
Klasická forma Ehlersova-Danlosova syndromu – popis případu
Classic Form of Ehlers-Danlos Syndrome
Ehlers-Danlos syndromes belong to the group of inherited connective tissue disorders characterized by increased skin elasticity, joint hypermobility, cutaneous fragility with formation of pseudotumors and gaping scars. Original classification by Villefranche (1997) divided Ehlers-Danlos syndrome into 6 groups. New classification (2017) supported by molecular analysis distinguishes 13 subtypes of Ehlers-Danlos syndromes. We present a case of 4-year old boy with classic EDS confirmed by histological skin examination and DNA analysis revealing yet non - published mutation (sequence variant) c.1048insG p.Ser350Cysfs*4 in the 7th exon of the COL5A1 gene.
Key words:
Ehlers-Danlos syndromes – skin hyperextensibility – generalised joint hypermobility – DNA analysis – new station (sequence variant) – new EDS classification 2017
Autoři:
J. Drochytková 1; L. Fajkusová 2; L. Kopečková 2; K. Veselý 3; R. Beharka 4; H. Bučková 1
Působiště autorů:
Dětské kožní oddělení PEK FN Brno a LF MU v Brně
prim. MUDr. Hana Bučková, Ph. D.
1; Centrum molekulární biologie a genové terapie IHOK FN Brno a LF MU v Brně
vedoucí sekce vrozených genetických chorob doc. RNDr. Lenka Fajkusová, CSc.
2; AeskuLab Patologie, k. s., Laboratoř Brno
vedoucí pracoviště MUDr. Karel Veselý, Ph. D.
3; Oddělení lékařské genetiky FN Brno a LF MU v Brně
prim. MUDr. Renata Gaillyová, Ph. D.
4
Vyšlo v časopise:
Čes-slov Derm, 92, 2017, No. 5, p. 221-228
Kategorie:
Kazuistiky
Souhrn
Vzácná onemocnění, Ehlersovy-Danlosovy syndromy, patří do skupiny dědičných chorob pojivové tkáně. Projevují se hypermobilitou kloubů, zvýšenou elasticitou a křehkostí kůže, vznikem pseudotumorů a zejících jizev. Původní klasifikace podle Villefranche (1997) dělila syndrom Ehlersův-Danlosův (EDS) do 6 skupin. Nová klasifikace 2017 vychází ze současných poznatků molekulární diagnostiky a rozlišuje 13 subtypů Ehlersových-Danlosových syndromů. Autoři prezentují čtyřletého pacienta s klasickým subtypem EDS, který byl potvrzen histologickým vyšetřením kůže pacienta a DNA analýzou, při které byla zjištěna dosud nepopsaná mutace (patogenní sekvenční varianta) c.1048insG p.Ser350Cysfs*4 v 7. exonu genu COL5A1. Současně představují novou klasifikaci.
Klíčová slova:
Ehlersovy-Danlosovy syndromy – kožní hyperextenzibilita – generalizovaná kloubní hypermobilita – DNA analýza – nová mutace (sekvenční varianta) – nová klasifikace 2017
ÚVOD
Ehlers-Danlos syndromy tvoří skupinu vrozených onemocnění pojivové tkáně s postižením kůže, kloubů, cév a dalších orgánů. Patogenetickým podkladem těchto vzácných onemocnění jsou mutace (sekvenční varianty) genů způsobující defekty syntézy kolagenu. Poprvé se o tomto onemocnění zmiňuje Hippokrates 400 let př. n. l. Jako samostatnou jednotku ji popsal dánský dermatolog Edvard Ehlers v roce 1901. Francouzský dermatolog Henri-Alexandre Danlos v roce 1908 uvedl hlavní znaky tohoto syndromu, zvýšenou pružnost a křehkost kůže [7]. Autoři prezentují případ Ehlersova-Danlosova syndromu u pacienta a jeho matky s nálezem dosud nepopsané patogenní sekvenční varianty v genu COL5A1.
POPIS PŘÍPADU
Pacient byl z 3. rizikové gravidity matky, které byl EDS diagnostikován v jejich 4 letech, ale nikde nebyla sledovaná. U rodičů matky ani bližšího příbuzenstva nebyly popsány projevy EDS. Porodila jednoho zdravého syna, 1krát došlo ke spontánnímu abortu dvojčat. Ve 3. graviditě u matky došlo od 6. týdne k opakovanému krvácení z rodidel, pro hrozící předčasný porod byla hospitalizována. Porod proběhl ve 38. týdnu gravidity spontánně záhlavím, porodní hmotnost byla 3 200g a délka 50 cm. Poporodní adaptace novorozence byla v normě. Pro výrazné krvácení během porodu byla matce podána transfuze. Matka pacienta měla typické znaky Ehlersova-Danlosova syndromu – těstovité podkoží, rozeklané jizvy, hematomy na bércích, hyperelasticitu kůže, hyperflexibilitu kloubů, kyfoskoliózu, chronickou žilní insuficienci.
Vzhledem k pozitivní rodinné anamnéze, tvorbě četných hematomů a vzniku atrofických jizev na kůži byl pacient ve 4 letech praktickým lékařem pro děti a dorost odeslán na Oddělení lékařské genetiky ve FN Brno. Odtud byl pacient k dalšímu vyšetření poslán na naši kožní ambulanci.
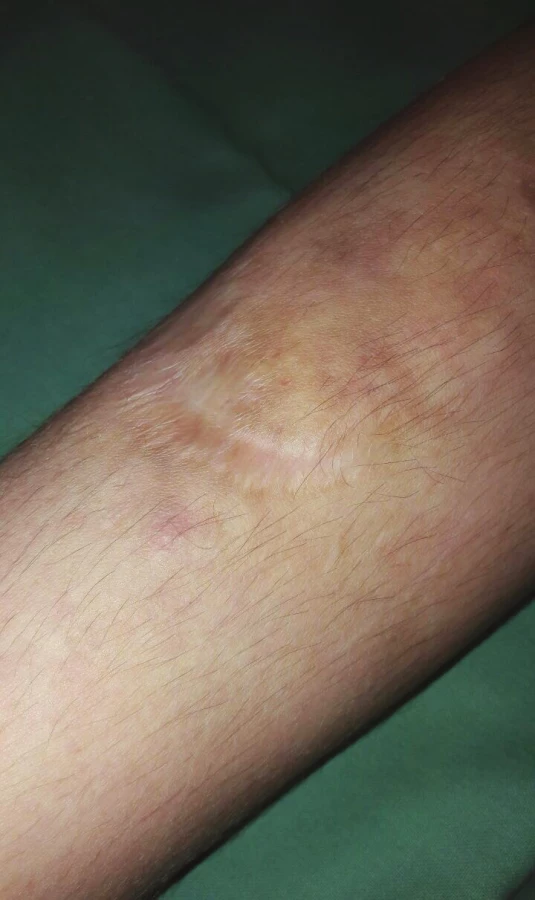
V kojeneckém věku byl pacient hospitalizován pro neprospívání. Ve 4 letech měl frakturu pravé klavikuly a v 5 letech si způsobil tržnou ránu na bérci vyžadující suturu, která se zhojila rozeklanou atrofickou jizvou. U pacienta byly typické známky klasické formy Ehlersova-Danlosova syndromu. Jemná, hebká kůže s nadměrnou kožní řasou, těstovité podkoží, hyperflexibilita kloubů, pozitivní příznak palce (obr. 1, 2). Na čele měl jizvy charakteru cigaretového papíru, na bércích byla patrna atrofická kůže v místě resorbujících se hematomů, hematomy průměru 2 x 3 cm na obou bércích (obr. 3, 4). Na pravém bérci ventrálně byla přítomna poloměsíčitá jizva velikosti cca 4 cm. Nad pravým zevním kotníkem měl okrouhlé ložisko hyperpigmentace velikosti asi 2 x 2 cm, pedes plani, vadné držení těla (obr. 5, 6).





Pacientovi byl odebrán vzorek kůže k histologickému vyšetření. Ultrastrukturální obraz prokázal nález kompatibilní s Ehlers-Danlos syndromem. U kolagenních vláken bylo místy patrno rozvolnění jednotlivých fibril s akumulací malého množství jemně granulárního materiálu. Nebyly přítomny „květinkové“ struktury či nepravidelně zesílené svazky kolagenu, ani zřetelné zvýšení množství amorfní extracelulární matrix (obr. 7).

U pacienta a jeho matky byla odebrána krev na DNA analýzu. Metodou Sequence Capture – NGS (next generation sequencing) byla provedena analýza genů spojených se syndromem Ehlerse-Danlose (COL1A1, COL1A2, COL3A1, COL5A1, COL5A2, PLOD1, TNXB). Následně byla analýza ověřena Sangerovým sekvenováním. U obou byla nalezena mutace (sekvenční varianta) c.1048insG p.Ser350Cysfs*4 v 7. exonu genu COL5A1. Jedná se o sekvenční variantu v literatuře dosud nepopsanou, která vede ke vzniku předčasného terminačního kodonu (obr. 8). Vyšetření u bratra a otce pacienta tuto mutaci neprokázala.

Laboratorní vyšetření krevního obrazu, základní biochemie i vyšetření moči bylo u pacienta v normě. UZ břicha byl rovněž s fyziologickým nálezem.
RTG Th-S pátěře v základní projekci prokázal, že páteř je v lehkém úklonu doprava a vpravo je hypoplastické XIII. žebro. Na bočním snímku byla popsána zachovalá hrudní kyfóza (21 st.) i bederní lordóza. Obratlová těla byla obvyklé výšky, tvaru i struktury, bez patologického ventrodorzálního posunu. Meziobratlové prostory nebyly sníženy.
Pacient byl vyšetřen u řady specialistů. Na ortopedii potvrdili výraznou hypermobilitu, planovalgózní postavení nohou až III. stupně. Podle rehabilitačního vyšetření byla patrná generalizovaná výrazná hypermobilita, slabé fixátory lopatek, protrakce ramen, hyperextenze v loktech a kolenních kloubech, těžká planovalgozita nohou s přetížením vnitřních hran nohou, snad více vlevo. Pacient se rychleji unavil při delší chůzi. Doporučena léčebná tělesná výchova klasická a na míči. Cévní vyšetření prokázalo prodloužené venózní plnící časy signalizující hraniční hodnoty žilní insuficience.
Kardiologické, neurologické a oční vyšetření byla s fyziologickým nálezem.
DISKUSE
Klasifikace EDS z roku 1997 podle Villefranche vycházela hlavně z klinických příznaků [6, 7]. Nová klasifikace z roku 2017 je založena na korelaci klinických příznaků s histologickým vyšetřením a s DNA analýzou (tab. 1, 2).
![Nová klasifikace Ehlers-Danlos syndromů (EDS) 2017 [5]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/c62ce24d42f5652aba52026b4538d6fa.png)



Klasický typ EDS je nejčastějším podtypem tohoto vzácného onemocnění. Dědičnost je autozomálně dominantní. Riziko přenosu na potomky je 50% bez rozdílu pohlaví. Prevalence je 2–5 na 100 000 obyvatel. Více než 50% případů klasického EDS je způsobeno sekvenčními variantami v genu COL5A1, což je případ našeho pacienta a jeho matky. Mnohé ze sekvenčních variant vážících se k EDS vedou k tvorbě nefunkčních nebo chybějících pro-alfa (V) řetězců. Kolagenní vlákna jsou nepravidelně uspořádaná a větší, než je obvyklé. Toto atypické uspořádání kolagenních vláken v kůži, kloubech, svalech, šlachách, cévách a viscerálních orgánech je příčinou klinických projevů EDS [1, 3, 4, 6].
U našeho pacienta a jeho matky jde o novou, v odborné literatuře dosud nepopsanou mutaci (patogenní sekvenční variantu), při které vzniká předčasný terminační kodon v 7. exonu genu. Gen COL5A1 má celkem 66 exonů. Dojde k předčasnému ukončení translace, vzniká tedy výrazně kratší protein, případně vlivem nestability protein ani nevznikne. U demonstrovaného případu se tedy jedná o patogenní sekvenční variantu, která způsobí závažnější klinické projevy klasického typu EDS.
Náš pacient splňuje obě hlavní kritéria podle nové mezinárodní klasifikace, která jsou nutná ke klinickému stanovení diagnózy tedy kožní hyperextenzibilita a atrofické jizvy a generalizovaná kloubní hypermobilita (tab. 3) a dále několik vedlejších kritérií jako snadná tvorba hematomů, měkká, těstovitá kůže, kožní fragilita, muskuloidní pseudotumory, komplikace z hypermobility kloubů (subluxace, plochá noha), pozitivní rodinná anamnéza u příbuzného prvního stupně splňující klinická kritéria (viz tab. 2) [5]. Postižení vnitřních orgánů nebo výskyt hernií zatím u našeho pacienta nebyly prokázány.

Pacienti s EDS často bývají nedonošení, v průběhu těhotenství dochází k předčasnému protržení plodových obalů [2]. U dospělých pacientek s vážnějšími formami klasického EDS se může vyskytnout také prolaps dělohy, nebo močového měchýře, větší poranění v oblasti perinea, samotný porod vaginální cestou je velkou zátěží na pánevní dno [2, 4, 6]. Matka našeho pacienta nebyla nikde sledovaná, preventivně necvičila pánevní dno, výrazné krvácení v průběhu dvou porodů nikdo nepovažoval za projev související s jejím základním onemocněním. Nebyl u ní indikován císařský řez, který je pro ni i pro novorozence s EDS šetrnější, neboť sníží riziko dislokace v oblasti ramenního nebo kyčelního kloubu [5].
Při kardiologickém vyšetření se může prokázat prolaps mitrální méně často trikuspidální chlopně a dilatace kořene aorty. Ruptury velkých tepen jsou spíše výjimkou a jsou četnější u cévního typu EDS [2, 4].
V diferenciální diagnóze je nutno zvážit ostatní typy EDS a také další vrozené poruchy pojivové tkáně jako např. Marfanův syndrom, cutis laxa, eventuálně někteří pacienti s neurofibromatózou 1 mají těstovité podkoží, hyperflexibilitu kloubů [1, 4, 6]. Tyto syndromy však vykazují další odlišné klinické příznaky. Tato onemocnění lze také diagnostikovat na molekulární úrovni.
Onemocnění s klasickým typem EDS obvykle nezkracuje délku života pacientů, může však mít vliv na kvalitu jejich života. Závažná orgánová postižení především pohybového aparátu s chronickými bolestmi mohou vést až k invaliditě [4].
ZÁVĚR
EDS patří mezi vzácná onemocnění. Existuje více než 8 000 různých vzácných onemocnění. Pro pacienty s EDS neexistuje kauzální léčba, terapie je pouze podpůrná. Důležitá je včasná diagnóza EDS, dispenzarizace pacientů, preventivní zaměření péče v centru, kde mají s pacienty zkušenosti. Komplexní péče vyžaduje mezioborový tým specialistů. Stanovení správného typu EDS je postaveno na klinických zkušenostech dermatologa, histologickém vyšetření kůže i v elektronovém mikroskopu, DNA molekulární analýze, kompletním genetickém poradenství a genetické prevenci. Jedná se o složitou problematiku, některé typy EDS mohou být spojeny s patologií ve více genech, jeden typ může mít více typů dědičnosti Nová mezinárodní klasifikace 2017 s jasně danými hlavními a vedlejšími kritérii, by měla usnadnit stanovení správného typu EDS.
Náš pacient je dispenzarizován v naší kožní ambulanci, pravidelně minimálně 1krát ročně je přešetřen na specializovaných ambulancích – kožní, ortopedie, rehabilitace, kardiologie, oční, neurologie, sonografie břicha a pánve, eventuálně cévní vyšetření. Matka pacienta byla odeslána ke sledování na dermatovenerologické pracoviště pro dospělé pacienty.
Systematickou preventivní péčí se předchází komplikacím u jednotlivých typů EDS. Nutná jsou opatření k minimalizaci poranění kůže především v batolecím věku. Zvláštní pozornost by měla být věnována péči o kůži, opatřením k ochraně kloubů, fyzioterapii, léčbě bolesti, psychologické podpoře, sledování v těhotenství [4, 6].
Je nutná doživotní observace a dispenzarizace ve specializovaných ambulancích, neboť některá orgánová postižení se mohou vyvíjet v průběhu růstu, např. postižení skeletu.
V reprodukčním věku pacientům doporučujeme prekoncepčně konzultaci i s partnerkou/partnerem u klinického genetika, kde mohou získat informace o možnostech genetické prevence – prenatální nebo preimplantační genetické diagnostiky s cílem snížit riziko narození dítěte s EDS v rodině, kde se již tato nemoc vyskytla.
Do redakce došlo dne 20. 9. 2017.
Adresa pro korespondenci:
MUDr. Jana Drochytková
Dětské kožní oddělení PeK FN a LF MU
Černopolní 9
662 63 Brno
e-mail: drochytkova.jana@fnbrno.cz
Zdroje
1. BOLOGNIA, J. L., JORIZZO, J. L., RAPINI, R. P. Dermatology. 2. vyd. Elsevier, 2008, p. 713. ISBN 9781416029991.
2. BOWEN, J. M., SOBEY, G. J., BURROWS, G. J. et al. Ehlers–Danlos syndrome, classical type. Am. J. Med. Genet. Part C Semin. Med. Genet., 2017, 175C, p. 27–39.
3. LANE, A. T., MORELLI, J. G., WESTON, W. L. Textbook of Pediatric Dermatology. 4. vyd. Philadelphia: Elsevier, 2007. p. 339–342. ISBN 9780323049092.
4. MALFAIT, F., WENSTRUP, R. J., DE PAEPE, A. Clinical and genetic aspects of Ehlers-Danlos syndrome, classic type. Genet. Med., 2010, 12, 10, p. 597–605.
5. MALFAIT, F., FRANCOMANO, C., BYERS, P. et al. The 2017 international classification of the Ehlers - -Danlos syndromes. Am. J. Med. Genet. Part C Semin. Med. Genet. 2017, 175, 1, p. 8–26.
6. PALLER, A. S., MANCINI, A. J. Hurwitz Clinical Pediatric Dermatology. 5. vyd., Elsevier, 2015, p. 489–491. ISBN 9780323244756.
7. PARAPIA, L. A., JACKSON, C. Ehlers-Danlos syndrome – a historical review. British Journal of Haematology, 2008, 141, p. 32–35.
Štítky
Dermatologie Dětská dermatologieČlánek vyšel v časopise
Česko-slovenská dermatologie

2017 Číslo 5
- Inosin pranobex v léčbě HPV infekcí děložního čípku
- Léčba chronické blefaritidy vyžaduje dlouhodobou péči
- Léčba zánětů spojivek a mazových žlázek víčka v primární péči
- Přidání perorálního acetazolamidu (Diluran) k topické léčbě dorzolamidem může vést k dalšímu snížení nitroočního tlaku u některých dětí s glaukomem
- Nová fixní kombinace pro léčbu akné – 1,2% klindamycinfosfát a 0,025% tretinoin
Nejčtenější v tomto čísle
- Klasická forma Ehlersova-Danlosova syndromu – popis případu
- Infantilní hemangiomy z pohledu dermatologa
- Sekce dětské dermatologie oslavila 20 let od založení
- Klinický případ: Noduly barvy kůže u dítěte
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy