
Zárodečné mutace v genech RAD51C a RAD51D a dědičná predispozice ke vzniku karcinomu ovaria
Germline mutations in RAD51C and RAD51D and hereditary predisposition to ovarian cancer
Ovarian cancer is one of the most common gynecologic cancers with the highest mortality rate over a long period. Genetic predisposition to ovarian cancer is unusually high. In the Czech Republic, causal mutation in any ovarian cancer predisposition gene is identified in approximately 30% of the ovarian cancer patients. Therefore, according to the current guidelines, all ovarian cancer patients should be provided with genetic testing. The BRCA1 and BRCA2 are the two major ovarian cancer predisposition genes. Nevertheless, mutations in other predisposition genes, including RAD51C and RAD51D, are associated with high ovarian cancer risk. Mutations in RAD51C and RAD51D are found in 1% of ovarian cancer patients in each respective gene. Currently, identification of germline mutation in RAD51C and RAD51D is primarily of preventive importance but it potentially could make a prognostic difference. The aim of this review is to summarize the recent RAD51C and RAD51D knowledge, including the biological function, cancer risks associated with germline mutations, and recommendations for mutation carriers.
Keywords:
ovarian cancer – DNA repair – cancer genes – next gen sequencing – Mutation – genetic testing
Autoři:
RNDr. Soukupová Jana, Ph.D. 1*; Mgr. Lhotová Klára 1*; RNDr. Janatová Markéta, Ph.D. 1; MUDr. Kleiblová Petra, Ph.D. 2; MUDr. Vočka Michal 3; doc. MUDr. Foretová Lenka, Ph.D. 4; prof. MUDr. Zikán Michal, Ph.D. 5; prof. MUDr. Kleibl Zdeněk, Ph.D. 1
Působiště autorů:
Ústav biochemie a experimentální onkologie, 1. LF UK, Praha
1; Ústav biologie a lékařské genetiky, 1. LF UK a VFN v Praze
2; Onkologická klinika 1. LF UK a VFN v Praze
3; Oddělení epidemiologie a genetiky nádorů, MOÚ, Brno
4; Gynekologicko-porodnická klinika 1. LF UK a Nemocnice Na Bulovce, Praha
5
Vyšlo v časopise:
Klin Onkol 2021; 34(1): 26-32
Kategorie:
Přehled
doi:
https://doi.org/10.48095/ccko202126
Souhrn
Karcinom ovaria patří k nejčastějším gynekologickým nádorovým onemocněním, mezi nimiž dlouhodobě zaujímá první místo v mortalitě. Genetická predispozice pro vznik karcinomu ovaria je neobvykle vysoká, v ČR dosahuje 30 % všech případů. Vysoká prevalence dědičných mutací v klinicky významných genech odůvodňuje dle současných doporučení testování přítomnosti zárodečných mutací u všech pacientek s karcinomem ovaria. Hlavními predispozičními geny dědičné formy karcinomu ovaria v ČR jsou BRCA1 a BRCA2, nicméně s vysokým rizikem vzniku tohoto onemocnění jsou spojeny zárodečné mutace i v dalších predispozičních genech vč. RAD51C a RAD51D. Mutace v každém z těchto dvou genů se u nás vyskytují u 1 % pacientek s karcinomem ovaria. Identifikace zárodečných mutací v genech RAD51C a RAD51D má kromě významu preventivního potenciálně také význam prognostický. Cílem této přehledové práce je shrnout dosavadní poznatky týkající se klinického významu RAD51C a RAD51D, rizika vzniku karcinomu ovaria a dalších nádorových onemocnění a klinických doporučení pro nosičky mutací.
Klíčová slova:
nádory vaječníků – oprava DNA – nádorové geny – sekvenování nové generace – mutace – genetické testování
Karcinom ovaria
Karcinom ovaria je pátým nejčastějším nádorovým onemocněním u žen [1–3] a zároveň onemocněním s nejvyšší mírou mortality v rámci gynekologických malignit [4]. Česká republika zaujímá v incidenci karcinomu ovaria 11. místo na světě [5]. V roce 2017 bylo v ČR diagnostikováno 982 nových případů; 641 žen v důsledku tohoto onemocnění zemřelo [5]. Vysoká mortalita je způsobena mimo jiné absencí specifických klinických příznaků u časných stadií onemocnění a jejich obtížnou diagnostikou. Téměř u dvou třetin nemocných byl karcinom ovaria diagnostikován ve stadiu III nebo IV [5].
Rizikovými faktory pro vznik karcinomu ovaria jsou kromě vyššího věku také hormonální faktory, životní styl a pozitivní rodinná anamnéza [6]. Celoživotní riziko se v rozvinutých zemích pohybuje do 2 % [7,8], pokud však má žena příbuznou I. stupně s diagnózou karcinomu ovaria, její riziko vzniku onemocnění se zvyšuje na 5 % [9]. Genetická predispozice pro vznik karcinomu ovaria je neobvykle častá a je odhalena až u 25–30 % nemocných [10–12].
Hlavními predispozičními geny pro vznik karcinomu ovaria jsou stejně jako v případě dědičného karcinomu prsu tumor supresorové geny BRCA1 a BRCA2 (OMIM 604370 a 612555) [13–16]. V naší populaci jsou nalezeny zárodečné mutace u 25,1 % pacientek s karcinomem ovaria [12]. Vysoké riziko – relativní riziko (RR) > 5 – vzniku karcinomu ovaria bylo prokázáno rovněž u nosiček mutací v genech Lynchova syndromu, STK11, RAD51C a RAD51D [12, 17–20]. Vysoké či středně zvýšené riziko (RR 2–5) se předpokládá u nosiček mutací v BRIP1; výše rizika vzniku karcinomu ovaria spojeného s mutacemi v řadě jiných predispozičních genů (CHEK2, NBN, ATM, PALB2, BARD1, TP53) nebyla dosud přesně stanovena. Lze předpokládat, že v některých rodinách může být vznik onemocnění spojen s privátními mutacemi v dalších kandidátních genech.
Rozvoj nových technologií a především zavedení sekvenování nové generace do klinické praxe umožnilo rutinní testování všech známých predispozičních genů. Genetické testování je v současné době v ČR indikováno u všech pacientek s karcinomem ovaria [21]. Cílem tohoto článku je shrnout poznatky týkající se genů RAD51C a RAD51D, výše rizik spojených s mutacemi v těchto genech a doporučení péče o nosiče mutací.
Biologická funkce RAD51C a RAD51D
Gen RAD51C (OMIM 602774) je lokalizován na chromozomu 17q23 a podobně jako BRCA1 (FANCS) a BRCA2 (FANCD1) patří do rodiny 22 dosud identifikovaných genů [21], jejichž vrozené bialelické inaktivace způsobují syndromologicky se překrývající závažné vrozené onemocnění – Fanconiho anémii. Bialelické mutace v RAD51C jsou spojeny s Fanconiho anémií komplementační skupiny O (FANCO; 613390) [23]. Gen RAD51D (OMIM 02954) byl identifikován na konci 90. let na chromozomu 17 v pozici 17q12 [24] a mezi geny Fanconiho anémie řazen není.
Oba proteiny RAD51C a RAD51D patří (spolu s RAD51B, XRCC2, XRCC3 a SWSAP1) mezi paralogy proteinu RAD51. Jedná se o proteiny kódované geny, které pravděpodobně vznikly duplikací jednoho genu a v průběhu evoluce si udržely strukturní podobnost s RAD51 [25].
Proteiny RAD51C a RAD51D se účastní oprav dvouřetězcových zlomů DNA mechanizmem homologní rekombinace, která reprezentuje sice vysoce přesný, avšak energeticky a faktorově velmi náročný proces oprav DNA probíhající v terminální S fázi a G2 fázi buněčného cyklu mitoticky aktivních buněk. Proteiny podílející se na procesu homologní rekombinace vytvářejí v místě přerušení DNA dvoušroubovice ohromné (mikroskopicky viditelné) molekulární komplexy. Jejich úkolem je upravit místo přerušené DNA a na obou koncích vytvořit tisíce bází dlouhé jednořetězcové přesahy sloužící k hybridizaci s homologní sekvencí na sesterské chromatidě, která je následně využita jako předloha pro syntézu chybějícího úseku v místě přerušení DNA. Po opravě jednoho z řetězců dochází k jeho přemístění z oblasti sesterské chromatidy zpět. Syntéza komplementárního vlákna podle opraveného úseku a finální ligace obnovující celistvost dvouvlákna DNA dokončí opravu poškození DNA. Poruchy homologní rekombinace významně porušují genomovou stabilitu a zvyšují pravděpodobnost nádorové transformace. Dědičné mutace mnoha genů, které kódují iniciální senzorové proteiny rozpoznávající tento typ poškození DNA (MRE11, RAD50, NBN), kinázy (ATM, CHK2) regulující aktivitu proteinových faktorů reparace, proteiny odpovědi buňky na poškození DNA (TP53) nebo přímo rekombinační faktory (BRCA1, BRCA2) se podílejí na riziku vzniku dědičných forem nádorových onemocnění, především karcinomů prsu a ovaria [26]. Proteiny RAD51B, RAD51C, RAD51D a XRCC2 tvoří heteromerní BCDX2 komplexy stabilizující polymery vláken proteinu RAD51, které slouží pro přemístění jednořetězcových přesahů přerušené DNA do oblasti sesterské chromatidy [27]. Kromě toho protein RAD51C tvoří s XRCC3 komplex CX3 zapojený do pozdnějších fází procesu homologní rekombinace [28]. Společně s BRCA2, PALB2 a RAD51 je také součástí komplexu, který zajišťuje správné načasování tvorby vláken z proteinu RAD51 [29]. Uvedené komplexy s proteiny RAD51C a RAD51D pozitivně ovlivňují správnou funkci proteinu RAD51 v homologní rekombinaci, nicméně detailní funkce jednotlivých RAD51 paralogů v homologní rekombinaci není ještě přesně určena.
Oba proteiny, RAD51C i RAD51D, se patrně mohou podílet i na dalších procesech spojených s udržením integrity genomu. RAD51C se účastní aktivace CHK2 kinázy [30]. RAD51D se podílí na ochraně konců chromozomů a udržení integrity telomer [31].
RAD51C a RAD51D a riziko vzniku karcinomu ovaria
Asociace mutací v genu RAD51C se zvýšeným rizikem vzniku karcinomu ovaria byla poprvé publikována v roce 2010 [17]. V této práci byly trunkační mutace v RAD51C zachyceny u 1,3 % žen ze 480 německých pacientek s karcinomem prsu a ovaria, zatímco u 620 pacientek pouze s karcinomem prsu a u 2 912 populačních kontrol patogenní mutace zachyceny nebyly. Následovala řada prací potvrzující asociaci RAD51C s dědičnou predispozicí ke karcinomu ovaria v různých populacích. Frekvence mutací se u pacientek s karcinomem ovaria pohybovala mezi 0,3 % [32] až 2,9 % [33]. Zjištěné relativní riziko vzniku onemocnění řadí RAD51C mezi ovariální predispoziční geny vysokého rizika (tab. 1).
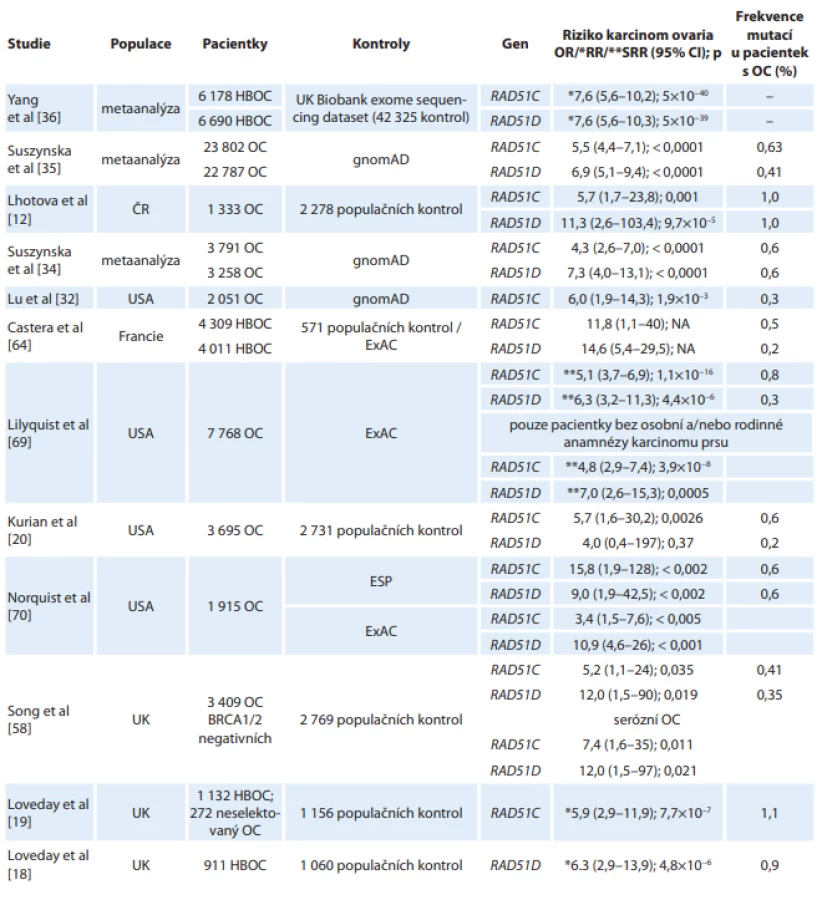
Záhy po identifikaci RAD51C jako genu predisponujícího ke karcinomu ovaria následovala práce Lovedaye et al zaměřená na analýzu RAD51D, která nalezla patogenní mutaci u 8 z 911 (0,9 %) pacientek (UK) s hereditárním karcinomem prsu a ovaria negativně testovaných na mutace v BRCA1/2 a v 1 z 1 060 (0,09 %) populačních kontrol [18], přičemž frekvence mutací byla nejvyšší v rodinách s výskytem jednoho a více případu karcinomu ovaria. Na základě těchto zjištění bylo stanoveno relativní riziko vzniku karcinomu ovaria pro nosičky mutace v genu RAD51D na 6,3 (95% CI 2,86–13,85; p = 4,8 × 10−6). Vysoké riziko vzniku karcinomu ovaria potvrdily i další studie (tab. 1).
Po potvrzení asociace zárodečných mutací v genech RAD51C a RAD51D s karcinomem ovaria následovaly metaanalýzy dostupných studií s cílem zpřesnit riziko vzniku tohoto onemocnění. Metaanalýza přibližně 3 500 pacientek stanovila odds ratio (OR) karcinomu ovaria v evropské populaci pro nosičky mutací v genu RAD51C na 4,24 (95% CI 2,56–7,02; p < 0,0001) a v genu RAD51D na 7,28 (95% CI 4,03–13,14; p < 0,0001) [34]. Aktuální metaanalýza case-control studií zahrnující přibližně 30 000 pacientek z různých populací zpřesnila OR pro karcinom ovaria u nosiček mutací v genu RAD51C na 5,5 (95% CI 4,42–7,07; p < 0,0001) a v genu RAD51D na 6,94 (95% CI 5,10–9,44; p < 0,0001), u majoritní podskupiny pacientek evropské kavkazské populace dosahovalo OR pro RAD51C 5,04 (95% CI 3,85–6,59; p < 0,0001) a pro RAD51D 7,60 (95% CI 5,29–10,93; p < 0,0001) [35]. Současně byla publikována další metaanalýza, jejímiž autory byli Yang et al, která s využitím britských kontrol stanovila relativní riziko vzniku karcinomu ovaria pro nosičky mutací v RAD51C na 7,55 (95% CI 5,60–10,19; p = 5 × 10–40) a v RAD51D na 7,60 (95% CI 5,61–10,30; p = 5 × 10–39) [36].
Pilotní analýza genů RAD51C/D u 171 BRCA1/2 negativních pacientek s karcinomem ovaria v české populaci identifikovala příčinné mutace v RAD51C u 1,2 % a v RAD51D u 1,8 % pacientek [37]. V aktuální práci zahrnující analýzu 1 333 pacientek s karcinomem ovaria vyšetřených panelem CZECANCA v rámci konzorcia českých laboratoří jsme nalezli patogenní mutace v každém z obou genů shodně u 1 % pacientek s karcinomem ovaria, přičemž frekvence mutací v kontrolní populaci byla přibližně 10× menší a OR dosahovalo 11,2 pro RAD51D a 5,6 pro RAD51C [12].
RAD51C a RAD51D a riziko vzniku karcinomu prsu a dalších nádorů
Ve většině studií, které zahrnovaly analýzu genů RAD51C a RAD51D u pacientek s hereditárním karcinomem prsu a ovaria, byly nalezené jasně patogenní mutace asociovány s karcinomem ovaria [17,19,38–45]. Rovněž bylo prokázáno, že prevalence mutací v genu RAD51D roste s počtem nemocných s karcinomem ovaria v rodině [18]. V uvedených studiích zároveň nebylo potvrzeno zvýšené riziko vzniku karcinomu prsu s výjimkou tří trunkačních mutací a missense varianty nejasného významu c.428A > G v RAD51C zachycených v rodinách s familiárním výskytem pouze karcinomu prsu [40–42,46,47]. Asociace mutací v genu RAD51D se zvýšeným rizikem vzniku karcinomu prsu je patrně ještě méně významná [47,48]. Význam patogenních mutací v RAD51C a RAD51D se pokusila objasnit americká studie na rozsáhlém souboru 65 057 pacientek s karcinomem prsu indikovaných ke genetickému vyšetření. Zjištěné OR dosahovalo pro mutace v RAD51D hodnoty 3,07 (95% CI 1,21–7,88; p = 0,01), zatímco patogenní alterace v RAD51C nebyly asociovány se zvýšeným rizikem vzniku karcinomu prsu [49]. Další rozsáhlá studie analyzující 95 561 pacientek s karcinomem prsu a/nebo ovaria však neprokázala významné zvýšení rizika vzniku karcinomu prsu pro nosičky mutací v RAD51C (1,43; 95% CI 0,97–2,12) ani RAD51D (1,37; 95% CI 0,76–2,49) [50]. Dle poslední metaanalýzy je relativní riziko vzniku karcinomu prsu pro nosičky mutací v RAD51C 1,99 (95% CI 1,39–2,85; p = 1,55 × 10–4) a v RAD51D 1,83 (95% CI 1,24–2,72; p = 0,002) [36]. Studie zabývající se významem zárodečných mutací v genech RAD51C a RAD51D pro vznik karcinomu prsu shrnuje tab. 2. Významné zvýšení rizika vzniku karcinomu prsu pro nosičky mutací v genech RAD51C a RAD51D nebylo spolehlivě prokázáno, ale nelze je (i vzhledem k šíři konfidenčních intervalů) v současné době zcela vyloučit.

Geny RAD51C a RAD51D byly rovněž navrženy jako možné kandidátní predispoziční geny pro karcinom prostaty [51,52], předchozí práce však asociaci RAD51C a RAD51D s významně zvýšeným rizikem vzniku karcinomu prostaty (a kolorekta) neprokázaly [53].
Klinický význam
Zárodečné mutace v genech RAD51C a RAD51D jsou v naší populaci zachyceny v každém z uvedených genů u 1 % pacientek s karcinomem ovaria. Zatímco riziko vzniku karcinomu ovaria v běžné populaci nepřesahuje 2 % [8], u nosiček mutací v genu RAD51C může být zvýšeno až 7×, u nosiček mutací v RAD51D i méně než 10× (tab. 1). Tato relativní rizika odpovídají celoživotnímu riziku přibližně 9 % pro nosičky mutací v RAD51C [45] a mohou přesáhnout 10 % pro RAD51D [54]. V recentní studii autorů Yang et al bylo celoživotní riziko vzniku karcinomu ovaria stanoveno na 11 % (RAD51C) a 13 % (RAD51D), avšak v případě výskytu ovariálního karcinomu u matky (matky a maternální babičky / matky a sestry) může dosáhnout 20 % (24/32 %) pro nosičky mutací v RAD51C a 23 % (27/36 %) pro nosičky mutací v RAD51D [36].
Dle současných názorů je u žen s rizikem vzniku karcinomu ovaria převyšujícím 4–5 % vhodné zvážit profylaktickou salpingooforektomii (riziko redukující salpingooforektomie – RRSO) [55]. S ohledem na výši rizika asociovaného se zárodečnými mutacemi v RAD51C/D je pro asymptomatické nosičky mutací RRSO doporučena [55–57]. Tento výkon je nutné vhodně načasovat. Průměrný věk v době diagnózy je přibližně 55–60 let u nosiček mutací v RAD51C a mírně vyšší u RAD51D [36,45,58]. Odhadované kumulativní riziko pro nosičky mutace v genu RAD51C ve věku 50 let je 1,3 % (95% CI 0,3–6,0 %) a pro RAD51D 3,0 % (95% CI 0,4–21 %), v 70 letech věku dosahuje 5,2 % (95% CI 1,1–22 %) pro RAD51C a 12,0 % (95% CI 1,5–60 %) pro RAD51D [58]. Aktuálně publikovaná metaanalýza uvádí, že kumulativní riziko vzniku karcinomu ovaria u nosiček mutací RAD51C/D dosáhne 4 % v 60. roce věku, v případě výskytu karcinomu ovaria také u matky a sestry již v 50 letech [36]. Dle současných doporučení je proto vhodné RRSO nabídnout nosičkám mutací v obou genech mezi 45. a 50. rokem [56], v rodinách s časným výskytem karcinomu ovaria individuálně dříve.
Relativní riziko vzniku karcinomu prsu pro nosičky zárodečných mutací v genech RAD51C/D řadí tyto geny mezi nízce až středně penetrantní. Celoživotní riziko vzniku karcinomu prsu dosahuje u nosiček mutací v RAD51C 21 % (95% CI 14–28 %) a v RAD51D 20 % (95% CI 15–29 %), v případě dvou příbuzných I. stupně s karcinomem prsu se riziko zvyšuje až na 44–46 % [36]. Doporučení pro sledování nosiček mutací v genech RAD51C/D by mělo vycházet z výše rizika ≥ 20 % [21]. Protože RAD51C patří do rodiny Fanconi anemia genů, mělo by genetické poradenství zahrnovat i informace o riziku případného autozomálně recesivního onemocnění u potomků [59].
Genetické testování DNA reparačních genů má v současné době význam především preventivní [55], má však i potenciální prognostický význam, jež v budoucnu pravděpodobně poroste. Mutace v DNA reparačních genech RAD51C a RAD51D jsou prediktivními markery odpovědi na léčbu platinovými deriváty, podobně jako mutace v BRCA1 a BRCA2. Protože mutace v RAD51C a RAD51D vedou k poškození procesu homologní rekombinace, lze předpokládat – podobně jako u nosiček mutací v BRCA1 a BRCA2 – citlivost k cílené léčbě PARP inhibitory [18,60–63].
Shrnutí
Analýza genů RAD51C a RAD51D je v současné době součástí rutinního genetického testování u všech pacientek s karcinomem ovaria. Mutace jsou identifikovány v každém z obou genů u přibližně 1 % nemocných. Nosičství patogenní mutace je spojeno s vysokým rizikem vzniku karcinomu ovaria; celoživotní riziko vzniku karcinomu ovaria se pohybuje okolo 11 % pro RAD51C a 13 % pro RAD51D, s vrcholem incidence mezi 55–60 lety věku. Riziko vzniku karcinomu prsu je pro nosičky mutací v RAD51C/D mírně až středně zvýšeno; celoživotní riziko dosahuje 20–21 %. V případě výskytu nádorů prsu a/nebo ovaria v rodině se riziko vzniku těchto malignit zvyšuje až na 32–36 % v případě karcinomu ovaria a na 44–46 % v případě karcinomu prsu. Zvýšené riziko dalších nádorových onemocnění nebylo spolehlivě prokázáno, nelze ho však v současné době vyloučit. Pro nosičky mutací je doporučena preventivní salpingooforektomie mezi 45. a 50. rokem, na základě rodinné anamnézy individuálně dříve. Preventivní sledování s ohledem na karcinom prsu se odvíjí od výše rizika ≥ 20 %.
Práce byla podpořena grantem Agentury pro zdravotnický výzkum MZ ČR NU20-03-00016.
This work was supported by grant of the Agency for Healthcare Research of the Ministry of Health of the Czech Republic No. NU20-03-00016.
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
The authors declare they have no potential confl icts of interest concerning drugs, products, or services used in the study.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do bi omedicínských časopisů.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
RNDr. Jana Soukupová, Ph.D.
Ústav biochemie a experimentální onkologie 1. LF UK Praha
Kateřinská 1660/32
121 08 Praha 2
e-mail: jana.soukupova@lf1.cuni.cz
Obdrženo/Submitted: 25. 6. 2020
Přijato/Accepted: 18. 8. 2020
Zdroje
1. Muinao T, Deka Boruah HP, Pal M. Multi-biomarker panel signature as the key to diagnosis of ovarian cancer. Heliyon 2019; 5 (12): e02826. doi: 10.1016/j.heliyon.2019.e02826.
2. Ferlay J, Parkin DM, Steliarova-Foucher E. Estimates of cancer incidence and mortality in Europe in 2008. Eur J Cancer 2010; 46 (4): 765–781. doi: 10.1016/j.ejca.2009. 12.014.
3. Goodman MT, Howe HL, Tung KH et al. Incidence of ovarian cancer by race and ethnicity in the United States, 1992–1997. Cancer 2003; 97 (10 Suppl): 2676–2685. doi: 10.1002/cncr.11349.
4. Momenimovahed Z, Tiznobaik A, Taheri S et al. Ovarian cancer in the world: epidemiology and risk factors. Int J Womens Health 2019; 11 : 287–299. doi: 10.2147/IJWH.S197604
5. Epidemiologie a incidence zhoubných nádorů v České republice. [online]. Dostupné z: www.svod.cz.
6. Colombo N, Van Gorp T, Parma G et al. Ovarian cancer. Crit Rev Oncol Hematol 2006; 60 (2): 159–179. doi: 10.1016/j.critrevonc.2006.03.004.
7. Wong AS, Auersperg N. Ovarian surface epithelium: family history and early events in ovarian cancer. Reprod Biol Endocrinol 2003; 1 : 70. doi: 10.1186/1477-7827 - 1-70.
8. Torre LA, Trabert B, DeSantis CE et al. Ovarian cancer statistics, 2018. CA Cancer J Clin 2018; 68 (4): 284–296. doi: 10.3322/caac.21456.
9. Piver MS. Hereditary ovarian cancer. Lessons from the first twenty years of the Gilda Radner Familial Ovarian Cancer Registry. Gynecol Oncol 2002; 85 (1): 9–17. doi: 10.1006/gyno.2001.6465.
10. Walsh T, Casadei S, Lee MK et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA 2011; 108 (44): 18032–18037. doi: 10.1073/pnas.1115052108.
11. Krivokuca A, Boljevic I, Jovandic S et al. Germline mutations in cancer susceptibility genes in high grade serous ovarian cancer in Serbia. J Hum Genet 2019; 64 (4): 281–290. doi: 10.1038/s10038-019-0562-z.
12. Lhotova K, Stolarova L, Zemankova P et al. Multigene panel germline testing of 1333 Czech patients with ovarian cancer. Cancers (Basel) 2020; 12 (4): 956. doi: 10.3390/cancers12040956.
13. Hall JM, Lee MK, Newman B et al. Linkage of early–onset familial breast cancer to chromosome 17q21. Science 1990; 250 (4988): 1684–1689. doi: 10.1126/science.2270482.
14. Miki Y, Swensen J, Shattuck-Eidens D et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994; 266 (5182): 66–71. doi: 10.1126/science.7545954.
15. Wooster R, Bignell G, Lancaster J et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995; 378 (6559): 789–792. doi: 10.1038/378789a0.
16. Wooster R, Neuhausen SL, Mangion J et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science 1994; 265 (5181): 2088–2090. doi: 10.1126/science.8091231.
17. Meindl A, Hellebrand H, Wiek C et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet 2010; 42 (5): 410–414. doi: 10.1038/ng.569.
18. Loveday C, Turnbull C, Ramsay E et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet 2011; 43 (9): 879–882. doi: 10.1038/ng.893.
19. Loveday C, Turnbull C, Ruark E et al. Germline RAD51C mutations confer susceptibility to ovarian cancer. Nat Genet 2012; 44 (5): 475–476. doi: 10.1038/ng.2224.
20. Kurian AW, Hughes E, Handorf EA et al. Breast and ovarian cancer penetrance estimates derived from germline multiple-gene sequencing results in women. JCO Precision Oncology 2017; (1): 1–12.
21. Foretova L, Navratilova M, Svoboda M et al. Recommendations for preventive care for women with rare genetic cause of breast and ovarian cancer. Klin Onkol 2019; 32 (Suppl 2): 2S6–2S13. doi: 10.14735/amko2019S6.
22. Niraj J, Farkkila A, D‘Andrea AD. The Fanconi anemia pathway in cancer. Annu Rev Cancer Biol 2019; 3 : 457–478. doi: 10.1038/nrm.2016.48.
23. Vaz F, Hanenberg H, Schuster B et al. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat Genet 2010; 42 (5): 406–409. doi: 10.1038/ng.570.
24. Pittman DL, Weinberg LR, Schimenti JC. Identification, characterization, and genetic mapping of RAD51D, a new mouse and human RAD51/RecA-related gene. Genomics 1998; 49 (1): 103–111. doi: 10.1006/geno.1998.5226.
25. Shibata T, Nishinaka T, Mikawa T et al. Homologous genetic recombination as an intrinsic dynamic property of a DNA structure induced by RecA/Rad51-family proteins: a possible advantage of DNA over RNA as genomic material. Proc Natl Acad Sci USA 2001; 98 (15): 8425–8432. doi: 10.1073/pnas.111005198.
26. Kleibl Z, Kristensen VN. Women at high risk of breast cancer: molecular characteristics, clinical presentation and management. Breast 2016; 28 : 136–144. doi: 10.1016/j.breast.2016.05.006.
27. Sun Y, McCorvie TJ, Yates LA et al. Structural basis of homologous recombination. Cell Mol Life Sci 2020; 77 (1): 3–18. doi: 10.1007/s00018-019-03365-1.
28. Chun J, Buechelmaier ES, Powell SN. Rad51 paralog complexes BCDX2 and CX3 act at different stages in the BRCA1-BRCA2-dependent homologous recombination pathway. Mol Cell Biol 2013; 33 (2): 387–395. doi: 10.1128/MCB.00465-12.
29. Sullivan MR, Bernstein KA. RAD-ical new insights into RAD51 regulation. Genes (Basel) 2018; 9 (12): 629. doi: 10.3390/genes9120629.
30. Badie S, Liao C, Thanasoula M et al. RAD51C facilitates checkpoint signaling by promoting CHK2 phosphorylation. J Cell Biol 2009; 185 (4): 587–600. doi: 10.1083/jcb.200811079.
31. Tarsounas M, Munoz P, Claas A et al. Telomere maintenance requires the RAD51D recombination/repair protein. Cell 2004; 117 (3): 337–347. doi: 10.1016/s0092-86 74 (04) 00337-x.
32. Lu HM, Li S, Black MH et al. Association of breast and ovarian cancers with predisposition genes identified by large-scale sequencing. JAMA Oncol 2019; 5 (1): 51–57. doi: 10.1001/jamaoncol.2018.2956.
33. Cunningham JM, Cicek MS, Larson NB et al. Clinical characteristics of ovarian cancer classified by BRCA1, BRCA2, and RAD51C status. Sci Rep 2014; 4 : 4026. doi: 10.1038/srep04026.
34. Suszynska M, Klonowska K, Jasinska AJ et al. Large-scale meta-analysis of mutations identified in panels of breast/ovarian cancer-related genes — providing evidence of cancer predisposition genes. Gynecol Oncol 2019; 153 (2): 452–462. doi: 10.1016/j.ygyno.2019.01.027.
35. Suszynska M, Ratajska M, Kozlowski P. BRIP1, RAD51C, and RAD51D mutations are associated with high susceptibility to ovarian cancer: mutation prevalence and precise risk estimates based on a pooled analysis of ~30,000 cases. J Ovarian Res 2020; 13 (1): 50. doi: 10.1186/s13048-020-00654-3.
36. Yang X, Song H, Leslie G et al. Ovarian and breast cancer risks associated with pathogenic variants in RAD51C and RAD51D. J Natl Cancer Inst 2020; 112 (12): 1242–1250. doi: 10.1093/jnci/djaa030.
37. Janatova M, Soukupova J, Stribrna J et al. Mutation analysis of the RAD51C and RAD51D genes in high-risk ovarian cancer patients and families from the Czech Republic. PloS One 2015; 10 (6): e0127711. doi: 10.1371/journal.pone.0127711.
38. Castera L, Krieger S, Rousselin A et al. Next-generation sequencing for the diagnosis of hereditary breast and ovarian cancer using genomic capture targeting multiple candidate genes. Eur J Hum Genet 2014; 22 (11): 1305–1313. doi: 10.1038/ejhg.2014.16.
39. Coulet F, Fajac A, Colas C et al. Germline RAD51C mutations in ovarian cancer susceptibility. Clin Genet 2013; 83 (4): 332–336. doi: 10.1111/j.1399-0004.2012.01917.x.
40. Osorio A, Endt D, Fernandez F et al. Predominance of pathogenic missense variants in the RAD51C gene occurring in breast and ovarian cancer families. Hum Mol Genet 2012; 21 (13): 2889–2898. doi: 10.1093/hmg/dds115.
41. Romero A, Perez-Segura P, Tosar A et al. A HRM-based screening method detects RAD51C germ-line deleterious mutations in Spanish breast and ovarian cancer families. Breast Cancer Res Treat 2011; 129 (3): 939–946. doi: 10.1007/s10549-011-1543-x.
42. Schnurbein G, Hauke J, Wappenschmidt B et al. RAD51C deletion screening identifies a recurrent gross deletion in breast cancer and ovarian cancer families. Breast Cancer Res 2013; 15 (6): R120. doi: 10.1186/bcr3589.
43. Thompson ER, Boyle SE, Johnson J et al. Analysis of RAD51C germline mutations in high-risk breast and ovarian cancer families and ovarian cancer patients. Hum Mutat 2012; 33 (1): 95–99. doi: 10.1002/humu.21625.
44. Li J, Meeks H, Feng B-J et al. Targeted massively parallel sequencing of a panel of putative breast cancer susceptibility genes in a large cohort of multiple-case breast and ovarian cancer families. J Med Genet 2016; 53 (1): 34–42. doi: 10.1136/jmedgenet-2015-103452.
45. Sopik V, Akbari MR, Narod SA. Genetic testing for RAD51C mutations: in the clinic and community. Clin Genet 2015; 88 (4): 303–312. doi: 10.1111/cge.12548.
46. Vuorela M, Pylkas K, Hartikainen JM et al. Further evidence for the contribution of the RAD51C gene in hereditary breast and ovarian cancer susceptibility. Breast Cancer Res Treat 2011; 130 (3): 1003–1010. doi: 10.1007/s10549-011-1677-x.
47. Golmard L, Castera L, Krieger S et al. Contribution of germline deleterious variants in the RAD51 paralogs to breast and ovarian cancers. Eur J Hum Genet 2017; 25 (12): 1345–1353. doi: 10.1007/s10549-011-1677-x.
48. Baker JL, Schwab RB, Wallace AM et al. Breast cancer in a RAD51D mutation carrier: case report and review of the literature. Clin Breast Cancer 2015; 15 (1): e71–e75. doi: 10.1016/j.clbc.2014.08.005.
49. Couch FJ, Shimelis H, Hu C et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol 2017; 3 (9): 1190–1196. doi: 10.1001/jamaoncol.2017.0424.
50. Kurian AW, Ward KC, Howlader N et al. Genetic testing and results in a population-based cohort of breast cancer patients and ovarian cancer patients. J Clin Oncol 2019; 37 (15): 1305–1315. doi: 10.1200/JCO.18.01854.
51. Paulo P, Maia S, Pinto C et al. Targeted next generation sequencing identifies functionally deleterious germline mutations in novel genes in early-onset/familial prostate cancer. PLoS Genet 2018; 14 (4): e1007355. doi: 10.1371/journal.pgen.1007355.
52. Pritchard CC, Mateo J, Walsh MF et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med 2016; 375 (5): 443–453. doi: 10.1056/NEJMoa1603144.
53. Pelttari LM, Kiiski J, Nurminen R et al. A Finnish founder mutation in RAD51D: analysis in breast, ovarian, prostate, and colorectal cancer. J Med Genet 2012; 49 (7): 429–432. doi: 10.1136/jmedgenet-2012-100852.
54. Osher DJ, De Leeneer K, Michils G et al. Mutation analysis of RAD51D in non-BRCA1/2 ovarian and breast cancer families. Br J Cancer 2012; 106 (8): 1460–1463. doi: 10.1038/bjc.2012.87.
55. Manchanda R, Menon U. Setting the threshold for surgical prevention in women at increased risk of ovarian cancer. Int J Gynecol Cancer 2018; 28 (1): 34–42. doi: 10.1097/IGC.0000000000001147.
56. Soukupova J, Lhotova K, Zemankova P et al. Contribution of massive parallel sequencing to diagnosis of hereditary ovarian cancer in the Czech Republic. Klin Onkol 2019; 32 (Suppl 2): 2S72–2S78. doi: 10.14735/amko2019S72.
57. Daly MB, Pilarski R, Berry M et al. NCCN Guidelines insights: genetic/familial high-risk assessment: breast and ovarian, Version 2.2017. J Natl Compr Canc Netw 2017; 15 (1): 9–20. doi: 10.6004/jnccn.2017.0003.
58. Song H, Dicks E, Ramus SJ et al. Contribution of germline mutations in the RAD51B, RAD51C, and RAD51D genes to ovarian cancer in the population. J Clin Oncol 2015; 33 (26): 2901–2907. doi: 10.1200/JCO.2015.61.2408.
59. Koudova M, Puchmajerova A. Risks of solid tumors in heterozygous carriers of recessive syndromes. Klin Onkol 2019; 32 (Suppl 2): 14–23. doi: 10.14735/amko2019S14.
60. Ngoi NY, Tay D, Heong V et al. Reversal of bowel obstruction with platinum-based chemotherapy and olaparib in recurrent, short platinum-free interval, RAD51C germline mutation–associated ovarian cancer. JCO Precision Oncology 2018; (2): 1–8.
61. George A, Kaye S, Banerjee S. Delivering widespread BRCA testing and PARP inhibition to patients with ovarian cancer. Nature Rev Clin Oncol 2017; 14 (5): 284–296. doi: 10.1038/nrclinonc.2016.191.
62. Ledermann J, Harter P, Gourley C et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. The Lancet Oncology 2014; 15 (8): 852–861. doi: 10.1016/S1470-2045 (14) 70228-1.
63. Chandran EA, Kennedy I. Significant tumor response to the poly (adp-ribose) polymerase inhibitor olaparib in heavily pretreated patient with ovarian carcinosarcoma harboring a germline RAD51D mutation. JCO Precision Oncology 2018; (2): 1–4.
64. Castera L, Harter V, Muller E et al. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet Med 2018; 20 (12): 1677–1686. doi: 10.1038/s41436-018-0005-9.
65. Li N, McInerny S, Zethoven M et al. Combined tumor sequencing and case-control analyses of RAD51C in breast cancer. J Natl Cancer Inst 2019; 111 (12): 1332–1338. doi: 10.1093/jnci/djz045.
66. Hauke J, Horvath J, Gross E et al. Gene panel testing of 5589 BRCA1/2-negative index patients with breast cancer in a routine diagnostic setting: results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancer Med 2018; 7 (4): 1349–1358. doi: 10.1002/cam4.1376.
67. Couch FJ, Shimelis H, Hu C et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol 2017; 3 (9): 1190–1196. doi: 10.1001/jamaoncol.2017.0424.
68. Slavin TP, Maxwell KN, Lilyquist J et al. The contribution of pathogenic variants in breast cancer susceptibility genes to familial breast cancer risk. NPJ Breast Cancer 2017; 3 : 22. doi: 10.1038/s41523-017-0024-8.
69. Lilyquist J, LaDuca H, Polley E et al. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol Oncol 2017; 147 (2): 375–380. doi: 10.1016/j.ygyno.2017.08.030.
70. Norquist BM, Harrell MI, Brady MF et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol 2016; 2 (4): 482–490. doi: 10.1001/jamaoncol.2015.5495.
Štítky
Dětská onkologie Chirurgie všeobecná OnkologieČlánek vyšel v časopise
Klinická onkologie

2021 Číslo 1
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Zkušenosti s axitinibem v léčbě metastatického renálního karcinomu
- Evaluace skóre m7-FLIPI u pacientů s folikulárním lymfomem odhalila slibný prediktivní biomarker k určení vhodné chemoterapie
- Neuropatie výrazně zpomaluje proces hojení u povrchových ran
- Zeolit-jodový komplex pomáhá v péči o infikované rány
Nejčtenější v tomto čísle
- Rehabilitácia pri rakovine prsníka
- Zárodečné mutace v genech RAD51C a RAD51D a dědičná predispozice ke vzniku karcinomu ovaria
- Droplet digitálna PCR ako nový diagnostický nástroj
- Skvamocelulárny karcinóm rekta u mladej ženy
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy