphorylation in a mitotically active tissue. The disparity in Akt phosphorylation may therefore reflect differences in negative feedback regulation of Akt at discrete stages of development and in distinct tissues. The recent observation that inhibition of TORC1 by rapamycin treatment in adult flies results in loss of S6K phosphorylation and, presumably, activity without eliciting changes in dAkt phosphorylation serves as a case in point [92]. The tissue specificity of dAkt feedback regulation will be an interesting topic of future investigation.
In the wing imaginal disc, the feedback-driven changes in dAkt phosphorylation occur in a manner that is uncoupled from changes in dAkt protein expression. Indeed, the genetic manipulations that resulted in changes in dAkt-TOR signaling activity left the dAkt expression levels unchanged. The only exception of a slight reduction in overall dAkt level, yet increased dAkt phosphorylation, was observed upon expression of PI3KCAAX (see Figure S3E and S3E'). We therefore propose that, in the wing imaginal disc, a change in the phosphorylation status of dAkt, but not in protein expression, represents the relevant regulatory event in vivo that is targeted by TORC1-dependent negative feedback. Changes in dAkt activity by manipulating the negative feedback can have significant biological effects [42], [93], [94]. In contrast to our findings in wing discs, we do observe a reduction of Akt protein levels in whole third instar larval extracts of s6Kl-1 mutants. Since in third instar larvae the wing imaginal discs represent only a minor and select fraction of cells and tissues, the mass disparity of larval vs. imaginal wing tissue may explain this differing result of dAkt in our western blot vs. the clonal wing imaginal disc analysis. Of note, our western blot analyses differed significantly from those reported by Radimerki et al. [65], [91], in the use of third versus second instar larval extracts, differences in protein extraction, antibodies, and normalization against total protein versus Akt levels. We interpret the divergent results as due to different assays and developmental stages analyzed. Importantly, a change of Akt protein levels under altered Akt-TOR signaling conditions is not unprecedented [95].
Although the TORC1-dependent feedback needs to be biochemically characterized, two mechanisms may be envisioned. First, TORC1 could participate in an inhibitory step required for downregulation of dAkt activity. This possibility may be supported by experimental evidence that TORC1 can elicit a direct inhibitory phosphorylation of IRS1 in mammalian cell culture [96], [97]. Second, disruption of TORC1 by RNAi knockdown against specific components of the complex may release the remaining components of TORC1, and might shift a mass-action equilibrium between TORC1 and TORC2 towards TORC2. While such equilibrium has been suggested [98], there is, to our knowledge, only experimental evidence for an equilibrium shift towards TORC1 when TORC2 is disrupted, but not vice versa [16], [50]. Lastly, in mammalian cells the Tsc1/Tsc2 complex is required for proper TORC2 activation, independently from its role in negative feedback signaling [99]. Nevertheless, we observe that RaptorRNAi hairpin expression reverses the decrease in dAkt phosphorylation in a tsc1 mutant clone, although not to the same extent as expression of the RaptorRNAi hairpin driven by ap-Gal4. These findings suggest that negative feedback is a central route of Tsc1/Tsc2 to regulate dAkt phosphorylation. However, we cannot exclude a functional role of Tsc1/Tsc2 in the activation of TORC2 [99].
dAkt–TOR pathway feedback in Drosophila cell culture versus in vivo
We also provide evidence for S6K-dependent negative feedback inhibiting the phosphorylation of dAkt in vivo. The S6K-dependent mode of feedback was previously proposed based on data in mammalian or Drosophila cell culture. Accordingly, we observed an S6K-dependent negative feedback circuit inhibiting the phosphorylation of dAkt in Drosophila Kc176 cells [65], [66], [67]. In vivo, however, this mode of feedback was observed only in cells with high TORC1 signaling, and was not seen in wild-type conditions, where the negative feedback mechanism is TORC1-dependent and does not depend on S6K. We therefore propose that in the wing imaginal disc, under conditions of high TORC1 signaling, the cells switch their feedback mechanism from a TORC1-dependent mode to an S6K-dependent mechanism similar to what is observed in Kc167 cell culture. We suggest that the constant presence of serum, insulin and high amino acid concentrations in the cell culture medium foster high TORC1 activity, favoring the S6K dependent negative feedback route. However, it is possible that in cultured Kc167 cells, both feedback mechanisms are simultaneously operative. Indeed, we found that, in Kc167 cells, RNAi-mediated knockdown of the TORC1 component Raptor triggers a stronger increase in dAkt phosphorylation than RNAi against S6K (see Figure S2B and Figure S8).
dAkt activity and cell size
The elevated dAkt phosphorylation observed in cells with increased Tsc1 and Tsc2 expression strongly supports a negative feedback regulation of dAkt phosphorylation by the Tsc1/Tsc2 complex in vivo. Nevertheless, this finding is surprising at two levels. First, dAkt has been described as a positive regulator of cell size, and aktq homozygous mutant clones show reduced cell size [100]. However, forced expression of Tsc1/Tsc2 results in reduced cell size, despite elevated dAkt phosphorylation [76], [77], [78]. The reciprocal experiment highlights the same paradox: tsc1Q87X or tsc2192 mutant cells have a larger size, despite decreased dAkt phosphorylation and activity [76], [77], [78]. These results may indicate that, for cell size, dAkt's function is to regulate Tsc1/Tsc2 activity, which is supported by the fact that so far no other dAkt substrate (e.g. FoxO, Gsk3beta) has been shown to elicit a cell size defect [81]. Correspondingly, the ability of Akt1 and Akt2 deficiency to suppress H-Ras mediated oncogenesis in mouse mammary glands is overcome by inactivation of tsc2, again supporting the hypothesis that a central function of dAkt in vivo is the regulation of TORC1 activity [101]. The consistency of the data in Drosophila with those in mice indicates that this function of dAkt in dAkt-TOR signaling is conserved. The second surprise relates to the elevated dAkt phosphorylation in the presence of ectopic Tsc1/Tsc2 expression, which may at first seem intuitive. The circular structure predicts that increased Tsc1/Tsc2 expression should inactivate the feedback inhibition of dAkt phosphorylation by repressing TORC1, thus releasing dAkt from negative feedback regulation, hence increasing dAkt phosphorylation. However, a perfectly circular dAkt-TOR pathway predicts that increased Tsc1/Tsc2 levels should trigger high dAkt activity, which in turn should inactivate the Tsc1/Tsc2 complex by direct phosphorylation of Tsc2 [75], [102], [103]. Thus, depending on the strength of the dAkt-Tsc2 connection, dAkt phosphorylation could either remain unchanged or even be reduced. However, the dAkt-Tsc2 link might be less physiologically relevant than initially suggested [75], [104], [105], pointing to additional regulatory connections of the InR-PI3K-dAkt and Tsc1/Tsc2-Rheb-TORC1 signaling branches [20], [99], [106], [107], [108], [109]. Alternatively, overexpressed Tsc1/Tsc2 may localize to a subcellular compartment where it escapes phosphorylation by active dAkt, yet can inhibit TORC1, or the derepressed activity level of dAkt might be insufficient to effectively control overexpressed Tsc1/Tsc2.
S6K serves as a sensor of TORC1 signaling load in vivo
In the developing Drosophila wing disc, S6K is a central mediator of TORC1 activity, especially as the fly 4E-BP1 ortholog, Thor, is not expressed [110]. Since ectopic expression of activated S6K, but not wild-type S6K, results in decreased dAkt phosphorylation, we conclude that S6K activation is sufficient to elicit a negative feedback on dAkt. In the activated S6K mutants, sites in the linker and autoinhibitory domain that are normally phosphorylated by TORC1 are replaced by phospho-mimetic acidic amino acids [90]. Our data therefore suggest that linker and the autoinhibitory domain phosphorylation of S6K may function as sensor for the TORC1 signaling load. Thus, only when TORC1 is highly active, S6K will become sufficiently phosphorylated to drive the negative feedback, a scenario that is mimicked by ectopic expression of activated S6K. Of note, the S6KTE and S6KSTDETE phospho-mimetic mutants, which have the linker site mutation, exert a visibly stronger inhibition of dAkt phosphorylation than the S6KSDTE phospho-mimetic mutant of the autoinhibitory domain only. These differences may suggest that the phosphorylation site in the linker region of S6K is the predominant site for transducing TORC1 activity. In Drosophila cell culture, supra-physiological levels of nutrients and amino acids may then trigger the high TORC1 activity required to drive S6K-mediated feedback on dAkt phosphorylation.
Since mTOR has recently been shown to be targeted for phosphorylation by S6K [111], it is tempting to speculate about a mechanism involving a feedback by S6K on TORC1 that then could drive the switch between TORC1 - and S6K-dependent feedback inhibition of Akt phosphorylation. However, the T2446 and S2448 sites in mTor that are phosphorylated by S6K are not conserved in Drosophila TOR.
In conclusion, we demonstrate that dependent on TORC1 signaling load, the negative feedback signal regulating dAkt activity is dynamically routed in vivo. It is either independent of S6K (under “normal” TORC1 activity) or dependent on S6K (when TORC1 activity is high). Therefore, we interpret the function of S6K as a sensor of TORC1 signaling that selectively provides additional dampening of the signaling input once TORC1 is highly active. These findings predict that pharmacological tools selectively impinging on S6K activity, in the context of obesity treatment or other conditions with high S6K activity, might carry the significant risk of uncontrollable TORC1 activity.
Material and Methods
Cell culture and RNAi in Drosophila Kc167 cells
For 384-well plate experiments, cells were uniformly dispensed into clear bottom black 384-well plates (Corning) containing 250ng of individual, arrayed dsRNAs using a MultiDrop liquid dispenser (Thermo). 8×103 cells per well in 10 ul of serum-free media per well were seeded. After 60 min of incubation, 70 ul of 10% serum-containing culture medium (Schneider's Medium, Invitrogen) per well was added. After three days of incubation at 25°C, cells were washed once and starved in 80 ul serum-free medium overnight (12 hrs). For insulin stimulation, cells were exposed to a final concentration of 387 nM bovine insulin (Sigma) for 10 min. Rapamycin was used at a final concentration of 50 nM for 4 hrs, amino acid free media (Atlanta Biologicals) was used for 8 hrs. For western blotting, six-well dishes wereused and the conditions were scaled accordingly. 10 ug of dsRNA per well was added to 1.5×106 cells per well in 1 ml of serum-free media, supplemented after 60 min with 5 ml of serum-containing media. For immunofluorescence, we used eight-well chamber slides, and cells were treated as described above, using 2 ug of dsRNA per well in 100 ul of serum-free media, complemented with 500 ul of serum-containing media after 60 min. All primer sequences of the genome-wide dsRNA library are available on the website of the Drosophila RNAi Screening Center (www.flyrnai.org). dsRNAs were generated from PCR-derived DNA templates by T7 RNA polymerase driven run-off transcription in vitro (Ambion). The generic T7 promoter sequence TAATACGACTCACTATAGG was added 5′ to all gene specific primers. All gene-specific primers were designed using Primer3 [112], and conceptual PCR products were controlled against off-target effects using SnapDragon (http://flyrnai.org/cgi-bin/RNAi_find_primers.pl). Gene-specific primer sequences used: GFP: CAAGGGCGAGGAGCTGTT, GTCGTCCTTGAAGAAGATGGTG; CSK: GAGGAAGCAGACGGCAAC, GGGACTTGGGCGAATGAT; MEKK1: AAGTGTGTGTTGGTGCTGGA, ATCTTCGGGAGGCAGGTC; Thread: GCTGGACTGGCTGGATAAAC, ATTCGGGATACTGGGGAAAA; InR: CAGCGCGAAAACTTCAATATCTTT, TGTTTTATCCAGTCCATCGGCTAT; Chico: CCAAGCATAGATTTGTCATTGTGC, GATCACCAGATCCCAAGACACTTT; PI3K92E: GAGGCACCAGATCCAAAATC, ATACAGCCGGAAGTCGTCAA; PI3K21B: GCTTTATCGAGACGGACCTG, GCATCCAGCAGATTGAGGAG; Pten: TGTATTATGCCAAGCGGAAGA, TCAATCGTTGGAGGGTTATGA, dAkt: GTCCACAAATCATCCGTTCC, ACCTCCTCCACCAAAATCAA; Tsc1: GAGGTAAACAATACGCGATGGAAG, AACTGAACTGACTCTGCTGGTCCT; Tsc2: CTAGACAGTCGTCAGGTGATCGTG, ACGCGACTAAGGATTTCTTCTTCA; S6K: TCTGCACCAAGACACTGAGG, GCAGTATGTTCTCGGGCTTC; Raptor: ACCTGGGTAAGGTGATTAGCAACA, AGGTGCAGAGCTTCTTAACGTCAT; Rheb: GCTAGGAGTGGTATTTCGGCTTC, CCAGTGCTTTGAAATAAATGGAGA; PDK1: CAAGGAGAAAGCATCAGCAA, GCCTATGTAACGACCGAAAATG.
Protein extracts and western bloting
Kc167 cells were rinsed once, scraped in PBS, and pelleted at low speed in a table top centrifuge. The cell pellet was lysed in standard SDS-PAGE loading buffer without dye. Extracts of third instar larvae were prepared by mechanical homogenization and lysis in 50 mM Tris, 120 mM NaCl, 30 mM NaF, 50 uM NaVO4, 1% Triton X100, 0.1% SDS with Complete protease inhibitor cocktail (Roche). Lysates were cleared from debris and lipids by 10 min centrifugation in a table top centrifuge. For all protein lysates, protein concentrations were determined using Protein DC Assay (Bio-Rad), and total protein concentrations of lysates were adjusted accordingly.
Cytoblot
The Cytoblot protocol for 384 well plates used here consists of 4 steps: (1) fixation, (2) permeabilization, (3) P-dAkt staining comprising of incubations with primary and secondary antibodies, and (4) DNA staining to assess total cell numbers in each individual well. Two versions of the cytoblot were used. The “first generation” cytoblot utilized HRP-conjugated secondary antibody and chemiluminescence to detect anti P-dAkt. This protocol was applied to the non-stimulated RNAi screen. The “second generation” cytoblot employed a fluorescently labeled secondary antibody and was used for the insulin-stimulated RNAi screen. The availability of a LiCor Aerius plate reader allowed the switch from luminescence to fluorescence based detection.
(1) Fixation: Tissue culture medium from the 384 well plates was removed and cells were fixed with 6% Formaldehyde for 90 minutes, followed by three washes with 80 ul of PBS. (2) Permeabilization: 0.1% Triton X-100 in PBS for 30 minutes (40 ul per well), followed by 3 washes in 80 ul PBS for 10 min. (3) P-dAkt staining: Cells were blocked in 5% non-fat milk in PBS for 60 minutes (90 ul per well). Anti-Drosophila P-dAkt Ser505 primary antibody (20 ul per well, 1∶800 diluted in 5% non-fat milk, Cell Signaling Technology, Beverley, MA) was added and incubated at 4°C overnight. After 3 washes with PBS (80 ul per well, 10 min), 20 ul secondary antibody (goat anti-Rabbit AlexaFluor 680, diluted 1∶2,500, Invitrogen, for “second generation Cytoblot”, used for insulin-stimulated RNAi screens) or a 1∶1,200 dilution of goat anti-Rabbit HRP (“first generation Cytoblot”, used for insulin-stimulated RNAi screen, Jackson Laboratories), was added in 5% non-fat milk and incubated for 4 hrs, followed by 3 washes with 80 ul of PBS, 10 min and 20 ul PBS was added to each well. Signal was developed by 20 ul SuperSignal West Pico Chemiluminescent Substrate (Pierce). HRP luminescence was read in Molecular Devices plate reader. Alexa 680 fluorescence was measured using a LiCor Aerius plate reader (680/720 nm). HRP luminescence and Alexa 680 fluorescence were interpreted as amounts of P-dAkt per well. (4) DNA staining was performed with Sytox Green (Invitrogen) 1∶20,000 in PBS for 30 min (40 ul per well). After 3 additional washes with PBS, plates were filled with 20 ul PBS per well and the fluorescent value of the Sytox dye DNA stain were read in a Molecular Devices plate reader (520/560 nm). This value, referred hereafter to as nuclear fluorescence (NucFl), is interpreted as the value representing relative cell numbers per well. All liquid manipulation steps were performed using a MultiDrop liquid handling device (Thermo).
Data analysis
All individual values quantifying amounts of P-dAkt were normalized to the cell number per well using the nuclear fluorescent value from the DNA stain. For the insulin-stimulated screen, linear regression was performed on the log2(P-dAkt/NucFl) values of each individual screen plate, and residuals from the log2(P-dAkt/NucFl) values to the regression line were calculated. All residuals of each genome-wide screen were pooled and a cell number dependent error model was developed to determine locally weighted standard deviations (SD) and averages in dependence of cell number. Z-Scores using these two parameters were calculated, expressing the deviation from the local average value in SDs. All Z-Scores were corrected against position effects by setting the Mean Z-Score of each individual well position across one genome-wide screen replicate to zero. All dsRNAs with predicted off-target effects (homologies to non-target genes of 19 bp or more) were excluded from data processing and the result file. Results from the two screening replicates were averaged and a cut-off value of +/ − 2.5 applied. Due to high variation within each 384 well screening plate caused by individual 96-well source plates (each 384-well screening plate is composed of aliquots from four 96-well source plates), Z-Scores for the baseline (no insulin stimulation) genome wide RNAi screen were calculated as follows: The 384 (P-dAkt/NucFl) values of each single screening plate were decomposed into the four 96 well groups, each defined by a single source plate, and the mean and SD was set to zero and one, respectively. This step compensated these inequalities, and data were recombined to 384 well plate data sets. Mean and SD for each individual 384 well plate were calculated, averaged between screens, and a cut-off value of three SDs was applied. For non-genome-wide RNAi experiments, an external standard consisting of 768 values of non-RNAi treated cells covering the whole spectrum of cell densities was used to determine cell number dependent averages and SDs to calculate experimental Z-Scores of RNAi treated wells. All P-dAkt values of non-stimulated cells were normalized using a baseline standard curve (the average non-treated, non-stimulated experiment scores zero). For the insulin-stimulated data set, the P-dAkt values of insulin-treated cells were normalized using a standard curve derived from insulin-stimulated cells (the average non-treated, insulin-stimulated experiment scores zero).
Antibodies
All P-dAkt indirect immunofluorescence images, Cytoblots and western blots were performed using anti-Drosophila P-dAkt Ser505 (Lot 1 and Lot 2, Cell Signaling Technology) using a 1∶200, 1∶800 and 1∶200 dilution, respectively. For immunofluorescence, AlexaFluor594 and AlexaFluor488 conjugated secondary antibodies against Rabbit, Mouse and Goat were used 1∶500 (Invitrogen). Western blotting was performed using HRP conjugated anti-rabbit and anti-mouse antisera (Amersham). Pan-dAkt and P-S6K Ser398 (Cell Signaling Technology) were used 1∶200. Anti-GFP was purchased from Cappel and used at 1∶4000. Mouse anti alpha-Tubulin (Sigma) was used 1∶2000 for immunofluorescence. Rabbit anti S6K was a generous gift from Mary Steward and used 1∶10,000 for western blotting.
Immunofluorescence, confocal microscopy, and image processing
Imaginal discs and Drosophila Kc167 cells were fixed using 6% Formaldyhyde in PBS (cells 10 min at room temperature, imaginal discs at 4°C overnight), permeabilized in 0.1% Triton X-100 (10 min for cells, 2 hrs for imaginal discs) and blocked with 5% BSA in PBS (1 hr). Primary antibody incubation was performed overnight at 4°C with antibody dilutions as indicated above, using 5% BSA. After 3 washes with PBS, secondary antibody was incubated overnight in 5% BSA, followed by 3 washes in PBS. Specimens were mounted using Vectrashield mounting medium with DAPI (Vector Co.). All data were acquired using a Leica SP2 confocal microscope, a 63x lens, digital zoom factor of four, 1024×1024 pixel detector setting and processed using Adobe Photoshop software. Images of experimental and control cells were processed identically.
Genetics
Mutant wing imaginal disc clones were generated by FLP/FRT-mediated mitotic recombination using the following chromosomes: FRT82B, aktq [72]; FRT82B, aktq, tsc1Q87X [76]; FRT82B, aktEX4 (derived by imprecise excision from aktP04226, Bloomington Stock center) FRT82, tsc1Q87X [77]; FRT82B, tsc1W243X [77]; FRT82B, foxo25 [81]; FRT82B, foxo25, aktq [81]; tsc2192, FRT80B [77]; tsc2*, FRT80B (generous gift from I.K. Hariharan); s6Kl-1, FRT80B [24]; s6Kl-1, tsc2192, FRT80B [71]; s6Kl-1, tsc2*, FRT80B. Males of the respective genotypes were crossed to y,w,hs-FLP,UAS-mCD8::GFP; tub-Gal4; FRT82B,tub-Gal80/TM6B or y,w,hs-FLP, ubi-GFP, FRT80B females and larvae were heat shocked 60 hrs +/ − 12 hrs after egg laying (unless otherwise specified) at 37°C for 45 min. Overexpression of PI3KCAAX [113], InRDN (Bloomington Drosophila Stock Center), Tsc1 and Tsc2 [77], Foxo™ [80], S6KWT [78], S6KTE, S6KSTDE and S6KSTDETE [90] in the dorsal compartment of the wing imaginal disc was performed using the Gal4-UAS system [58] with y,w; ap-Gal4,UAS-mCD8::GFP (gift from C. Micchelli). The RaptorRNAi hairpin and transgenic line was generated using the VALIUM1 vector [114] as part of the transgenic RNAi project (TRIP, http://flyrnai.org/TRiP-HOME.html).
Gene names
Based on BLAST searches, information in the public ortholog databases InParanoid [115], and Homologene [116] published sequence homologies [117], CG3004 (Fbgn0030142), and CG10105 (Fbgn0033935) were referred to as Lst8 and Sin1 [29], [50], respectively.
Supporting Information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Zdroje
1. GhiglioneC
CarrawayKL3rd
AmundadottirLT
BoswellRE
PerrimonN
1999 The transmembrane molecule kekkon 1 acts in a feedback loop to negatively regulate the activity of the Drosophila EGF receptor during oogenesis. Cell 96 847 856
2. KleinDE
NappiVM
ReevesGT
ShvartsmanSY
LemmonMA
2004 Argos inhibits epidermal growth factor receptor signalling by ligand sequestration. Nature 430 1040 1044
3. YooAS
BaisC
GreenwaldI
2004 Crosstalk between the EGFR and LIN-12/Notch pathways in C. elegans vulval development. Science 303 663 666
4. PerrimonN
McMahonAP
1999 Negative feedback mechanisms and their roles during pattern formation. Cell 97 13 16
5. FreemanM
2000 Feedback control of intercellular signalling in development. Nature 408 313 319
6. RohatgiR
ScottMP
2007 Patching the gaps in Hedgehog signalling. Nat Cell Biol 9 1005 1009
7. NiehrsC
2006 Function and biological roles of the Dickkopf family of Wnt modulators. Oncogene 25 7469 7481
8. Pendas-FrancoN
GarciaJM
PenaC
ValleN
PalmerHG
2008 DICKKOPF-4 is induced by TCF/beta-catenin and upregulated in human colon cancer, promotes tumour cell invasion and angiogenesis and is repressed by 1alpha,25-dihydroxyvitamin D3. Oncogene 27 4467 4477
9. DhillonAS
von KriegsheimA
GrindlayJ
KolchW
2007 Phosphatase and feedback regulation of Raf-1 signaling. Cell Cycle 6 3 7
10. KeyseSM
2000 Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr Opin Cell Biol 12 186 192
11. LoTL
FongCW
YusoffP
McKieAB
ChuaMS
2006 Sprouty and cancer: the first terms report. Cancer Lett 242 141 150
12. MasonJM
MorrisonDJ
BassonMA
LichtJD
2006 Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol 16 45 54
13. ValentinoL
PierreJ
2006 JAK/STAT signal transduction: regulators and implication in hematological malignancies. Biochem Pharmacol 71 713 721
14. LetterioJJ
2005 TGF-beta signaling in T cells: roles in lymphoid and epithelial neoplasia. Oncogene 24 5701 5712
15. CourtoisG
GilmoreTD
2006 Mutations in the NF-kappaB signaling pathway: implications for human disease. Oncogene 25 6831 6843
16. SarbassovDD
GuertinDA
AliSM
SabatiniDM
2005 Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307 1098 1101
17. GuertinDA
SabatiniDM
2007 Defining the role of mTOR in cancer. Cancer Cell 12 9 22
18. ShawRJ
CantleyLC
2006 Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 441 424 430
19. PearceLR
HuangX
BoudeauJ
PawlowskiR
WullschlegerS
2007 Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem J 405 513 522
20. ThedieckK
PolakP
KimML
MolleKD
CohenA
2007 PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS ONE 2 e1217 doi:10.1371/journal.pone.0001217
21. CalnanDR
BrunetA
2008 The FoxO code. Oncogene 27 2276 2288
22. InokiK
CorradettiMN
GuanKL
2005 Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet 37 19 24
23. ManningBD
CantleyLC
2003 Rheb fills a GAP between TSC and TOR. Trends Biochem Sci 28 573 576
24. MontagneJ
StewartMJ
StockerH
HafenE
KozmaSC
1999 Drosophila S6 kinase: a regulator of cell size. Science 285 2126 2129
25. RichardsonCJ
SchalmSS
BlenisJ
2004 PI3-kinase and TOR: PIKTORing cell growth. Semin Cell Dev Biol 15 147 159
26. JacintoE
FacchinettiV
LiuD
SotoN
WeiS
2006 SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127 125 137
27. GuertinDA
StevensDM
ThoreenCC
BurdsAA
KalaanyNY
2006 Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 11 859 871
28. ShiotaC
WooJT
LindnerJ
SheltonKD
MagnusonMA
2006 Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev Cell 11 583 589
29. FriasMA
ThoreenCC
JaffeJD
SchroderW
SculleyT
2006 mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr Biol 16 1865 1870
30. ShawRJ
KosmatkaM
BardeesyN
HurleyRL
WittersLA
2004 The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A 101 3329 3335
31. ShawRJ
BardeesyN
ManningBD
LopezL
KosmatkaM
2004 The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 6 91 99
32. InokiK
ZhuT
GuanKL
2003 TSC2 mediates cellular energy response to control cell growth and survival. Cell 115 577 590
33. DannSG
ThomasG
2006 The amino acid sensitive TOR pathway from yeast to mammals. FEBS Lett 580 2821 2829
34. BruggeJ
HungMC
MillsGB
2007 A new mutational AKTivation in the PI3K pathway. Cancer Cell 12 104 107
35. DannSG
SelvarajA
ThomasG
2007 mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med 13 252 259
36. NiYG
WangN
CaoDJ
SachanN
MorrisDJ
2007 FoxO transcription factors activate Akt and attenuate insulin signaling in heart by inhibiting protein phosphatases. Proc Natl Acad Sci U S A 104 20517 20522
37. NiYG
BerenjiK
WangN
OhM
SachanN
2006 Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation 114 1159 1168
38. PuigO
MarrMT
RuhfML
TjianR
2003 Control of cell number by Drosophila FOXO: downstream and feedback regulation of the insulin receptor pathway. Genes Dev 17 2006 2020
39. PuigO
TjianR
2005 Transcriptional feedback control of insulin receptor by dFOXO/FOXO1. Genes Dev 19 2435 2446
40. MarrMT2nd
D'AlessioJA
PuigO
TjianR
2007 IRES-mediated functional coupling of transcription and translation amplifies insulin receptor feedback. Genes Dev 21 175 183
41. ShahOJ
HunterT
2005 Tuberous sclerosis and insulin resistance. Unlikely bedfellows reveal a TORrid affair. Cell Cycle 4 46 51
42. ManningBD
LogsdonMN
LipovskyAI
AbbottD
KwiatkowskiDJ
2005 Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev 19 1773 1778
43. HarutaT
UnoT
KawaharaJ
TakanoA
EgawaK
2000 A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrate-1. Mol Endocrinol 14 783 794
44. RuiL
FisherTL
ThomasJ
WhiteMF
2001 Regulation of insulin/insulin-like growth factor-1 signaling by proteasome-mediated degradation of insulin receptor substrate-2. J Biol Chem 276 40362 40367
45. EdgarBA
2006 How flies get their size: genetics meets physiology. Nat Rev Genet 7 907 916
46. WullschlegerS
LoewithR
HallMN
2006 TOR signaling in growth and metabolism. Cell 124 471 484
47. LeeversSJ
aHE
2004 Growth Regulation by Insulin and Tor Signaling in Drosophila.
HallM
Raff
Martin
Thomas
George
Cell Growth: Control of Cell Size New York Cold Spring Harbor Press Laboratory Press
48. HietakangasV
CohenSM
2007 Re-evaluating AKT regulation: role of TOR complex 2 in tissue growth. Genes Dev 21 632 637
49. LeeG
ChungJ
2007 Discrete functions of rictor and raptor in cell growth regulation in Drosophila. Biochem Biophys Res Commun 357 1154 1159
50. YangQ
InokiK
KimE
GuanKL
2006 TSC1/TSC2 and Rheb have different effects on TORC1 and TORC2 activity. Proc Natl Acad Sci U S A 103 6811 6816
51. GuertinDA
StevensDM
SaitohM
KinkelS
CrosbyK
2009 mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 15 148 159
52. LuoJ
ManningBD
CantleyLC
2003 Targeting the PI3K-Akt pathway in human cancer: rationale and promise. Cancer Cell 4 257 262
53. ManningBD
CantleyLC
2007 AKT/PKB signaling: navigating downstream. Cell 129 1261 1274
54. UmSH
D'AlessioD
ThomasG
2006 Nutrient overload, insulin resistance, and ribosomal protein S6 kinase 1, S6K1. Cell Metab 3 393 402
55. WeinkoveD
NeufeldTP
TwardzikT
WaterfieldMD
LeeversSJ
1999 Regulation of imaginal disc cell size, cell number and organ size by Drosophila class I(A) phosphoinositide 3-kinase and its adaptor. Curr Biol 9 1019 1029
56. BohniR
Riesgo-EscovarJ
OldhamS
BrogioloW
StockerH
1999 Autonomous control of cell and organ size by CHICO, a Drosophila homolog of vertebrate IRS1-4. Cell 97 865 875
57. GoberdhanDC
ParicioN
GoodmanEC
MlodzikM
WilsonC
1999 Drosophila tumor suppressor PTEN controls cell size and number by antagonizing the Chico/PI3-kinase signaling pathway. Genes Dev 13 3244 3258
58. BrandAH
PerrimonN
1993 Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118 401 415
59. BoutrosM
KigerAA
ArmknechtS
KerrK
HildM
2004 Genome-wide RNAi analysis of growth and viability in Drosophila cells. Science 303 832 835
60. HsuYC
ChernJJ
CaiY
LiuM
ChoiKW
2007 Drosophila TCTP is essential for growth and proliferation through regulation of dRheb GTPase. Nature 445 785 788
61. RehmannH
BruningM
BerghausC
SchwartenM
KohlerK
2008 Biochemical characterisation of TCTP questions its function as a guanine nucleotide exchange factor for Rheb. FEBS Lett 582 3005 3010
62. WangX
FonsecaBD
TangH
LiuR
EliaA
2008 Re-evaluating the roles of proposed modulators of mammalian target of rapamycin complex 1 (mTORC1) signaling. J Biol Chem 283 30482 30492
63. KwiatkowskiDJ
2003 Rhebbing up mTOR: new insights on TSC1 and TSC2, and the pathogenesis of tuberous sclerosis. Cancer Biol Ther 2 471 476
64. PanD
DongJ
ZhangY
GaoX
2004 Tuberous sclerosis complex: from Drosophila to human disease. Trends Cell Biol 14 78 85
65. RadimerskiT
MontagneJ
Hemmings-MieszczakM
ThomasG
2002 Lethality of Drosophila lacking TSC tumor suppressor function rescued by reducing dS6K signaling. Genes Dev 16 2627 2632
66. ShahOJ
WangZ
HunterT
2004 Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol 14 1650 1656
67. HarringtonLS
FindlayGM
GrayA
TolkachevaT
WigfieldS
2004 The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol 166 213 223
68. HidalgoM
RowinskyEK
2000 The rapamycin-sensitive signal transduction pathway as a target for cancer therapy. Oncogene 19 6680 6686
69. MitaMM
MitaA
RowinskyEK
2003 The molecular target of rapamycin (mTOR) as a therapeutic target against cancer. Cancer Biol Ther 2 S169 177
70. SawyersCL
2003 Will mTOR inhibitors make it as cancer drugs? Cancer Cell 4 343 348
71. GaoX
ZhangY
ArrazolaP
HinoO
KobayashiT
2002 Tsc tumour suppressor proteins antagonize amino-acid-TOR signalling. Nat Cell Biol 4 699 704
72. StaveleyBE
RuelL
JinJ
StambolicV
MastronardiFG
1998 Genetic analysis of protein kinase B (AKT) in Drosophila. Curr Biol 8 599 602
73. WuJS
LuoL
2006 A protocol for mosaic analysis with a repressible cell marker (MARCM) in Drosophila. Nat Protoc 1 2583 2589
74. BlairSS
2003 Genetic mosaic techniques for studying Drosophila development. Development 130 5065 5072
75. PotterCJ
PedrazaLG
XuT
2002 Akt regulates growth by directly phosphorylating Tsc2. Nat Cell Biol 4 658 665
76. GaoX
PanD
2001 TSC1 and TSC2 tumor suppressors antagonize insulin signaling in cell growth. Genes Dev 15 1383 1392
77. TaponN
ItoN
DicksonBJ
TreismanJE
HariharanIK
2001 The Drosophila tuberous sclerosis complex gene homologs restrict cell growth and cell proliferation. Cell 105 345 355
78. PotterCJ
HuangH
XuT
2001 Drosophila Tsc1 functions with Tsc2 to antagonize insulin signaling in regulating cell growth, cell proliferation, and organ size. Cell 105 357 368
79. DattaSR
BrunetA
GreenbergME
1999 Cellular survival: a play in three Akts. Genes Dev 13 2905 2927
80. HwangboDS
GershmanB
TuMP
PalmerM
TatarM
2004 Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature 429 562 566
81. JungerMA
RintelenF
StockerH
WassermanJD
VeghM
2003 The Drosophila forkhead transcription factor FOXO mediates the reduction in cell number associated with reduced insulin signaling. J Biol 2 20
82. LongX
LinY
Ortiz-VegaS
YonezawaK
AvruchJ
2005 Rheb Binds and Regulates the mTOR Kinase. Curr Biol 15 702 713
83. LongX
Ortiz-VegaS
LinY
AvruchJ
2005 Rheb binding to mTOR is regulated by amino acid sufficiency. J Biol Chem
84. SaucedoLJ
GaoX
ChiarelliDA
LiL
PanD
2003 Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol 5 566 571
85. StockerH
RadimerskiT
SchindelholzB
WittwerF
BelawatP
2003 Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol 5 559 565
86. BhaskarPT
HayN
2007 The two TORCs and Akt. Dev Cell 12 487 502
87. TremblayF
LavigneC
JacquesH
MaretteA
2007 Role of dietary proteins and amino acids in the pathogenesis of insulin resistance. Annu Rev Nutr 27 293 310
88. DennisPB
PullenN
PearsonRB
KozmaSC
ThomasG
1998 Phosphorylation sites in the autoinhibitory domain participate in p70(s6k) activation loop phosphorylation. J Biol Chem 273 14845 14852
89. DennisPB
PullenN
KozmaSC
ThomasG
1996 The principal rapamycin-sensitive p70(s6k) phosphorylation sites, T-229 and T-389, are differentially regulated by rapamycin-insensitive kinase kinases. Mol Cell Biol 16 6242 6251
90. BarceloH
StewartMJ
2002 Altering Drosophila S6 kinase activity is consistent with a role for S6 kinase in growth. Genesis 34 83 85
91. RadimerskiT
MontagneJ
RintelenF
StockerH
van der KaayJ
2002 dS6K-regulated cell growth is dPKB/dPI(3)K-independent, but requires dPDK1. Nat Cell Biol 4 251 255
92. BjedovI
ToivonenJM
KerrF
SlackC
JacobsonJ
2010 Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metab 11 35 46
93. MartinKA
MerenickBL
DingM
FetalveroKM
RzucidloEM
2007 Rapamycin promotes vascular smooth muscle cell differentiation through insulin receptor substrate-1/phosphatidylinositol 3-kinase/Akt2 feedback signaling. J Biol Chem 282 36112 36120
94. MaL
Teruya-FeldsteinJ
BehrendtN
ChenZ
NodaT
2005 Genetic analysis of Pten and Tsc2 functional interactions in the mouse reveals asymmetrical haploinsufficiency in tumor suppression. Genes Dev 19 1779 1786
95. KalaanyNY
SabatiniDM
2009 Tumours with PI3K activation are resistant to dietary restriction. Nature 458 725 731
96. ShahOJ
HunterT
2006 Turnover of the active fraction of IRS1 involves raptor-mTOR - and S6K1-dependent serine phosphorylation in cell culture models of tuberous sclerosis. Mol Cell Biol 26 6425 6434
97. TzatsosA
KandrorKV
2006 Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol Cell Biol 26 63 76
98. HayN
2005 The Akt-mTOR tango and its relevance to cancer. Cancer Cell 8 179 183
99. HuangJ
DibbleCC
MatsuzakiM
ManningBD
2008 The TSC1-TSC2 complex is required for proper activation of mTOR complex 2. Mol Cell Biol 28 4104 4115
100. VerduJ
BuratovichMA
WilderEL
BirnbaumMJ
1999 Cell-autonomous regulation of cell and organ growth in Drosophila by Akt/PKB. Nat Cell Biol 1 500 506
101. SkeenJE
BhaskarPT
ChenCC
ChenWS
PengXD
2006 Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner. Cancer Cell 10 269 280
102. ManningBD
TeeAR
LogsdonMN
BlenisJ
CantleyLC
2002 Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 10 151 162
103. InokiK
LiY
ZhuT
WuJ
GuanKL
2002 TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4 648 657
104. TavazoieSF
AlvarezVA
RidenourDA
KwiatkowskiDJ
SabatiniBL
2005 Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci 8 1727 1734
105. DongJ
PanD
2004 Tsc2 is not a critical target of Akt during normal Drosophila development. Genes Dev 18 2479 2484
106. Vander HaarE
LeeSI
BandhakaviS
GriffinTJ
KimDH
2007 Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 9 316 323
107. SancakY
ThoreenCC
PetersonTR
LindquistRA
KangSA
2007 PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 25 903 915
108. FonsecaBD
SmithEM
LeeVH
MacKintoshC
ProudCG
2007 PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J Biol Chem 282 24514 24524
109. HanEK
LeversonJD
McGonigalT
ShahOJ
WoodsKW
2007 Akt inhibitor A-443654 induces rapid Akt Ser-473 phosphorylation independent of mTORC1 inhibition. Oncogene 26 5655 5661
110. TelemanAA
ChenYW
CohenSM
2005 4E-BP functions as a metabolic brake used under stress conditions but not during normal growth. Genes Dev 19 1844 1848
111. HolzMK
BlenisJ
2005 Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase. J Biol Chem 280 26089 26093
112. RozenS
SkaletskyH
2000 Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132 365 386
113. LeeversSJ
WeinkoveD
MacDougallLK
HafenE
WaterfieldMD
1996 The Drosophila phosphoinositide 3-kinase Dp110 promotes cell growth. Embo J 15 6584 6594
114. NiJQ
MarksteinM
BinariR
PfeifferB
LiuLP
2008 Vector and parameters for targeted transgenic RNA interference in Drosophila melanogaster. Nat Methods 5 49 51
115. O'BrienKP
RemmM
SonnhammerEL
2005 Inparanoid: a comprehensive database of eukaryotic orthologs. Nucleic Acids Res 33 D476 480
116. WheelerDL
BarrettT
BensonDA
BryantSH
CaneseK
2005 Database resources of the National Center for Biotechnology Information. Nucleic Acids Res 33 Database Issue D39 45
117. KimDH
SarbassovDD
AliSM
LatekRR
GunturKV
2003 GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell 11 895 904
Štítky
Genetika Reprodukční medicínaČlánek vyšel v časopise
PLOS Genetics
2010 Číslo 6
- Kazuistika – Perspektivy využití precizované medicíny v rámci personalizované specifické terapie onkologických pacientů
- Nobelova cena za chemii pro genetické nůžky: Objev, který změní naši budoucnost?
- Technologie na bázi RNA v klinické praxi: od přebarvených petúnií k terapii vzácných a dosud jen obtížně léčitelných chorob u lidí
- „Nepředstavovali jsme si, že náš výzkum povede přímo ke vzniku nových léků, dokonce ještě za našeho života“
- Bezplatné služby pro diagnostiku ATTRv amyloidózy pro kardiology
Nejčtenější v tomto čísle
- Web-Based, Participant-Driven Studies Yield Novel Genetic Associations for Common Traits
- The IG-DMR and the -DMR at Human Chromosome 14q32.2: Hierarchical Interaction and Distinct Functional Properties as Imprinting Control Centers
- Cushing's Syndrome and Fetal Features Resurgence in Adrenal Cortex–Specific Knockout Mice
- Amplification of a Cytochrome P450 Gene Is Associated with Resistance to Neonicotinoid Insecticides in the Aphid
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy