
Dědičné mutace v genu CHEK2 jako příčina dispozice k nádorům prsu – typy mutací, jejich biologická a klinická relevance
Germline CHEK2 Gene Mutations in Hereditary Breast Cancer Predisposition – Mutation Types and their Biological and Clinical Relevance
Background: Hereditary mutations in the CHEK2 gene (which encodes CHK2 kinase) contribute to a moderately increased risk of breast cancer (BC) and other cancers. Large variations in the frequency of CHEK2 mutations and the occurrence of variants of unknown clinical significance (VUS) complicate estimation of cancer risk in carriers of germline CHEK2 mutations.
Patients and methods: We performed mutation analysis of 1,526 high-risk Czech BC patients and 3,360 Czech controls. Functional analysis was performed for identified VUS using a model system based on a human RPE1-CHEK2-KO cell line harboring biallelic inactivation of endogenous CHEK2.
Results: The frequency of ten truncating CHEK2 variants differed markedly between BC patients (2.26%) and controls (0.11%; p = 4.1 × 10−12). We also found 23 different missense variants in 4.5% patients and in 4.0% of controls. The most common was p.I157T, which was found in patients and controls with the same frequency. Functional analysis identified nine functionally deleterious VUS, another nine functionally neutral VUS, and four intermediate VUS (including p.I157T). We found that carriers of truncating CHEK2 mutations had a high BC risk (OR 8.19; 95% CI 4.11–17.75), and that carriers of functionally deleterious missense variants had a moderate risk (OR 4.06; 95% CI, 1.37–13.39). Carriers of these mutations developed BC at 44.4 and 50.7 years, respectively. Functionally neutral and functionally intermediate missense variants did not increase the BC risk. BC in CHEK2 mutation carriers was frequently ER-positive and of higher grade. Notably, carriers of CHEK2 mutations developed second cancers more frequently than BRCA1/BRCA2/PALB2/p53 or mutation non-carriers.
Conclusion: Hereditary CHEK2 mutations contribute to the development of hereditary BC. The associated cancer risk in mutation carriers increases with the number of affected individuals in a family. Annual follow-up with breast ultrasound, mammography, or magnetic resonance imaging is recommended for asymptomatic mutation carriers from the age of 40. Surgical prevention and specific follow-up of other tumors should be considered based on family cancer history.
The work was supported by grants from the Czech Health Research Council of the Ministry of Health of the Czech Republic NR 15-28830A, 16-29959A, NV19-03-00279, projects of the PROGRES Q28/LF1, GAUK 762216, SVV2019 / 260367, PRIMUS/17/MED/9, UNCE/MED/016, Progress Q26, LQ1604 NPU II and project AVČR Qualitas. The analysis of a set of unselected controls was made possible by the existence and support of the scientific infrastructure of the National Center for Medical Genomics (LM2015091) and its project aimed at creating a reference database of genetic variants of the Czech Republic (CZ.02.1.01/0.0/0.0/16_013/0001634).
The authors declare they have no potential conflicts of interest concerning drugs, products, or services used in the study.
The Editorial Board declares that the manuscript met the ICMJE recommendation for biomedical papers.
Submitted: 2. 4. 2019
Accepted: 14. 5. 2019
Keywords:
breast cancer – CHEK2 – hereditary mutations – variants of unknown significance – functional analysis
Autoři:
Petra Kleiblová 1,2; Lenka Stolařová 1; Křížová Křížová 3; Filip Lhota 1; Jan Hojný 1; Petra Zemánková 1; Ondřej Havránek 4,5; Michal Vočka 6; Marta Černá 1; Klára Lhotová 1; Marianna Borecká 1; Markéta Janatová 1; Jana Soukupová 1; Jan Ševčík 1; Martina Zimovjanová 6; Jaroslav Kotlas 2; Aleš Panczak 2,7; Kamila Veselá 2; Jana Červenková 8; Michaela Schneiderová 9; Monika Burócziová 3; Kamila Burdová 3; Viktor Stránecký 10; Lenka Foretová 1; Eva Macháčková 11; Spiros Tavandzis 12; Stanislav Kmoch 10; Libor Macůrek 3; Zdeněk Kleibl 1
Působiště autorů:
Laboratoř onkogenetiky, Ústav biochemie a experimentální onkologie, 1. LF UK v Praze
1; Ústav biologie a lékařské genetiky, 1. LF UK a VFN v Praze
2; Laboratoř biologie nádorové buňky, Ústav molekulární genetiky AV ČR v. v. i., Praha
3; BIOCEV, 1. LF UK v Praze 5 I. interní klinika 1. LF UK a VFN v Praze
4; Onkologická klinika 1. LF UK a VFN v Praze
6; Radiologická klinika 1. LF UK a VFN v Praze
7; Radioterapeutická a onkologická klinika FN Královské Vinohrady, Praha
8; I. chirurgická klinika 1. LF UK a VFN v Praze
9; Laboratoř pro studium vzácných nemocí, Klinika dětského a dorostového lékařství 1. LF UK a VFN v Praze
10; Oddělení nádorové epidemiologie, Masarykův onkologický ústav, Brno
11; Oddělení lékařské genetiky, Laboratoře AGEL, Praha
12
Vyšlo v časopise:
Klin Onkol 2019; 32(Supplementum2): 36-50
Kategorie:
Původní práce
doi:
https://doi.org/10.14735/amko2019S36
Souhrn
Východiska: Dědičné mutace v genu CHEK2 kódujícím CHK2 proteinkinázu způsobují středně zvýšené riziko vzniku karcinomu prsu (breast cancer – BC) a dalších nádorových onemocnění. Vysoká populační variabilita CHEK2 mutací a výskyt vzácných missense variant nejasného významu (variants of unknown clinical significance – VUS) komplikuje odhad rizika vzniku nádorových onemocnění u nosičů germinálních variant.
Soubor pacientů a metody: Mutační analýzu CHEK2, vč. analýzy velkých přestaveb, jsme provedli u 1 526 vysoce rizikových pacientek s BC a 3 360 kontrol z ČR. Nalezené VUS jsme klasifikovali pomocí funkční analýzy v modelovém systému lidské buněčné linie RPE1-CHEK2-KO, ve které byly obě endogenní alely inaktivovány metodou CRISPR/Cas9.
Výsledky: Četnost 10 různých trunkačních mutací CHEK2 byla významně vyšší u pacientek s BC (2,62 %) než u kontrol (0,11 %; p = 4,1 × 10−12), 23 různých missense variant jsme nalezli u 4,5 % pacientek a 4,0 % kontrol. Nejčastější alteraci představovala p.I157T se srovnatelnou četností u pacientek a kontrol (3,08 vs. 3,10 %). Funkční analýza identifikovala u 9 VUS zásadní poruchu kinázové aktivity, zatímco u dalších 9 zachovanou kinázovou aktivitu. Zbývající VUS a p.I157T byly částečně funkční. Riziko BC zvyšovaly trunkační mutace (OR 8,19; 95% CI 4,11–17,75) a nefunkční missense mutace (OR 4,06; 95% CI 1,37–13,39). Částečně funkční (vč. p.I157T) a plně funkční missense varianty riziko neovlivňovaly. Pacientky s trunkačními a funkčně-defektními missense variantami CHEK2 vyvinuly BC (převážně ER-pozitivní s vyšším gradingem) v průměrném věku 44,4 a 50,7 roku a signifikantně častěji vyvinuly sekundární tumory než nosičky mutací v BRCA1/BRCA2/PALB2/p53 a nenosičky.
Závěr: Dědičné mutace v genu CHEK2 představují významnou komponentu dědičného BC v ČR. Riziko vzniku onemocnění u nosičů patogenních mutací CHEK2 se zvyšuje s počtem příbuzných s BC a dalšími nádory v rodině. U asymptomatických nosičů je indikována dispenzarizace (jednou ročně ultrazvuk, mamografie nebo magnetická rezonance) od 40 let věku a chirurgická prevence v závislosti na rodinné anamnéze. Prevence vzniku dalších nádorů je ke zvážení dle výskytu nádorových onemocnění v rodině.
Klíčová slova:
karcinom prsu – CHEK2 – dědičné mutace – varianty nejasného významu – funkční analýza
Východiska
Karcinom prsu (breast cancer – BC) patří mezi nejčastější onkologické diagnózy v populaci žen v ČR. Přibližně 10 % případů se vyvíjí na základě genetické příčiny způsobené přítomností patogenní mutace v některém z genů predisponujících ke vzniku dědičné formy BC. Charakteristickou okolností vzniku většiny dědičných forem BC je jejich asociace s hereditárními mutacemi v genech kódujících proteiny podílející se na opravách genomové DNA, především na reparacích dvouřetězcových zlomů DNA cestou homologní rekombinace [1].
Dědičná forma BC se se zvýšenou četností vyskytuje v rámci syndromu dědičného karcinomu prsu a ovaria způsobeného vysoce penetrantními mutacemi v hlavních predispozičních genech BRCA1 a BRCA2. Vzhledem k vysoké frekvenci patogenních mutací v těchto genech a dostatečnému množství identifikovaných nosičů existují již v současné době kvalitní indikační kritéria pro genetické testování, jakož i doporučení pro klinický management osob s mutací [2,3]. Třetím klinicky nejvýznačnějším genem s vysokou penetrancí pro vznik dědičné formy karcinomu prsu je PALB2 [4]. Mezi další vysoce penetrantní predispoziční geny podmiňující vznik dědičné formy BC patří TP53, PTEN, CDH1 s extrémně vzácným výskytem zárodečných mutací. Řádově četnější jsou mutace v genech se střední penetrancí (s relativním rizikem < 4), mezi nimiž u pacientek s BC dominují alterace genů ATM a CHEK2 [5]. Četnost zárodečných mutací v genu CHEK2 je u pacientek s BC v naší populaci třetí nejvyšší ze známých nádorových predispozičních genů. Mutace v CHEK2 jsou spojeny i se zvýšeným rizikem výskytu dalších malignit [6].
Gen CHEK2 (checkpoint kinase 2; OMIM: 604373) je lokalizován na chromozomu 22q12.1 a kóduje serin/treoninovou proteinkinázu CHK2 tvořenou 543 aminokyselinami. CHK2 kináza je aktivována fosforylací tyrozinu 68 zprostředkovanou proteinem ATM v rámci buněčné odpovědi na přítomnost dvouřetězcových zlomů DNA [7]. Aktivační fosforylace katalyzuje homodimerizaci dvou CHK2 molekul v oblasti FHA (forkhead-associated) domény a umožňuje následnou autofosforylaci kinázové domény podmiňující plnou katalytickou aktivitu CHK2. Aktivovaná CHK2 kináza fosforyluje řadu jaderných proteinů zapojených do oprav poškození DNA a do buněčné odpovědi na přítomnost těchto alterací směřujících k zástavě buněčného cyklu, indukci apoptózy a změnám genové exprese [8]. Mezi substráty CHK2 patří BRCA1 a BRCA2 – klíčové proteiny reparace dvouřetězcových zlomů v DNA, transkripční faktor p53 – ovlivňující expresi genů regulujících průběh buněčného cyklu a apoptózy nebo transkripční represor KAP1 (KRAB-asociovaný protein 1, alias TIF1, TRIM28) – protein řídící genovou expresi rozsáhlé skupiny genů vč. genů kódujících proteiny zapojené do oprav genomové DNA [9].
Asociace zárodečných mutací v genu CHEK2 s karcinomem prsu byla identifikována začátkem milénia [10], ale většina zejména původních studií se zaměřila pouze na analýzu přítomnosti několika vybraných zakladatelských (founder) mutací. Mezi nejčastěji analyzované dědičné alterace patří západoevropská trunkační mutace c.1100delC (p.T367Mfs*15) a dále missense varianta c.470T>C (p.I157T), která se ve zvýšené míře vyskytuje ve slovanské populaci. Výsledky metaanalýz těchto variant u pacientek s BC opakovaně ukázaly, že v případě c.1100delC se jedná o středně penetrantní alelu [11,12], zatímco význam varianty p.I157T u BC je na úrovni nízce rizikových alterací s OR~1,5 [13]. Další typicky slovanské rekurentní mutace vyskytující se v naší populaci zahrnují sestřihovou variantu c.444+1G>A (IVS2+1G>A; p.E149Ifs*6) a velkou intragenovou deleci postihující exony 9 a 10 (c.909-2028_1095+330del5395; p.M304Lfs*15), poprvé identifikovanou u pacientů z ČR a SR [14].
Mutační analýzy celé kódující oblasti genu CHEK2 byly u starších prací provedeny pouze výjimečně [15–18]. Jejich výsledky však naznačily, že nejčastěji studované mutace představují pouze část dědičných variant CHEK2 u pacientek s BC. S nástupem sekvenování nové generace (next generation sequencing – NGS) do rutinní diagnostiky se mutační analýza genu CHEK2 stala standardní součástí vyšetření. Výsledky NGS analýz na velkých souborech odhalily, že prevalence zárodečných CHEK2 variant patří mezi nejvyšší mezi dalšími nádorovými predispozičními geny (mimo BRCA1 a BRCA2), zejména v evropské a židovské populaci [19–22]. V rámci NGS analýz však dochází nejen k identifikaci jednoznačně patogenních mutací, ale také variant nejasného významu (variant of unknown signifikance – VUS), jejichž biologický a potažmo i klinický význam musí být následně vyhodnocen testováním na modelových organizmech in vitro a/nebo důkladnou genetickou analýzou v rodinách nosičů VUS. Tyto přístupy dalece přesahují možnosti rutinní diagnostiky a informace o přítomnosti VUS značně komplikuje klinickou využitelnost NGS analýz [23]. Nejednotné hodnocení VUS způsobuje, že téměř třetina diagnostikovaných CHEK2 variant je reportována diskrepantně [24].
V předkládané práci jsme se zaměřili na identifikaci spektra dědičných variant genu CHEK2 v naší populaci, které jsme analyzovali v souboru vzorků získaných od 1 526 vysoce rizikových pacientek s BC a 3 360 kontrol. Nalezené VUS jsme podrobili funkční analýze využívající modelový systém založený na kvantifikaci fosforylace serinu 473 v proteinu KAP1 v lidské buněčné linii RPE1 s delecí endogenního CHEK2 genu.
Soubor pacientů a metody
Vyšetřovaný soubor pacientek tvořily vzorky od 1 526 žen s BC odeslaných ke genetickému vyšetření nádorové predispozice do Laboratoře onkogenetiky (Ústav biochemie a experimentální onkologie, 1. LF UK v Praze) v letech 1997–2017. Všechny pacientky podepsaly informovaný souhlas schválený etickou komisí 1. LF UK a VFN v Praze. Anamnestická a klinicko-patologická data byla získána ze zdravotnické dokumentace (tab. 1). Kromě souboru pacientek bylo vyšetřeno 3 360 kontrol, z nichž 1 329 vzorků bylo získáno od osob bez nádorového onemocnění v osobní anamnéze a 2 031 vzorků tvořila skupina neselektovaných jedinců (369 vzorků anonymních dárců krve a 1 662 anonymních vzorků osob vyšetřených exomovým sekvenováním v Národním centru lékařské genomiky – bez detailnějších údajů o věku, pohlaví a zdravotním stavu). Soubor pacientů i kontrol tvořily osoby české národnosti.

#563426
Vstupním materiálem pro vyšetření byla DNA izolovaná z leukocytů periferní nesrážlivé krve. S ohledem na dobu trvání projektu byla mutační analýza genu CHEK2 prováděna s využitím různých metodických přístupů, které však u všech vzorků zahrnovaly mutační analýzu celé kódující sekvence genu i analýzu přítomnosti velkých přestaveb (u všech pacientů a 2 271/3 360 kontrol; u zbývajících 1 089 vzorků kontrolního souboru byla provedena pouze analýza přítomnosti dvou velkých přestaveb CHEK2 nalezených v naší populaci). Vzorky od všech pacientek s identifikovanou germinální variantou CHEK2 genu byly vyšetřeny pomocí panelového NGS (CZECANCA) z důvodu identifikace přítomnosti případných dalších variant v dalších nádorových predispozičních genech [25,26].
U všech pacientek byla vyšetřena přítomnost mutací v genech BRCA1, BRCA2, PALB2 a TP53, jejichž zárodečné mutace jsou spojeny s vyšším rizikem vzniku BC než mutace v genu CHEK2. Z 1 526 analyzovaných pacientek s BC bylo 1 209 pacientek bez mutací v těchto nádorových predispozičních genech, 317 pacientek tvořily nosičky mutací v BRCA1/2, PALB2 nebo TP53.
Varianty identifikované u pacientů byly ověřeny na úrovni RNA, resp. cDNA. Pro in silico predikci významu missense variant byly využity nástroje Align GVGD, MutationTaster, CADD, SIFT, PolyPhen-2, Spidex a GERP.
Funkční testy byly provedeny na lidské buněčné linii RPE1 s delecí (knock-out, KO) endogenního CHEK2 genu dosažené pomocí CRISPR/Cas9 systému (RPE1-CHEK2-KO). Jako unikátní cíl specifické CHK2 fosforylace byl identifikován serin 473 (S473) proteinu KAP1. Míra fosforylace S473 KAP1 (stanovená pomocí specifické protilátky a kvantifikovaná Scan^R mikroskopií) vyjadřovala enzymovou aktivitu tranzientně exprimovaných testovaných variant CHK2 v RPE1-CHEK2-KO buňkách [27]. Míra fosforylace S473 KAP1 byla vyjádřena jako relativní poměr kinázové aktivity analyzované varianty k aktivitě nemutované (wild-type) formy CHK2 (100 %). Aktivita posunové mutace c.1100delC (p.T367Mfs*15) byla použita jako kontrola bez kinázové aktivity. Testované varianty byly na základě jejich kinázové aktivity klasifikovány jako nefunkční (s aktivitou < 25 % aktivity wild-type CHK2), částečně funkční (s aktivitou v rozmezí 25–50 % aktivity wild-type CHK2) a plně funkční (s aktivitou > 50 % aktivity wild-type CHK2).
Pro statistické analýzy byl soubor pacientek rozdělen do skupin v závislosti na mutačním stavu genu CHEK2, funkčním významu CHEK2 variant a s ohledem na přítomnost mutací ve vysoce penetrantních národových predispozičních genech. Statistické analýzy byly provedeny s využitím Fisherova exaktního testu. Míra asociace mutací v genu CHEK2 se sledovanými proměnnými byla vyjádřena pomocí míry rizika (odds ratio – OR). Za statisticky signifikantní byly považovány výsledky s p < 0,05.
Výsledky a diskuze
Četnost výskytu variant CHEK2 u pacientek s BC a kontrol
Ve všech vzorcích jsme nalezli celkem 33 různých nesynonymních variant genu CHEK2 (velké intragenové přestavby, nonsense, frame-shift, sestřihové nebo missense varianty), které se nacházely u 106/1 526 pacientek s BC (6,95 %) a u 142/3 360 kontrol (4,23 %; p = 0,0001; tab. 2). Významný rozdíl v četnosti výskytu jsme zaznamenali pro skupinu 10 mutací vedoucích k syntéze zkráceného CHK2 proteinu (trunkace; ověřeno na úrovni RNA). Tyto mutace jsme nalezli u 40/1 526 pacientek (2,62 %) a u 11/3 360 kontrol (0,33 %; p = 4,1 × 10−12). Pokud jsme ze souboru 1 526 pacientek vyloučili 317 nosiček mutací ve vysoce penetrantních národových predispozičních genech (BRCA1, BRCA2, PALB2 nebo TP53), dosáhla ve zbývajícím souboru četnost trunkačních mutací CHEK2 2,89 % (35/1 209 pacientek s BC).

# vč. dvou homozygotů p.I157T (1× unilat. a 1× bilat. BC před 50. rokem); tři další ženy s unilat. BC byly složení heterozygoti pro:
p.D265_H282del+p.D438Y, c.5601del+p.I157T, c.1100delC+p.I157T. Referenční sekvence CHEK2: NM_007194.3, transkripční varianta A.
BC – karcinom prsu, kontr. – populačně specifické kontroly, wt – wild-type, n – počet
# vč. dvou homozygotů p.I157T (1× unilat. a 1× bilat. BC před 50. rokem); tři další ženy s unilat. BC byly složení heterozygoti pro:
p.D265_H282del+p.D438Y, c.5601del+p.I157T, c.1100delC+p.I157T. Referenční sekvence CHEK2: NM_007194.3, transkripční varianta A.
BC – karcinom prsu, kontr. – populačně specifické kontroly, wt – wild-type, n – počet

# vč. dvou homozygotů p.I157T (1× unilat. a 1× bilat. BC před 50. rokem); tři další ženy s unilat. BC byly složení heterozygoti pro:
p.D265_H282del+p.D438Y, c.5601del+p.I157T, c.1100delC+p.I157T. Referenční sekvence CHEK2: NM_007194.3, transkripční varianta A.
BC – karcinom prsu, kontr. – populačně specifi cké kontroly, wt – wild-type, n – počet
Frekvence trunkačních mutací CHEK2 u pacientek s BC v různých regionech světa vykazuje značnou variabilitu. Frekvence mutací CHEK2 srovnatelná s frekvencí v našem souboru byla zaznamenána u 507 pacientek s BC bez mutací v BRCA1/BRCA2 z Francie (2,9 %) [15] a u 1 007 židovských pacientek z USA (2,88 %) [28]. Nižší záchyt mutací CHEK2 byl nalezen u pacientek z Německa (1,74 % v souboru 516 pacientek [17] a 1,84 % v souboru 5 589 pacientek [22]). Je třeba zdůraznit, že ani v jedné z těchto prací nebyly hodnoceny velké intragenové přestavby CHEK2. Frekvence mutací v CHEK2 je výrazně nižší v populacích neevropského původu. V souboru 7 657 čínských pacientek s BC byla zachycena frekvence nosiček mutací v CHEK2 pouze 0,34 % [21].
Přes výrazně nižší frekvenci patogenních mutací v našem kontrolním souboru oproti souboru pacientek s BC byla zachována podobná proporce v zastoupení jednotlivých typů trunkací tvořených přibližně stejnoměrně velkými intragenovými delecemi (37 % u pacientek, 36 % u kontrol), mutacemi postihujícími vysoce konzervativní sestřihová místa (32 % u pacientek, 27 % u kontrol) a krátkými delecemi/non-sense mutacemi (32 % u pacientek; 36 % u kontrol). Velké intragenové delece zahrnovaly u pacientek rekurentní deleci 5395 bp postihující exony 9 a 10 a nově popsanou deleci 5601 bp s oblastí kódující exon 8 (tab. 2). S ohledem na jejich frekvenci musí být hodnocení velkých přestaveb CHEK2 v naší populaci nezbytnou součástí analýzy CHEK2 genu. Varianty způsobující aberantní sestřih mRNA zahrnovaly kromě známé mutace c.444+1G>A variantu c.1260-8A>G (způsobující inzerci sedmi nukleotidů z přilehlé intronové oblasti do mRNA s posunem čtecího rámce; p.L421Ifs*4) a opakující se variantu c.846+4_846+7delAGTA (způsobující na úrovni mRNA potvrzený in-frame výpadek exonu 7 kódujícího 18 aminokyselin v oblasti kinázové domény proteinu CHK2; p.D265_H282del). Identický dopad na mRNA (delece exonu 7) má i recentně popsaná rozsáhlá (~7,5 kb) rekurentní intragenová delece úseku CHEK2 genu s exonem 7 u řeckých pacientek s BC [29]. Nejčastější krátké delece v našem souboru tvořily mutace c.1100delC a c.277delT. U jedné pacientky s unilaterálním, HER2 pozitivním BC (dg. v 41 letech) byly přítomny dvě patogenní mutace (c.277delT a c.444+1G>A) v heterozygotním stavu.
Rozdíly ve frekvenci germinálních mutací CHEK2 v různých populacích jsou patrné rovněž z přítomnosti různých founder mutací. Výskyt nejvíce studované CHEK2 varianty c.1100delC dominuje v Německu (s frekvencí 1,41 % všech pacientek s BC bez mutace v BRCA1/BRCA2 a 0,37 % v populačních kontrolách) [22], Francii [15], Nizozemí, Finsku, Velké Británii [30], Rusku [31] a u pacientek evropského původu v USA [20]. V našem souboru pacientek s BC jsme identifikovali pouze šest nosiček mutace c.1100delC (0,39 % všech pacientek), které tak představovaly pouze 15 % pacientek s trunkační mutací CHEK2. Přesto byl výskyt varianty c.1100delC v kontrolním souboru signifikantně a více než 4× nižší (0,09 %; p = 0,02). Velmi raritní je c.1100delC v jižní Evropě [32] a přítomna není v Koreji [33] nebo Číně [21], kde byla nově popsána u 30 % nosiček mutací v CHEK2 jinde neznámá founder mutace p.Y139*. V Polsku je nejčastější mutací CHEK2 c.444+1G>A [34], která se rekurentně vyskytuje i u nás (společně s variantou c.444+1G>T [35,36]).
Z 23 zaznamenaných missense variant byla v naší populaci nejčastější varianta c.470T>C (p.I157T), přítomná u 47/1 526 (3,08 %) pacientek (u dvou z nich byla v homozygotním stavu) a u 104/3 360 (3,10 %) kontrol. Z porovnání s ostatními studiemi analyzujícími varianty CHEK2 genu je patrné, že i spektrum missense variant má výraznou populační variabilitu. Z 18 různých missense variant CHEK2, které jsme nalezli v naší studii u pacientek s BC, jich 11 bylo identifikováno mezi 56 missense variantami zachycenými při analýze 5 589 pacientek s BC z Německa [22], 6 mezi 28 missense variantami nalezenými při vyšetření 1 303 pacientek s BC z USA a Austrálie [16] a 2 mezi 9 missense variantami přítomnými u 507 pacientek s BC z Francie [15].
Výsledky in silico analýzy missense variant nalezených v našem souboru byly pro většinu variant diskrepantní. Jejich klinický význam a interpretace v databázi ClinVar byly rovněž nejasné (16× VUS, class 3) nebo konfliktní (u šesti variant); varianta c.1309A>G nebyla v databázi popsána (tab. 2). Z uvedených důvodů jsme přistoupili k provedení funkčních analýz.
Funkční klasifikace variant CHEK2
Funkční analýzy jsme provedli u všech 23 missense variant nalezených v souboru pacientek s BC a v kontrolách a pro sestřihovou variantu c.846+4_846+7delAGTA. Pro funkční analýzu jsme vyvinuli systém umožňující testování kinázové aktivity studovaných missense variant v modelu lidské buněčné linie RPE1 s inaktivací endogenního CHEK2 genu (RPE1-CHEK2-KO). V porovnání s jinými přístupy pro funkční analýzu variant v CHEK2 na úrovni purifikovaných proteinů in vitro [15,18] nebo v modelech na kvasinkách [37,38] je námi vytvořený systém vhodný pro studium kinázové aktivity testovaných CHEK2 variant v přirozeném intracelulárním prostředí lidských buněk, s přítomností aktivátorů (ATM) a substrátů (KAP1) kinázy CHK2. Stanovení fosforylace S473 proteinu KAP1 zprostředkované tranzientně exprimovanými, analyzovanými variantami v buňkách linie RPE1-CHEK2-KO umožnilo kvantifikaci funkční kapacity nalezených VUS s určením relativní kinázové aktivity k aktivitě wild-type CHK2 (tab. 2). Systém do budoucna umožní analyzovat i další VUS identifikované v CHEK2, kterých je v databázi ClinVar doposud (březen 2019) popsáno 882 [39].
Funkční analýzou v RPE1-CHEK2-KO buňkách jsme prokázali úplnou ztrátu katalytické aktivity u varianty c.846+4_846+7delAGTA způsobující in-frame deleci 18 aminokyselin v kinázové doméně. Podstatnou redukci (snížení na < 25 % aktivity wild-type proteinu CHK2) až ztrátu kinázové aktivity jsme dále zaznamenali u 9/23 testovaných missense variant, které byly klasifikovány jako nefunkční. Naopak pouze mírně sníženou (> 50 % aktivity wild-type proteinu CHK2) až plně zachovanou funkci mělo dalších 9/23 missense variant, které byly klasifikovány jako plně funkční. Mezi pěti variantami, jejichž aktivita se pohybovala v rozmezí 25–50 % wild-type formy proteinu CHK2 a které byly klasifikovány jako částečně funkční, byla také nejčastější missense varianta p.I157T. Tento výsledek je v souladu s předchozími studiemi ukazujícími, že p.I157T si sice zachovává plnou katalytickou účinnost, ale variantní izoforma vykazuje poruchu ve vazbě substrátů [40,41]. Funkční kinázová kapacita této varianty na úrovni 48,8 % wild-type CHK2 naznačuje, že u heterozygotních nosičů by reziduální celková kapacita CHK2 kinázy exprimované z obou alel (kódujících 100% aktivní wild-type CHK2 a přibližně 50% aktivní p.I157T) měla poskytovat dostatečnou funkční rezervu, avšak u homozygotních nosičů p.I157T varianty je celková funkční kapacita CHK2 snížena na úroveň aktivity u heterozygotních nosičů trunkačních mutací, jako je c.1100delC.
Asociace CHEK2 mutací s rizikem vzniku karcinomu prsu u žen
Riziko vzniku BC u nosiček dědičných alterací v genu CHEK2 klasifikovaných na základě typu alterace a výsledků funkčních analýz bylo hodnoceno ve srovnání s kontrolní populací jak v souboru všech 1 526 žen s BC, tak v podskupině 1 209 žen s BC bez přítomnosti příčinné mutace v ostatních vyšetřených nádorových predispozičních genech (BRCA1, BRCA2, PALB2 nebo TP53). Soubor bez mutací v těchto predispozičních genech sloužil k porovnání s výsledky zahraničních prací, které obvykle analyzují populace pacientek s BC bez mutací v hlavních predispozičních genech.
Nejvyšší riziko vzniku BC jsme zaznamenali pro varianty vedoucí ke zkrácení proteinového produktu (trunkace), a to jak při hodnocení v celém souboru 1 526 vyšetřovaných žen (OR 8,19; 95% CI 4,11–17,75; p = 4,1 × 10−12), tak při hodnocení v podskupině 1 209 pacientek bez mutací v ostatních vyšetřených nádorových predispozičních genech (OR 9,07; 95% CI 4,49–19,87; p = 2,4 × 10−12) (tab. 3). Missense varianty klasifikované pomocí funkčního vyšetření jako nefunkční byly rovněž statisticky významně spojeny se zvýšeným rizikem vzniku karcinomu prsu u žen (OR > 4), bez ohledu na přítomnost dalších mutací v ostatních predispozičních genech (tab. 3). Naproti tomu missense varianty klasifikované ve funkční analýze jako skupina variant s částečnou poruchou funkce a skupina plně funkčních variant nezvyšovaly riziko vzniku BC ve skupině všech pacientek ani v podskupině pacientek bez mutací v genech BRCA1, BRCA2, PALB2 nebo TP53 (tab. 3).

BC – karcinom prsu, OR – odds ratio, CI – interval spolehlivosti, n – počet
Rizika vzniku karcinomu prsu spojená s nosičstvím alterací v CHEK2, která jsme vyčíslili v našem souboru u všech pacientek s BC a v podsouboru pacientek s BC bez mutací v hlavních predispozičních genech, jsou vyšší, než je tomu v ostatních studiích analyzujících celý gen CHEK2 nebo vybrané founder mutace (tab. 4). Příčin tohoto rozdílu je několik.
- Výskyt variant CHEK2 genu vykazuje významné populační rozdíly. Z přehledu vyplývá, že práce, ve kterých byly k výpočtu rizik použity populační kontroly, dospěly k vyššímu odhadu OR než práce, ve kterých byly použity frekvence alterací CHEK2 z databáze variant Exome Aggregation Consortia (ExAC) od osob z evropské populace mimo Finsko (European non-Finnish – NFE) [42]. Důvodem je pravděpodobně vysoké zastoupení vzorků od osob ze severní Evropy, kde je celosvětově nejvyšší zastoupení c.1100delC v populaci (v kohortě 33 370 vzorků ExAC-NFE je více než třetina vzorků (12 119) tvořena souborem Swedish Schizophrenia & Bipolar Studies, přičemž frekvence c.1100delC v kontrolách ve Švédsku dosahuje 0,7 % [43]).
- Převážná většina i recentních analýz CHEK2 genu nezahrnuje do vyšetření velké přestavby, které v našem souboru tvořily třetinu trunkačních variant a které jsme nalezli významně častěji u pacientů než u kontrol.
- Odhad rizika ovlivňuje i složení (výběr) vyšetřovaných pacientů a kontrol – zatímco vyšší četnost mladých pacientek s BC, pacientek s pozitivní rodinnou anamnézou a osob bez mutací v BRCA1/BRCA2 zvyšuje záchyt patogenních variant CHEK2 genu ve skupině pacientů, naopak výběr starších osob bez nádorového onemocnění snižuje výskyt patogenních variant v kontrolním souboru. Vyšší četnost patogenních mutací CHEK2 nalezená v námi vyšetřovaném souboru může být ovlivněna vyšším zastoupením pacientek z rodin s pozitivní onkologickou anamnézou. Z publikovaných prací systematicky vyplývá vyšší riziko vzniku BC pro familiární formu onemocnění než pro neselektované nebo sporadické případy (tab. 4). Naopak nižší výskyt nalezených variant CHEK2 v kontrolním souboru může být ovlivněn vyšším podílem nenádorových a starších osob. Oba protichůdné faktory se pravděpodobně částečně spolupodílejí na vyšším OR zjištěném v naší práci, než je tomu v jiných studiích.

pop. – populace, OR – odds ratio, CI – interval spolehlivosti, FI – Finsko, FR – Francie, DE – Německo, USA – Spojené státy americké,
UK – Spojené království Velké Británie a Severního Irska, PL – Polsko, AU – Austrálie, CN – Čína, DK – Dánsko

pop. – populace, OR – odds ratio, CI – interval spolehlivosti, meta – metaanalýza, PL – Polsko, DE – Německo, UK – Spojené království
Velké Británie a Severního Irska, NL – Nizozemí, FI – Finsko, AU – Austrálie
Pro přesnější určení rizika bude nezbytné provedení rozsáhlých analýz v rámci mezinárodních konsorcií zahrnujících neselektované soubory pacientek s BC a jejich porovnání se vzorky populačně-specifických kontrol. Nicméně z výsledků našich analýz a přehledu publikovaných výsledků CHEK2 analýz u pacientek s BC je nepochybné, že nosičství patogenních trunkačních mutací genu CHEK2 je obecně spojeno s nejméně trojnásobným zvýšením rizika vzniku karcinomu prsu. Je pravděpodobné, že vyšší riziko vzniku BC bude spojeno s nosičstvím mutací CHEK2 u pacientek s pozitivní rodinnou anamnézou BC [44] (ale pravděpodobně i dalších nádorových onemocnění), naopak nižší riziko může představovat nosičství mutací CHEK2 u osob bez nádorových onemocnění u příbuzných. Celoživotní riziko vzniku BC u nosiček bez pozitivní rodinné anamnézy se pohybuje kolem 20 %, zatímco u nosiček s pozitivní rodinnou anamnézou bylo vyčísleno na 40 % [45]. Odlišná míra rizika je modifikována přítomností dalších genetických faktorů, např. nízkopenetrantními 313 SNP polygenně ovlivňujícími riziko vzniku BC, vyjadřujících tzv. polygenic risk score [46]. Jejich zařazení do diagnostiky může v budoucnu umožnit i lepší predikci rizika u nosičů mutací CHEK2 [44,47].
Nedořešenou otázkou zůstává určení rizika spojeného s nosičstvím funkčně-defektních vzácných missense mutací, které v naší studii i v několika publikovaných analýzách (tab. 4) (které však definovaly „potenciálně“ patogenní missense varianty především na úrovni in silico predikcí) vykazovaly nižší míru rizika vzniku BC (v pásmu variant středního významu s OR~2) než trunkační mutace (s OR > 3). Předpokladem dalších analýz vzácných missense variant je robustní funkční in vitro analýza, kterou plánujeme provést pro všechny missense varianty a in-frame delece/inzerce identifikované u pacientů a kontrol v ČR.
Asociace CHEK2 mutací s histologickým typem BC a s dalšími nádorovými onemocněními
Hodnocení vztahu přítomnosti germinální CHEK2 mutace (trunkací a nefunkčních missense variant) k histopatologickým charakteristikám BC bylo provedeno ve skupině 1 209 pacientek bez přítomnosti příčinné mutace v genech BRCA1, BRCA2, PALB2 nebo TP53, ve které bylo identifikováno 44 nosiček patogenních trunkačních CHEK2 variant nebo funkčně-defektních missense variant. U těchto nosiček jsme pozorovali vyšší četnost luminal A subtypu BC oproti pacientkám bez CHEK2 mutace (51,4 vs. 29,4 %; p = 6 × 10−3). Výrazný rozdíl jsme zachytili ve výskytu triple-negativního (ER-, PR-, HER2-negativní) BC, který byl zachycen pouze u jedné nosičky CHEK2 mutace (2,7 %) oproti 186/868 pacientkám (21,4 %) bez mutace v CHEK2 a dalších predispozičních genech (p = 3,0 × 10−3). Histologický typ BC, menopauzální status ani indikační kritéria ke genetickému testování se ve vyšetřovaném souboru 1 209 pacientek bez mutací v genech BRCA1, BRCA2, PALB2 nebo TP53 nelišily mezi pacientkami s/bez CHEK2 mutací, avšak u nosiček CHEK2 mutací byl častěji přítomen grade 2 nádoru proti ženám bez mutace (67,5 vs. 44,4 %; p = 5,3 × 10−3).
Výsledky našich analýz jsou v souladu s dříve publikovanými studiemi ukazujícími, že nosičky CHEK2 mutací vykazují silnou asociaci s ER-pozitivními BC, časnějším nástupem onemocnění a pokročilejším gradingem, nosičky p.I157T (ale ne c.1100delC) také s lobulárním BC [48]. Přestože prognóza ER-pozitivních nádorů je u neselektovaného BC lepší, nosičství mutací v CHEK2 je spojeno s horším celkovým přežitím u pacientek s BC [49,50].
Zajímavým pozorováním v našem souboru byla významně vyšší četnost výskytu sekundárních nádorů dalších typů (jiných než BC nebo ovaria), vč. karcinomu kolorekta, ledviny, štítné žlázy nebo hematologických malignit u nosiček CHEK2 mutací, které jsme zaznamenali v 9/44 (20,5 %) případů nosiček trunkačních a funkčně-defektních missense variant CHEK2, v porovnání se 17/317 (5,4 %) případů u nosiček mutací v ostatních predispozičních genech (p = 0,002), 2/38 (5,3 %) případů u nosiček p.I157T a 80/1127 (7,1 %) případů u pacientek s BC bez mutací v BRCA1, BRCA2, PALB2, TP53 nebo CHEK2 (p = 0,004). Zvýšená četnost výskytu nádorových onemocnění v dalších lokalitách dokumentuje podíl mutací v CHEK2 genu na vzniku dědičné predispozice i k jiným typům nádorových onemocnění. Od první práce Cybulského et al [6], ve které byly trunkační varianty (c.1100delC a c.444+1G>A) asociovány se zvýšeným rizikem vzniku kar-cinomu štítné žlázy (OR 4,9; p = 0,0006), prsu (OR 2,2; p = 0,02) a prostaty (OR 2,2; p = 0,04) a varianta p.I157T se zvýšeným rizikem BC (OR 1,4; p = 0,02), kolorekta (OR 2,0; p = 0,001), ledviny (OR 2,1; p = 0,006), prostaty (OR 1,7; p = 0,002) a štítné žlázy (OR 1,9; p = 0,04) v polské populaci, byla publikována řada prací a metaanalýz ukazujících na zvýšený výskyt těchto onemocnění u nosičů alterací genu CHEK2.
Přesnější odhady relativních rizik pro vznik dalších malignit u nosičů mutací CHEK2 zatím chybějí, zejména z důvodu nedostatečného množství vyšetřených pacientů. Nicméně i další práce (tab. 5) poukazují na asociaci mutací CHEK2 s jinými tumory, jako jsou karcinom ledviny, prostaty, štítné žlázy a kolorekta, non-hodgkinské lymfomy, maligní melanom nebo karcinom prsu u mužů.
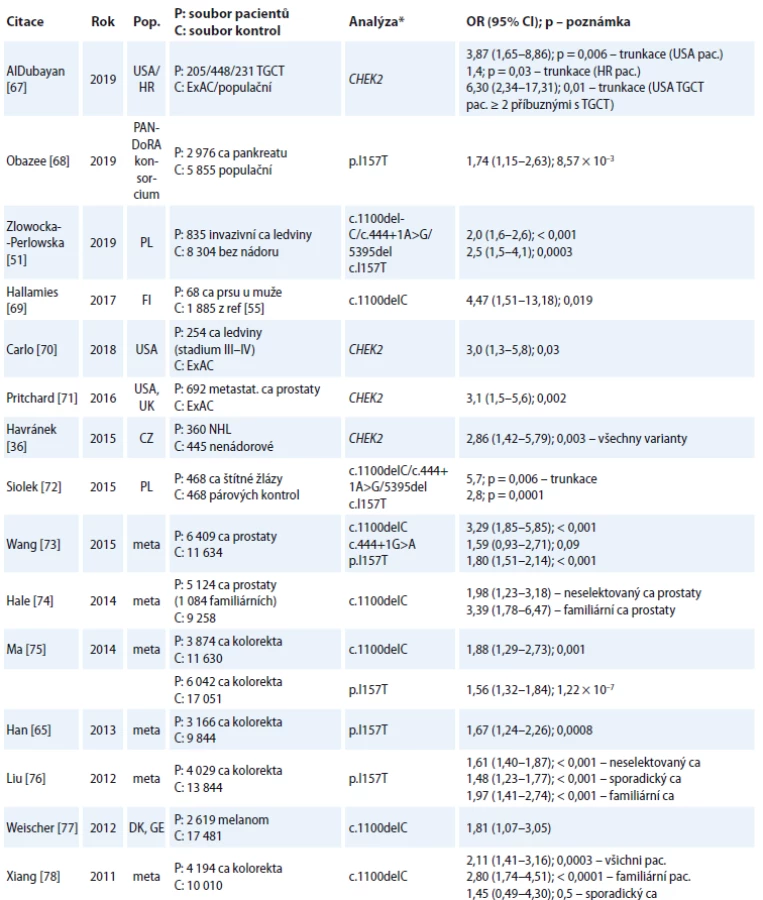
Integrace těchto poznatků do klinických doporučení je předmětem diskuzí [51] a pro preventivní sledování nosičů CHEK2 mutací zatím není plošně možná a ošetřující lékaři jsou odkázáni na indikaci sledování dalších nádorů na základě výskytu onkologických onemocnění v rodině nosičů mutací či na využití dostupných screeningových vyšetření.
Současný stav klinických doporučení
Současný stav klinických doporučení pro nosiče patogenních mutací v genu CHEK2 vychází z aktuálních mezinárodních doporučení a zahrnuje zejména přístupy umožňující časnou detekci malignit (pravidelné samovyšetření prsů, mamografie a ultrasonografie nebo magnetická rezonance jednou ročně od věku 40 let, příp. od věku 10 let, před nástupem onemocnění v rodině) [52,53].
Pro nosičky patogenních a pravděpodobně patogenních variant (class 4 a 5) v heterozygotním stavu způsobujících zkrácení proteinu CHK2 (vč. nově charakterizovaných sestřihových mutací c.846+4_846+7delAGTA a c.1260-8A>G) s absencí části či celé kinázové domény je doporučeno zařazení do preventivních gynekologických a onkologických programů zohledňujících rizika nádorů asociovaných s mutacemi v genu CHEK2. Prevence v tomto případě odpovídá schématu pro nejčastěji diskutovanou variantu CHEK2 c.1100delC.
U nosiček homozygotních mutací [54] nebo složených heterozygotů (dvě prokazatelně patogenní trunkace v genu CHEK2, každá v heterozygotním stavu) je možné nabídnout také preventivní chirurgické zákroky snižující riziko vzniku karcinomu prsu (bilaterální mastektomie, podle závažnosti rodinné anamnézy případně i profylaktická salpingo-ooforektomie). Preventivní chirurgické výkony je vhodné zvážit i u heterozygotních nosiček trunkačních variant, s ohledem na rodinnou anamnézu a segregaci varianty CHEK2 v rodině.
Nosičky missense variant vedoucích ke ztrátě funkce proteinu CHK2 dle dostupných funkčních vyšetření by měly mít stejné preventivní sledovací schéma jako v případě variant trunkačních. Pro indikaci k preventivním chirurgickým výkonům není zatím dostatek informací. Je vhodné provádět v rodinách segregační analýzy, je možné prediktivně (ale s omezeným výstupem) testovat zdravé příbuzné. Nosiče zařadit do adekvátních preventivních programů, ovšem v případě negativně testovaných osob zatím i nadále ponechat riziko vzniku nádorových onemocnění plynoucí z rodinné anamnézy.
Interpretace nálezu missense variant v genu CHEK2 je obtížnější. Rekurentní missense varianta p.I157T je ve veřejných databázích klasifikována rozporuplně ve spektru class 3–5. V naší populaci však alelická frekvence této varianty převyšuje 1 %, což ze své podstaty vylučuje možnost, že by se mohlo jednat o vysoce nebo i středně penetrantní nádorovou predispoziční variantu. Přestože se dle funkčních analýz jedná o variantu s částečně omezenou funkcí proteinu CHK2, nebylo pozorováno klinicky významné zvýšení rizika vzniku BC u žen spojené s jejím výskytem oproti populačně specifickým kontrolám (OR~1,5). Pokud v rámci genetického testování dojde k identifikaci nosičky p.I157T v homozygotním stavu, je vhodné ji o skutečnosti informovat, avšak klinická doporučení nejsou jednoznačná. Tato skutečnost sama o sobě není důvodem k preventivním chirurgickým výkonům, probandku je však možno zařadit do preventivních sledovacích programů. Přítomnost varianty p.I157T v rodině není indikací pro prediktivní testování zdravých příbuzných.
Ostatní raritní missense varianty klasifikované funkčně jako plně či částečně funkční a v dostupných databázích klasifikované jako VUS, je nutno podrobit dalšímu testování. Jejich klinické uplatnění je v současnosti omezené.
Varianty zařazené jako benigní nebo pravděpodobně benigní (class 1 a 2) v dostupných databázích jsou bez klinického využití a obvykle nejsou pro-bandům v rámci genetické konzultace reportovány ani nebývají reportovány laboratoři provádějící genetické vyšetření.
Práce byla podpořena granty Agentury pro zdravotnický výzkum MZČR NR 15-28830A, 16-29959A, NV19-03-00279, projekty Univerzity PROGRES Q28/LF1, GAUK 762216, SVV2019/260367, PRIMUS/17/MED/9, UNCE/MED/016, Progres Q26, LQ1604 NPU II a projektem AVČR Qualitas. Analýza souboru neselektovaných kontrol byla umožněna díky existenci a podpoře vědecké infrastruktury Národního centra lékařské genomiky (LM2015091) a jeho projektu zaměřeného na vytvoření referenční databáze genetických variant České republiky (CZ.02.1.01/0.0/0.0/16_013/0001634).
Autoři deklarují, že v souvislosti s předmětem studie nemají žádné komerční zájmy.
Redakční rada potvrzuje, že rukopis práce splnil ICMJE kritéria pro publikace zasílané do biomedicínských časopisů.
MUDr. Petra Kleiblová, Ph.D.
Ústav biochemie a experimentální onkologie
1. LF UK
U Nemocnice 5
120 00 Praha 2
e-mail: pekleje@lf1.cuni.cz
prof. MUDr. Zdeněk Kleibl, Ph.D.
Ústav biochemie a experimentální onkologie
1. LF UK
U Nemocnice 5
120 00 Praha 2
e-mail: zdekleje@lf1.cuni.cz
Obdrženo: 2. 4. 2019
Přijato: 14. 5. 2019
Zdroje
1. Kleibl Z, Kristensen VN. Women at high risk of breast cancer: Molecular characteristics, clinical presentation and management. Breast 2016; 28 : 136–144. doi: 10.1016/j.breast.2016.05.006.
2. Foretová L, Macháčková E, Palácová M et al. Doporučení rozšíření indikačních kriterií ke genetickému testování mutací v genech BRCA1 a BRCA2 u hereditárního syndromu nádorů prsu a ovarií. Klin Onkol 2016; 29 (Suppl 1): 9–13.
3. Petráková K, Palácová M, Schneiderová M et al. Syndrom hereditárního karcinomu prsu a ovarií. Klin Onkol 2016; 29 (Suppl 1): 14–21. doi: 10.14735/amko2016S14.
4. Janatová M, Borecká M, Soukupová J et al. PALB2 jako další kandidátní gen pro genetické testování u pacientů s hereditárním karcinomem prsu v České republice. Klin Onkol 2016; 29 (Suppl 1): 31–34. doi: 10.14735/amko2016S31.
5. Pohlreich P, Kleibl Z, Kleiblova P et al. Klinický význam analýz genů středního rizika pro hodnocení rizika vzniku karcinomu prsu a dalších nádorů v České republice. Klin Onkol 2012; 25 (Suppl): 59–66. doi: 10.14735/amko20121S59.
6. Cybulski C, Gorski B, Huzarski T et al. CHEK2 is a multiorgan cancer susceptibility gene. Am J Hum Genet 2004; 75 (6): 1131–1135. doi: 10.1086/426403.
7. Matsuoka S, Rotman G, Ogawa A et al. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci USA 2000; 97 (19): 10389–10394. doi: 10.1073/pnas.190030497.
8. Zannini L, Delia D, Buscemi G. CHK2 kinase in the DNA damage response and beyond. J Mol Cell Biol 2014; 6 (6): 442–457. doi: 10.1093/jmcb/mju045.
9. Hu C, Zhang S, Gao X et al. Roles of Kruppel-associated Box (KRAB) -associated Co-repressor KAP1 Ser-473 Phosphorylation in DNA Damage Response. J Biol Chem 2012; 287 (23): 18937–18952. doi: 10.1074/jbc.M111.313262.
10. Meijers-Heijboer H, van den OA, Klijn J et al. Low-penetrance susceptibility to breast cancer due to CHEK2 (*) 1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat Genet 2002; 31 (1): 55–59. doi: 10.1038/ng879.
11. Weischer M, Bojesen SE, Ellervik C et al. CHEK2*1100delC genotyping for clinical assessment of breast cancer risk: meta-analyses of 26,000 patient cases and 27,000 controls. J Clin Oncol 2008; 26 (4): 542–548. doi: 10.1200/JCO.2007.12.5922.
12. Yang Y, Zhang F, Wang Y et al. CHEK2 1100delC variant and breast cancer risk in Caucasians: a meta-analysis based on 25 studies with 29,154 cases and 37,064 controls. Asian Pac J Cancer Prev 2012; 13 (7): 3501–3505.
13. Liu C, Wang Y, Wang QS et al. The CHEK2 I157T variant and breast cancer susceptibility: a systematic review and meta-analysis. Asian Pac J Cancer Prev 2012; 13 (14): 1355–1360.
14. Walsh T, Casadei S, Coats KH et al. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA 2006; 295 (12): 1379–1388. doi: 10.1001/jama.295.12.1379.
15. Desrichard A, Bidet Y, Uhrhammer N et al. CHEK2 contribution to hereditary breast cancer in non-BRCA families. Breast Cancer Res 2011; 13 (6): R119. doi: 10.1186/bcr3062.
16. Le Calvez-Kelm F, Lesueur F, Damiola F et al. Rare, evolutionarily unlikely missense substitutions in CHEK2 contribute to breast cancer susceptibility: results from a breast cancer family registry case-control mutation-screening study. Breast Cancer Res 2011; 13 (1): R6. doi: 10.1186/bcr2810.
17. Dufault MR, Betz B, Wappenschmidt B et al. Limited relevance of the CHEK2 gene in hereditary breast cancer. Int J Cancer 2004; 110 : 320–325. doi: 10.1002/ijc.20073.
18. Bell DW, Kim SH, Godwin AK et al. Genetic and functional analysis of CHEK2 (CHK2) variants in multiethnic cohorts. Int J Cancer 2007; 121 (12): 2661–2667. doi: 10.1002/ijc.23026.
19. Leedom TP, LaDuca H, McFarland R et al. Breast cancer risk is similar for CHEK2 founder and non-founder mutation carriers. Cancer Genet 2016; 209 (9): 403–407. doi: 10.1016/j.cancergen.2016.08.005.
20. Couch FJ, Shimelis H, Hu C et al. Associations between cancer predisposition testing panel genes and breast cancer. JAMA Oncol 2017; 3 (9): 1190–1196. doi: 10.1001/jamaoncol.2017.0424.
21. Fan Z, Ouyang T, Li J et al. Identification and analysis of CHEK2 germline mutations in Chinese BRCA1/2-negative breast cancer patients. Breast Cancer Res Treat 2018; 169 (1): 59–67. doi: 10.1007/s10549-018-4673-6.
22. Hauke J, Horvath J, Gross E et al. Gene panel testing of 5589 BRCA1/2-negative index patients with breast cancer in a routine diagnostic setting: results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancer Med 2018; 7 (4): 1349–1358. doi: 10.1002/cam4. 1376.
23. Young EL, Feng BJ, Stark AW et al. Multigene testing of moderate-risk genes: be mindful of the missense. J Med Genet 2016; 53 (6): 366–376. doi: 10.1136/jmedgenet-2015-103398.
24. Espenschied C, Kleiblova P, Richardson M et al. Classifying variants in the CHEK2 gene: the importance of collaboration. Eur J Cancer 2017; 72 (Suppl 1): S25. doi: 10.1016/S0959-8049 (17) 30161-2.
25. Soukupova J, Zemankova P, Lhotova K et al. Validation of CZECANCA (CZEch CAncer paNel for Clinical Application) for targeted NGS-based analysis of hereditary cancer syndromes. PLoS One 2018; 13 (4): e0195761. doi: 10.1371/journal.pone.019576.
26. Soukupová J, Zemanková P, Kleiblová P et al. CZECANCA: CZEch CAncer paNel for Clinical Application – návrh a příprava cíleného sekvenačního panelu pro identifikaci nádorové predispozice u rizikových osob v České republice. Klin Onkol 2016; 29 (Suppl 1): 46–54. doi: 10.14735/amko2016S46.
27. Kleiblova P, Stolarova L, Krizova K et al. Identification of deleterious germline CHEK2 mutations and their association with breast and ovarian cancer. Int J Cancer 2019. doi: 10.1002/ijc.32385.
28. Walsh T, Mandell JB, Norquist BM et al. genetic predisposition to breast cancer due to mutations other than BRCA1 and BRCA2 founder alleles among Ashkenazi Jewish women. JAMA Oncol 2017; 3 (12): 1647–1653. doi: 10.1001/jamaoncol.2017.1996.
29. Apostolou P, Fostira F, Mollaki V et al. Characterization and prevalence of two novel CHEK2 large deletions in Greek breast cancer patients. J Hum Genet 2018; 63 (8): 877–886. doi: 10.1038/s10038-018-0466-3.
30. Consortium CBCC-C. CHEK2*1100delC and susceptibility to breast cancer: a collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from 10 studies. Am J Hum Genet 2004; 74 (6): 1175–1182. doi: 10.1086/421251.
31. Chekmariova EV, Sokolenko AP, Buslov KG et al. CHEK2 1100delC mutation is frequent among Russian breast cancer patients. Breast Cancer Res Treat 2006; 100 (1): 99–102. doi: 10.1007/s10549-006-9227-7.
32. Kleibl Z, Novotny J, Bezdickova D et al. The CHEK2 c.1100delC germline mutation rarely contributes to breast cancer development in the Czech Republic. Breast Cancer Res Treat 2005; 90 (2): 165–167. doi: 10.1007/s10549-004-4023-8.
33. Choi DH, Cho DY, Lee MH et al. The CHEK2 1100delC mutation is not present in Korean patients with breast cancer cases tested for BRCA1 and BRCA2 mutation. Breast Cancer Res Treat 2008; 112 (3): 569–573. doi: 10.1007/s10549-007-9878-z.
34. Cybulski C, Wokołorczyk D, Jakubowska A et al. Risk of breast cancer in women with a CHEK2 mutation with and without a family history of breast cancer. J Clin Oncol 2011; 29 (28): 3747–3752. doi: 10.1200/JCO.2010.34.0778.
35. Kleibl Z, Havranek O, Novotny J et al. Analysis of CHEK2 FHA domain in Czech patients with sporadic breast cancer revealed distinct rare genetic alterations. Breast Cancer Res Treat 2008; 112 (1): 159–164. doi: 10.1007/s10549-007-9838-7.
36. Havranek O, Kleiblova P, Hojny J et al. Association of Germline CHEK2 gene variants with risk and prognosis of non-Hodgkin lymphoma. Plos One 2015; 10 (10): e0140819. doi: 10.1371/journal.pone.0140819.
37. Roeb W, Higgins J, King MC. Response to DNA damage of CHEK2 missense mutations in familial breast cancer. Hum Mol Genet 2012; 21 (12): 2738–2744. doi: 10.1093/hmg/dds101.
38. Delimitsou A, Fostira F, Kalfakakou D et al. Functional characterization of CHEK2 variants in a Saccharomyces cerevisiae system. Hum Mutat 2019; 40 (5): 631–648. doi: 10.1002/humu.23728.
39. ClinVar. [online]. Available from: www.ncbi.nlm.nih.gov/clinvar?term=CHEK2.
40. Li J, Williams BL, Haire LF et al. Structural and functional versatility of the FHA domain in DNA-damage signaling by the tumor suppressor kinase Chk2. Mol Cell 2002; 9 (5): 1045–1054.
41. Falck J, Mailand N, Syljuasen RG et al. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001; 410 (6830): 842–847. doi: 10.1038/35071124.
42. Lek M, Karczewski J, Minikel EV et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016; 536 (7616): 285–291. doi: 10.1038/nature19057.
43. Margolin S, Eiberg H, Lindblom A et al. CHEK2 1100delC is prevalent in Swedish early onset familial breast cancer. BMC Cancer 2007; 7 : 163. doi: 10.1186/1471-2407-7-163.
44. Lee A, Mavaddat N, Wilcox AN et al. BOADICEA: a comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet Med 2019. doi: 10.1038/s41436-018-0406-9.
45. Pelttari LM, Kiiski J, Nurminen R et al. A Finnish founder mutation in RAD51D: analysis in breast, ovarian, prostate, and colorectal cancer. J Med Genet 2012; 49 (7): 429–432. doi: 10.1136/jmedgenet-2012-100852.
46. Mavaddat N, Michailidou K, Dennis J et al. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am J Hum Genet 2019; 104 (1): 21–34. doi: 10.1016/j.ajhg.2018.11.002.
47. Muranen TA, Greco D, Blomqvist C et al. Genetic modifiers of CHEK2*1100delC-associated breast cancer risk. Genet Med 2017; 19 (5): 599–603. doi: 10.1038/gim.2016.147.
48. Muranen TA, Blomqvist C, Dork T et al. Patient survival and tumor characteristics associated with CHEK2: p.I157T – findings from the Breast Cancer Association Consortium. Breast Cancer Res 2016; 18 (1): 98. doi: 10.1186/s13058-016-0758-5.
49. Liu C, Chang H, Li XH et al. Network meta-analysis on the effects of DNA damage response-related gene mutations on overall survival of breast cancer based on TCGA database. J Cell Biochem 2017; 118 (12): 4728–4734. doi: 10.1002/jcb.26140.
50. Weischer M, Nordestgaard BG, Pharoah P et al. CHEK2*1100delC heterozygosity in women with breast cancer associated with early death, breast cancer-specific death, and increased risk of a second breast cancer. J Clin Oncol 2012; 30 (35): 4308–4316. doi: 10.1200/JCO.2012.42. 7336.
51. Zlowocka-Perlowska E, Narod SA, Cybulski C. CHEK2 alleles predispose to renal cancer in poland. JAMA Oncol 2019. doi: 10.1001/jamaoncol.2019. 0022.
52. Huzarski T, Gorecka-Szyld B, Huzarska J et al. Screening with magnetic resonance imaging, mammography and ultrasound in women at average and intermediate risk of breast cancer. Hered Cancer Clin Pract 2017; 15 : 4. doi: 10.1186/s13053-017-0064-y.
53. Macklin S, Gass J, Mitri G et al. The role of screening MRI in the era of next generation sequencing and moderate-risk genetic mutations. Fam Cancer 2018; 17 (1): 167–173. doi: 10.1007/s10689-017-0007-9.
54. Huijts PE, Hollestelle A, Balliu B et al. CHEK2*1100delC homozygosity in the Netherlands-prevalence and risk of breast and lung cancer. Eur J Hum Genet 2014; 22 (1): 46–51. doi: 10.1038/ejhg.2013.85.
55. Nurmi A, Muranen TA, Pelttari LM et al. Recurrent moderate-risk mutations in Finnish breast and ovarian cancer patients. Int J Cancer 2019. doi: 10.1002/ijc.32309.
56. Girard E, Eon-Marchais S, Olaso R et al. Familial breast cancer and DNA repair genes: Insights into known and novel susceptibility genes from the GENESIS study, and implications for multigene panel testing. Int J Cancer 2019; 144 (8): 1962–1974. doi: 10.1002/ijc.31921.
57. Decker B, Allen J, Luccarini C et al. Rare, protein-truncating variants in ATM, CHEK2 and PALB2, but not XRCC2, are associated with increased breast cancer risks. J Med Genet 2017; 54 (11): 732–741. doi: 10.1136/jmedgenet-2017-104588.
58. Slavin TP, Maxwell KN, Lilyquist J et al. The contribution of pathogenic variants in breast cancer susceptibility genes to familial breast cancer risk. NPJ Breast Cancer 2017; 3 : 22. doi: 10.1038/s41523-017-0024-8.
59. Schmidt MK, Hogervorst F, van Hien R et al. Age-and tumor subtype-specific breast cancer risk estimates for CHEK2*1100delC carriers. J Clin Oncol 2016; 34 (23): 2750–2760. doi: 10.1200/JCO.2016.66.5844.
60. Southey MC, Goldgar DE, Winqvist R et al. PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. J Med Genet 2016; 53 (12): 800–811. doi: 10.1136/jmedgenet-2016-103839.
61. Liu Y, Liao J, Xu Y et al. A recurrent CHEK2 p.H371Y mutation is associated with breast cancer risk in Chinese women. Hum Mutat 2011; 32 (9): 1000–1003. doi: 10.1002/humu.21538.
62. Weischer M, Bojesen SE, Tybjaerg-Hansen A et al. Increased risk of breast cancer associated with CHEK2*1100delC. J Clin Oncol 2007; 25 (1): 57–63. doi: 10.1200/JCO.2005.05.5160.
63. Vahteristo P, Bartkova J, Eerola H et al. A CHEK2 genetic variant contributing to a substantial fraction of familial breast cancer. Am J Hum Genet 2002; 71 (2): 432–438. doi: 10.1086/341943.
64. Liang M, Zhang Y, Sun C et al. Association between CHEK2*1100delC and breast cancer: a systematic review and meta-analysis. Mol Diagn Ther 2018; 22 (4): 397–407. doi: 10.1007/s40291-018-0344-x.
65. Han FF, Guo CL, Liu LH. The effect of CHEK2 variant I157T on cancer susceptibility: evidence from a meta-analysis. DNA Cell Biol 2013; 32 (6): 329–335. doi: 10.1089/dna.2013.1970.
66. Zhang B, Beeghly-Fadiel A, Long J et al. Genetic variants associated with breast-cancer risk: comprehensive research synopsis, meta-analysis, and epidemiological evidence. Lancet Oncol 2011; 12 (5): 477–488. doi: 10.1016/S1470-2045 (11) 70076-6.
67. AlDubayan SH, Pyle LC, Gamulin M et al. association of inherited pathogenic variants in checkpoint kinase 2 (CHEK2) with susceptibility to testicular germ cell tumors. JAMA Oncol 2019. doi: 10.1001/jamaoncol.2018.6477.
68. Obazee O, Archibugi L, Andriulli A et al. Germline BRCA2 K3326X and CHEK2 I157T mutations increase risk for sporadic pancreatic ductal adenocarcinoma. Int J Cancer 2019; 145 (3): 686–693. doi: 10.1002/ijc.32127.
69. Hallamies S, Pelttari LM, Poikonen-Saksela P et al. CHEK2 c.1100delC mutation is associated with an increased risk for male breast cancer in Finnish patient population. BMC Cancer 2017; 17 (1): 620. doi: 10.1186/s12885-017-3631-8.
70. Carlo MI, Mukherjee S, Mandelker D et al. Prevalence of germline mutations in cancer susceptibility genes in patients with advanced renal cell carcinoma. JAMA Oncol 2018; 4 (9): 1228–1235. doi: 10.1001/jamaoncol.2018.1986.
71. Pritchard CC, Mateo J, Walsh MF et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med 2016; 375 (5): 443–453. doi: 10.1056/NEJMoa1603144.
72. Siolek M, Cybulski C, Gasior-Perczak D et al. CHEK2 mutations and the risk of papillary thyroid cancer. Int J Cancer 2015; 137 (3): 548–552. doi: 10.1002/ijc.29426.
73. Wang Y, Dai B, Ye D. CHEK2 mutation and risk of prostate cancer: a systematic review and meta-analysis. Int J Clin Exp Med 2015; 8 (9): 15708–15715.
74. Hale V, Weischer M, Park JY. CHEK2 (*) 1100delC mutation and risk of prostate cancer. prostate cancer 2014; 2014 : 294575. doi: 10.1155/2014/294575.
75. Ma X, Zhang B, Zheng W. Genetic variants associated with colorectal cancer risk: comprehensive research synopsis, meta-analysis, and epidemiological evidence. Gut 2014; 63 (2): 326–336. doi: 10.1136/gutjnl-2012-304 121.
76. Liu C, Wang QS, Wang YJ. The CHEK2 I157T variant and colorectal cancer susceptibility: a systematic review and meta-analysis. Asian Pac J Cancer Prev 2012; 13 (5): 2051–2055. doi: 10.7314/apjcp.2012.13.5.2051.
77. Weischer M, Heerfordt IM, Bojesen SE et al. CHEK2*1100delC and risk of malignant melanoma: Danish and German studies and meta-analysis. J Invest Dermatol 2012; 132 (2): 299–303. doi: 10.1038/jid.2011.303.
78. Xiang HP, Geng XP, Ge WW et al. Meta-analysis of CHEK2 1100delC variant and colorectal cancer susceptibility. Eur J Cancer 2011; 47 (17): 2546–2551. doi: 10.1016/j.ejca.2011.03.025.
Štítky
Dětská onkologie Chirurgie všeobecná OnkologieČlánek vyšel v časopise
Klinická onkologie

2019 Číslo Supplementum2
- Incidence mozkových metastáz a intrakraniální aktivita sotorasibu u pacientů s NSCLC s mutací G12C onkogenu KRAS
- Přibývající důkazy o přínosu léčebného konopí u pacientů s chronickou bolestí
- Předchozí zánětlivá onemocnění plic zvyšují riziko rozvoje nádoru plic
- "Železem saturovaný" hovězí laktoferin zlepšuje chemoterapeutický účinek tamoxifenu u léčby basal-like karcinomu prsu u myší
- Časná fyzioterapie může zabránit lymfedému po operaci karcinomu prsu
Nejčtenější v tomto čísle
- Dědičné mutace v genu CHEK2 jako příčina dispozice k nádorům prsu – typy mutací, jejich biologická a klinická relevance
- Rizika solidních nádorů u heterozygotních přenašečů recesivních syndromů
- Doporučení pro sledování žen se vzácnějšími genetickými příčinami nádorů prsu a ovarií
- GAPPS – syndrom adenokarcinomu žaludku a mnohočetné polypózy žaludku v 8 rodinách testovaných v Masarykově onkologickém ústavu – prevence vč. profylaktické gastrektomie
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy