
Polypózy zažívacího traktu a Lynchův syndrom z pohledu patologa
Gastrointestinal Polyposes and Lynch Syndrome – a Pathologist’s Perspective
Gastrointestinal polyposes and Lynch syndrome are a group of heterogenous hereditary tumor syndromes associated with an increased risk of developing colorectal carcinoma and other malignancies. Typical early manifestations of gastrointestinal polyposes include multiple polyps in the gastrointestinal tract. Early recognition of these syndromes enables patients carrying a pathogenic mutation to undergo screening and to instigate precautions to minimize the risk of developing tumors. In some cases, gastrointestinal lesions could be an early indicator of tumor syndrome and histopathologic examination could lead to a recommendation for genetic testing of patients and their families.
Supported by Ministry of Health, Czech Republic – Conceptual Development of Research Organization (MMCI, 00209805).
The authors declare they have no potential conflicts of interest concerning drugs, products,or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted: 16. 4. 2019
Accepted: 6. 6. 2019
Keywords:
Lynch syndrome – gastrointestinal polyposes
Autoři:
Šárka Pokorová; Pavel Fabian
Působiště autorů:
Oddělení onkologické patologie, Masarykův onkologický ústav, Brno
Vyšlo v časopise:
Klin Onkol 2019; 32(Supplementum2): 92-96
Kategorie:
Přehled
doi:
https://doi.org/10.14735/amko2019S92
Souhrn
Gastrointestinální polypózy a Lynchův syndrom jsou heterogenní skupinou dědičných nádorových syndromů spojených se zvýšeným rizikem kolorektálního karcinomu i jiných zhoubných nádorů, přičemž jejich typickou časnou klinickou manifestací je – s výjimkou Lynchova syndromu – výskyt mnohočetných polypů v zažívacím traktu. Včasné rozpoznání umožňuje zařazení nosičů patogenních mutací do screeningových programů a přijetí preventivních opatření směřujících k minimalizaci rizik spojených s rozvojem nádorů. Histopatologické vyšetření gastrointestinálních lézí může v některých případech být časným indikátorem nádorového syndromu a nasměrovat pacienty a jejich rodiny k vyšetření lékařským genetikem.
Klíčová slova:
Lynchův syndrom – gastrointestinální polypózy
Přehled morfologie polypů GIT spojených s polypózami [1–4]
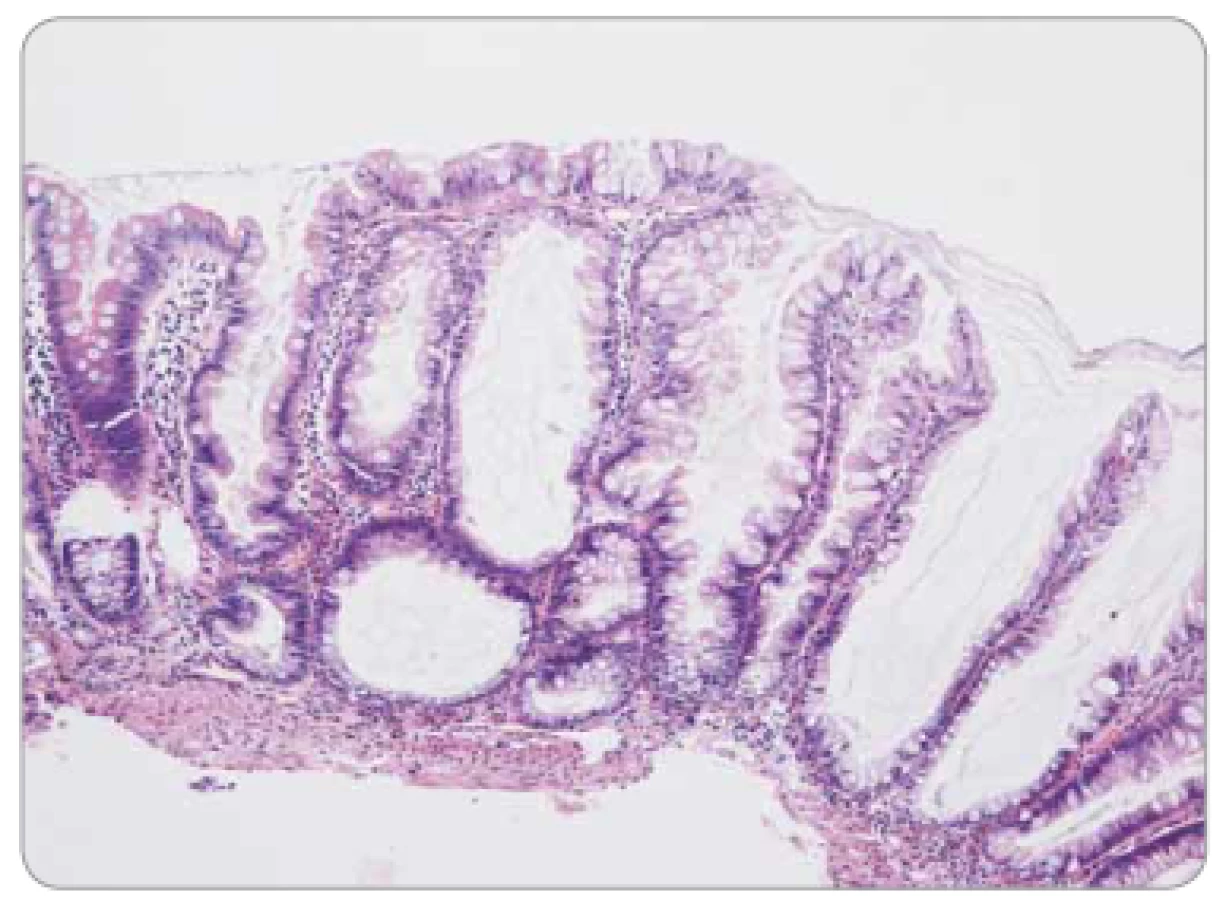
Konvenční adenomy jsou charakterizovány tubulárním (obr. 1) či vilózním (obr. 2) uspořádáním a epiteliální dysplazií převážně nízkého (low-grade – LG) či méně často vysokého (high-grade – HG) stupně. Morfologicky jsou adenomy u polypóz zcela totožné se sporadickými lézemi. Na možnost polypózy tak upozorní spíše jejich výskyt v neobvykle mladém věku či mnohočetnost. Je potřeba zmínit, že u Lynchova syndromu se mohou též vyskytovat polypózní léze – typicky jsou to v mladším věku a v proximálním colon vznikající vilózní adenomy, často s HG dysplazií. Analogicky s invazivními nádory u Lynchova syndromu (LS) jsou charakteristické ztrátou exprese některého z mismatch repair (MMR) proteinů.


Pilovité (serrated) léze jsou charakterizovány epiteliální proliferací tvořící četné jemné mikropapilární projekce, takže v podélném řezu připomínají zuby pily. Do spektra serrated lézí patří: a) hyperplastické polypy – morfologicky lze definovat tři subtypy, pravděpodobně bez klinického významu – jsou tvořeny paralelně uspořádanými kryptami s povrchově lokalizovaným pilovitým reliéfem non-dysplastického epitelu (obr. 3); b) sesilní serrated adenomy/polypy (SSA/P) s podobným nekomplexním uspořádáním, pilovitá kontura je ale lokalizována zejména v bázích krypt, které se navíc nepravidelně dilatují a deformují (obr. 4), odlišení hyperplastického polypu od SSA/P je v některých případech obtížné až nemožné; c) tradiční serrated adenomy (TSA), jež se svou komplexní větvenou strukturou více podobají tradičním adenomům, zachovávají si ale pilovitou konturu výstelky (obr. 5). Epiteliální dysplazie v hyperplastickém polypu a SSA/P obvykle chybí, naopak v TSA je spíše pravidlem. Tato dysplazie může mít specifický vzhled (tzv. dysplazie serrated typu), ale také může mít morfologii shodnou s konvenčními adenomy (dříve se serrated léze s konvenční dysplazií považovaly za kombinované či smíšené léze), a proto také může být v individuálním případě obtížné až nemožné rozlišit dysplastickou serrated lézi od konvenčního adenomu.


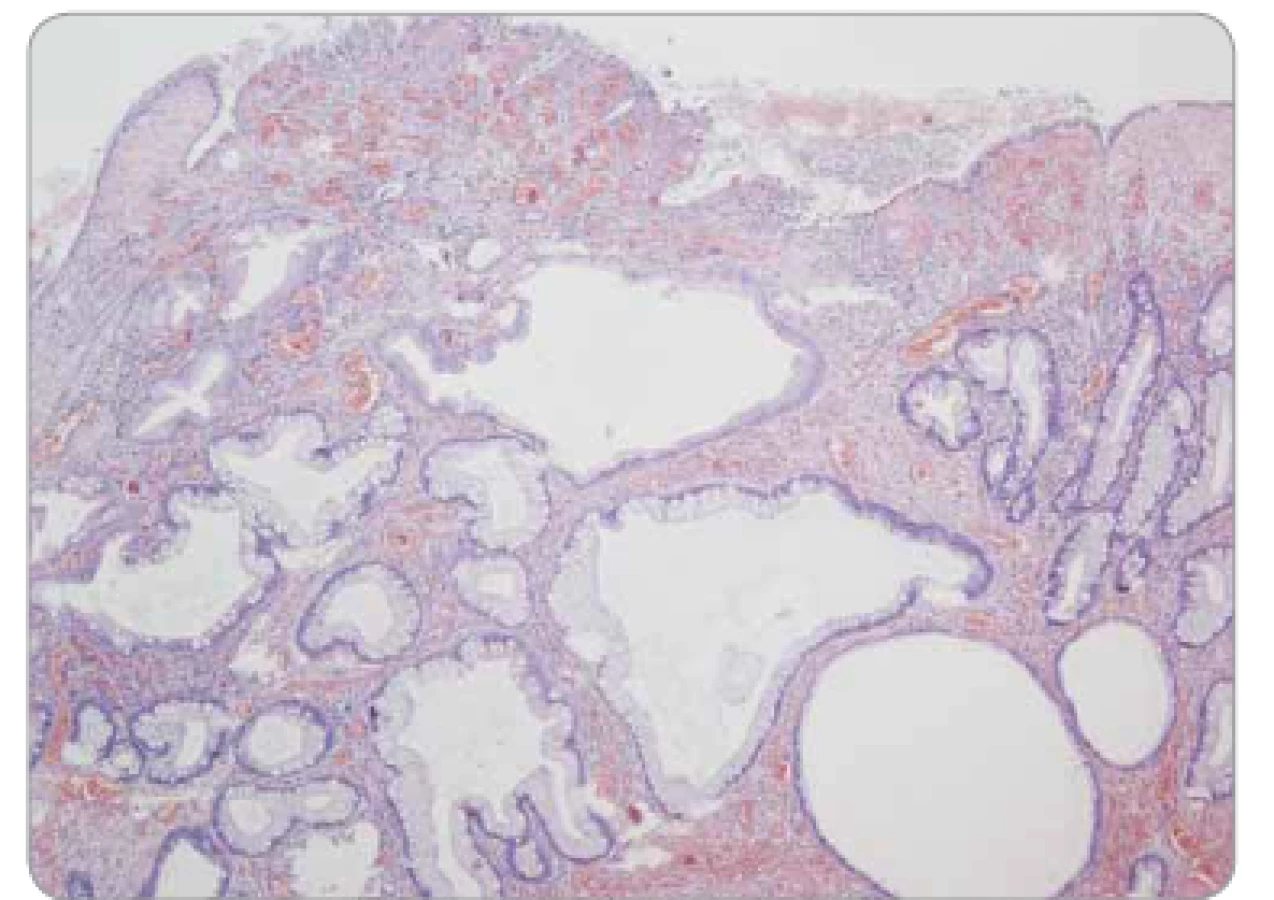
Polypy z fundických žlázek (fundic gland polyps – FGP) jsou charakteristické mikrocystickou dilatací gastrických žlázek a výskytem v oxyntické (korporální) žaludeční sliznici (obr. 6). Sporadické případy FGP (často spojené s dlouhodobým užíváním inhibitorů protonové pumpy) jsou morfologicky totožné se syndromickými. Epiteliální dysplazie ve sporadických případech je naprosto výjimečná, zatímco u syndromických se může vyskytnout; její přítomnost sama o sobě je tedy znakem signalizujícím suspekci z gastrointestinální (GIT) polypózy.

Peutzovy-Jeghersovy polypy (PJP) jsou hamartomy vyskytující se napříč celým GIT. Jsou tvořeny laločnatě uspořádanou místně příslušnou sliznicí, často se znaky hyperplazie, ale obvykle bez epiteliální dysplazie. Typickým znakem je arboreskující větvení výběžků muscularis mucosae, jež ale nemusí být nutně vždy vyjádřeno (obr. 7). Přítomnost epiteliální dysplazie zvyšuje pravděpodobnost Peutzova-Jeghersova syndromu (PJS), nikoliv ale absolutně, jako jsme zmiňovali v případě dysplazie ve FGP.

Juvenilní polypy jsou hamartomy vyskytující se napříč celým GIT, jsou tvořeny místně příslušnou sliznicí s dominující edematózní a zánětlivě infiltrovanou lamina propria, typické jsou mikrocystické žlázové struktury. Časté jsou povrchové eroze či ulcerace (obr. 8). Odlišení od zánětlivých pseudopolypů a v případě žaludeční sliznice i od hyperplastického gastrického polypu je obtížné, často až nemožné.
PTEN-hamartomové polypy jsou polypy vznikající v souvislosti s PTEN hamartomatózním syndromem, jehož typickým zástupcem je syndrom Cowdenové. Jejich stavba může být totožná s juvenilními polypy či navíc obsahují příměs tukové tkáně, organoidně uspořádanou lymfoidní tkáň či ganglioneuromatózní okrsky. V takovémto případě histologický obraz přímo svědčí pro vysokou suspekci z PTEN hamartomatózního syndromu.
Úskalí histopatologické diagnostiky
Výše popsané léze mají některé společné rysy a jak již bylo naznačeno, jejich přesná klasifikace není vždy jednoduchá a přímočará. Vyskytují se hraniční stavy a v kombinaci s objektivními technickými obtížemi (málo reprezentativní povrchové odběry, špatně topograficky orientované vzorky) lze některé léze zaměnit. Týká se to zejména odlišení hyperplastických polypů od SSA/P, odlišení TSA od konvenčního vilózního adenomu a odlišení juvenilního polypu od zánětlivých a reaktivních lézí (hyperplastický polyp žaludku, inflamatorní pseudopolyp) a též od inflamatorního fibroidního polypu (tzv. Vaňkův tumor). Dále je třeba upozornit, že v hodnocení stupně epiteliální dysplazie (LG vs. HG) je obecně dosahováno spíše vyšších úrovní „interobserver variability“ [5,6]. Jde o změny představující morfologické kontinuum, kde oba konce plynulého spektra jsou jasně definovány a relativně snadno diagnostikovány, avšak nastavení hranice mezi LG a HG dysplazií je do značné míry subjektivní.
Příspěvek patologa k časnému rozpoznání polypózních nádorových syndromů
Histologická klasifikace příslušného polypu sama o sobě neumožňuje diagnózu hereditárního nádorového syndromu, neboť všechny zmiňované léze se vyskytují též sporadicky a tyto non-syndromické nálezy v rutinní praxi gastroenterologa i patologa dominují, může však vyvolat podezření na některou z polypóz, a to především v případě hamartomových polypů. Zejména již zmíněné (a zcela raritní) hamartomové polypy s obsahem tukové, lymfoidní či ganglioneuromatózní tkáně se mimo PTEN hamartomatózní syndrom vyskytují jen vzácně [4,7]. Vodítkem k suspekci na polypózu tak může být mnohočetný výskyt polypů, nízký věk pacienta a samozřejmě rodinná anamnéza. V situaci, kdy je klinicky zjevně přítomna polypóza, patolog přispěje alespoň klasifikováním typu polypu, a tím může pomoci nasměrovat další diagnostiku.
Rozpoznání dysplazie ve FGP je znakem, který je suspektní z familiární adenomové polypózy (FAP) či jejích variant vč. GAPPS (gastric adenocarcioma and proximal polyposis syndrome) [1,2,8]. Méně signifikantně dysplazie v juvenilním polypu a PJP ukazuje na možnost juvenilní polypózy, resp. PJS [1,2,4,7,9,10].
Za pozornost stojí ta skutečnost, že – patrně v souvislosti s hodnocením úspěšnosti programu screeningu kolorektálního karcinomu (colorectal carcinoma – CRC) – došlo k zařazení kolorektálních adenomů mezi povinně hlášené novotvary. Ústav zdravotnických informací a statistiky (ÚZIS) a Národní onkologický registr (NOR) by tak do budoucna mohly sloužit jako platforma k zachycení pacientů s vícečetnými polypy, v současné podobě tedy alespoň v tračníku. Po překonání přechodného období bude hlášení probíhat automaticky, elektronickým propojením nemocničních a histologických databází s ÚZIS, takže nebude přinášet zvýšené úsilí ani náklady. Obě organizace se nebrání smysluplnému rozšíření hlášení novotvarů o další položky. Nastavení algoritmů k vyhledání osob v riziku polypózy by pak mohlo být již relativně snadným dílem, obtížnější jistě bude vybalancovat etické aspekty vytipování osob s podezřením na dědičný nádorový syndrom bez jejich vědomí, ochranu osobních dat a obdobné problémy.
Kromě samotných polypů je vhodné zmínit jiné změny či novotvary, které jsou silně asociované s polypózními nádorovými syndromy a LS. V tab. 1 s přehledem zde diskutovaných syndromů je nazýváme signálními lézemi. Pro FAP silně svědčí přítomnost Gardnerova fibromu, méně silně pak výskyt abdominální fibromatózy [1,2,8,11–13]. Mnohočetné kožní nádory se sebaceózní diferenciací vyskytující se u jedinců před 50. rokem života nebo postihující tělní partie mimo obličej, stejně tak mnohočetné keratoakantomy vznikající na místech chráněných před sluncem u mladých osob jsou silným indikátorem Muir-Torre syndromu (varianta LS) [14]. Ovariální sex-cord tumor s anulárními tubuly je velmi vzácný nádor, velmi silně asociovaný s PJS [1,4,8]. Dysplastický gangliocytom mozečku („Lhermitte Duclos disease“) je obdobně raritní nádorovitá léze, prakticky patognomická pro PTEN hamartomatózní syndrom [1,4,7–10]. Všechny jmenované léze by měly vést přinejmenším k pátrání po dalších projevech příslušných syndromů, ať už u daného jedince, či v jeho rodinné anamnéze, optimálně ke směřování pacienta k lékařskému genetikovi.
Tzv. aberantní kryptální fokus (jde o mikroskopický monokryptální či oligokryptální konvenční adenom) se může vyskytnout jako ojedinělá, většinou náhodně zachycená změna, vyskytnou-li se u jedné osoby ale dva a více takových fokusů, je vysoce suspektní, že jde o osobu s FAP [1,2,8,12]. Obdobnou a velmi zajímavou lézí jsou relativně nově popsané prekurzory novotvarů u LS – tzv. MMR-deficientní kryptální fokusy. Jde o léze, které jsou na úrovni endoskopické i mikroskopické morfologicky neodlišitelné od normální sliznice, při imunohistochemickém průkazu MMR proteinů však s absencí nejméně jednoho z nich. V zatím publikovaných studiích se u pacientů bez LS tyto fokusy ve zdravé sliznici v okolí karcinomů tračníku nenašly ani v jednom případě, zatímco u pacientů s LS a karcinomem se našly v hustotě přibližně 1/1 cm2 [15,16]. Tato nízká hustota, a tedy velmi malá pravděpodobnost záchytu v endoskopických biopsiích značně limituje praktickou aplikovatelnost tohoto fenoménu. Snad by mohl být alternativní metodou detekce LS u pacientů, kteří chtějí vědět, zda jsou postiženi LS, a současně odmítají vyšetření germinální DNA.
Screening Lynchova syndromu
Lynchův syndrom je ze zde diskutovaných syndromů daleko nejčastější. Odhaduje se, že incidence LS je 1/200 až 1/2 000. Považuje se za prokázané, že až 5 % kolorektálních karcinomů a cca 3 % endometriálních karcinomů vzniká právě na podkladě LS [2,17,18]. Přitom relativně vyšší medián věku v době vzniku malignit (ve srovnání např. s FAP) není dostatečně alarmujícím příznakem, který by vedl k suspekci na hereditu. Nádory vzniklé při LS se vyznačují defektem v mismatch repair systému, kdy mutace v příslušném genu vede téměř vždy k absenci jednoho či více MMR proteinů (tato absence je detekovatelná imunohistochemicky) a z toho vyplývající vysokou mírou mikrosatelitní nestability (microsatellite instability-high – MSI-H). Stejné charakteristiky mají i sporadické CRC s epigeneticky inaktivovaným MMR systémem, a to mechanizmem hypermetylace promotoru genu MLH1, současně bývá prakticky vždy přítomna v nádorové populaci mutace BRAF genu – obě tyto změny umožňují velmi spolehlivé odlišení „lynchovského“ a sporadického CRC [19,20]. Z pohledu onkologů je vyšetření MSI-H (defekt v MMR) u CRC důležitým ukazatelem prognózy CRC (MSI-H nádory mají lepší prognózu) a v některých případech i vhodnosti adjuvantní chemoterapie a také velmi silným prediktorem účinnosti protinádorové imunoterapie tzv. checkpoint inhibitory, a to napříč všemi histologickými typy nádorů. Vyšetření MMR proteinů se tedy nezávisle na hereditárních aspektech postupně stává (nebo brzy stane) standardem péče o pacienty s CRC. Ve shodě s dalšími [17–20] považujeme za racionální zavedení plošného screeningu LS imunohistochemickou detekcí defektu MMR (doplněnou v indikovaných případech o stanovení BRAF mutace a metylace promotoru MLH1) u všech nově diagnostikovaných CRC a endometriálních karcinomů, neboť tato metoda naplňuje požadavky kladené na screening – jde o metodu relativně levnou (v řádu jednotek tisíc Kč), s vysokou senzitivitou i specificitou, aplikovanou na jasně definovanou cílovou populaci. Odhalení rodin s LS má významné pozitivní dopady jak na samotné osoby – nosiče, tak na celý systém zdravotní a sociální péče.
Shrnutí
Znalost klinických a histomorfologických projevů polypózních nádorových syndromů napříč odbornostmi je základním předpokladem jejich správné a včasné diagnostiky. Některé chorobné procesy (v GIT i mimo něj) jsou silně asociované s hereditárními nádorovými syndromy a jejich výskyt by měl být signálem pro další cílená vyšetření. Využití dat o kolorektálních polypech z NOR by mohlo pomoci k vyhledání osob s rizikem polypóz – tato úvaha nepochybně má mnohé problematické eticko-právní aspekty. Imunohistochemické vyšetření MMR proteinů ve všech kolorektálních a endometriálních karcinomech se jeví jako vhodný screeningový nástroj pro detekci LS.
Podpořeno MZ ČR RVO (MOÚ, 00209805).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Pavel Fabian, Ph.D.
Oddělení onkologické patologie
Masarykův onkologický ústav
Žlutý kopec 7
656 53 Brno
e-mail: fabian@mou.cz
Obdrženo: 16. 4. 2019
Přijato: 6. 6. 2019
Zdroje
1. Bosman FT, Carneiro F, Hruban RH et al. WHO classification of tumours of the digestive system. 4. vyd. Lyon: IARC Press 2010.
2. Ma H, Brosens LA, Offerhaus GJ et al. Pathology and genetics of hereditary colorectal cancer. Pathology 2018; 50 (1): 49–59. doi: 10.1016/j.pathol.2017.09. 004.
3. Aust DE, Baretton GB et al. Serrated polyps of the colon and rectum (hyperplastic polyps, sessile serrated adenomas, traditional serrated adenomas, and mixed polyps) -proposal for diagnostic criteria. Virchows Arch 2010; 457 (3): 291–297. doi: 10.1007/s00428-010-0 45-1.
4. Shaco-Levy R, Jasperson KW, Martin K et al. Morphologic characterization of hamartomatous gastrointestinal polyps in Cowden syndrome, Peutz-Jeghers syndrome, and juvenile polyposis syndrome. Hum Pathol 2016; 49 : 39–48. doi: 10.1016/j.humpath.2015.10.002.
5. Osmond A, Li-Chang H, Kirsch R et al. Interobserver variability in assessing dysplasia and architecture in colorectal adenomas: a multicentre Canadian study. J Clin Pathol 2014; 67 (9): 781–786. doi: 10.1136/jclinpath-2014-202177.
6. Costantini M, Sciallero S, Giannini A et al. Interobserver agreement in the histologic diagnosis of colorectal polyps. The experience of the multicenter adenoma colorectal study (SMAC). J Clin Epidemiol 2003; 56 (3): 209–214.
7. Jelsig AM, Qvist N, Brusgaard K et al. Hamartomatous polyposis syndromes: a review. Orphanet J Rare Dis 2014; 9 : 101. doi: 10.1186/1750-1172-9-101.
8. Huber AR, Findeis-Hosey JJ, Whitney-Miller CL. Hereditary gastrointestinal polyposis syndromes: a review including newly identified syndromes. J Gastroint Dig Syst 2013; 3 : 155. doi: 10.4172/2161-069X.1000155.
9. Campos FG, Figueiredo MN, Martinez CA. Colorectal cancer risk in hamartomatous polyposis syndromes. World J Gastrointest Surg 2015; 7 (3): 25–32. doi: 10.4240/wjgs.v7.i3.25.
10. Chen HM, Fang JY. Genetics of the hamartomatous polyposis syndromes: a molecular review. Int J Colorectal Dis 2009; 24 (8): 865–874. doi: 10.1007/s00384-009-0714-2.
11. Lucci-Cordisco E, Risio M, Venesio T et al. The growing complexity of the intestinal polyposis syndromes. Am J Med Genet 2013; 161A (11): 2777–2787. doi: 10.1002/ajmg.a.36253.
12. Shussman N, Wexner SD. Colorectal polyps and polyposis syndromes. Gastroenterol Rep (Oxf) 2014; 2 (1): 1–15. doi: 10.1093/gastro/got041.
13. Brosens LA, van Hattem WA, Jansen M et al. Gastrointestinal polyposis syndromes. Curr Mol Med 2007; 7 (1): 29–46.
14. Kacerovská D, Kazakov DV, Cerná K et al. Muir-Torre syndrom – fenotypicka varianta Lynchova syndromu. Cesk Patol 2010; 46 (4): 86–94.
15. Kloor M, Huth C, Voigt AY et al. Prevalence of mismatch repair-deficient crypt foci in Lynch syndrome: a pathological study. Lancet Oncol 2012; 13 (6): 598–606. doi: 10.1016/S1470-2045 (12) 70109-2.
16. Staffa L, Echterdiek F, Nelius N et al. Mismatch repair-deficient crypt foci in Lynch Syndrome – molecular alterations and association with clinical parameters. PLoS ONE 2015; 10 (3): e0121980. doi: 10.1371/journal.pone.0121980.
17. Dušek M, Hadravský L, Stehlík J et al. Výsledky morfologické depistáže Lynchova syndromu v období 2013–2016. Cesk Patol 2018; 54 (2): 86–92.
18. Hampel H, Frankel W, Panescu J et al. Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res 2006; 66 (15): 7810–7817. doi: 10.1158/0008-5472.CAN-06-1114.
19. Dušek M, Hadravský L, Černá K et al. Diagnóza Lynchova syndromu od patologa. Klin Onkol 2016; 29 (3): 180–186. doi: 10.14735/amko2016180.
20. Daum O, Beneš Z, Hadravsky L et al. Lynchův syndrom v rukach patologa. Cesk Patol 2014; 50 (1): 18–24.
21. Chintalacheruvu LM, Shaw T, Buddam A et al. Major hereditary gastrointestinal cancer syndromes: a narrative review. J Gastrointestin Liver Dis 2017; 26 (2): 157–163. doi: 10.15403/jgld.2014.1121.262.maj.
22. Wells K, Wise PE. Hereditary colorectal cancer syndromes. Surg Clin North Am 2017; 97 (3): 605–625. doi: 10.1016/j.suc.2017.01.009.
Štítky
Dětská onkologie Chirurgie všeobecná OnkologieČlánek vyšel v časopise
Klinická onkologie

2019 Číslo Supplementum2
- Diosmin a hesperidin: Co ukazuje farmakokinetika?
- Zkušenosti s axitinibem v léčbě metastatického renálního karcinomu
- Evaluace skóre m7-FLIPI u pacientů s folikulárním lymfomem odhalila slibný prediktivní biomarker k určení vhodné chemoterapie
- Neuropatie výrazně zpomaluje proces hojení u povrchových ran
- Zeolit-jodový komplex pomáhá v péči o infikované rány
Nejčtenější v tomto čísle
- Dědičné mutace v genu CHEK2 jako příčina dispozice k nádorům prsu – typy mutací, jejich biologická a klinická relevance
- Rizika solidních nádorů u heterozygotních přenašečů recesivních syndromů
- Doporučení pro sledování žen se vzácnějšími genetickými příčinami nádorů prsu a ovarií
- GAPPS – syndrom adenokarcinomu žaludku a mnohočetné polypózy žaludku v 8 rodinách testovaných v Masarykově onkologickém ústavu – prevence vč. profylaktické gastrektomie
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy