
Neurofibromatóza z pohledu dermatologa
Neurofibromatosis from the View of Dermatologist
Neurofibromatosis is a relatively common autosomal dominant hereditary neuro-cutaneous disease caused by mutation in tumor-suppressor genes. It results in the development of multiple hamartomas, benign and malignant tumors. The disease is currently classified into 3 individual subunits with different clinical presentations based on mutations of different genes – neurofibromatosis type 1 (NF1), neurofibromatosis type 2 (NF2) and schwannomatosis. The most common type is NF1. The symptoms are age-dependent, the severity of clinical picture is variable. Cutaneous symptoms are present namely in NF1, they are less common in other types. They include multiple café-au-lait spots and axillary and inguinal freckling. Neurofibromas, plexiform neurofibromas and optic gliomas are present in NF1, Lisch nodules in the iris are pathognomonic. Vestibular schwannomas are typical for NF2, and multiple schwannomas in peripheral nerves are present in schwannomatosis. The prognosis differs and is based on the clinical picture and complications. Diagnostic criteria play a key role in differentiating various types of neurofibromatosis and dermatological examination plays a key role. Patients should be concentrated in specialized centers. The treatment of neurofibromatosis is symptomatic.
Key words:
neurofibromatosis – café-au-lait spot – freckling – neurofibroma – schwannoma – glioma – specialized centers specialized care
Autoři:
D. Humhejová 1; B. Petrák 2
Působiště autorů:
Kožní oddělení, Masarykova nemocnice v Ústí nad Labem o. z., Krajská zdravotní a. s.
primář MUDr. Olga Filipovská
1; Klinika dětské neurologie 2. LF UK a FN Motol, Praha
přednosta prof. MUDr. Vladimír Komárek, CSc.
2
Vyšlo v časopise:
Čes-slov Derm, 90, 2015, No. 3, p. 95-109
Kategorie:
Souborné referáty (doškolování lékařů)
Souhrn
Neurofibromatóza je relativně časté autozomálně dominantně dědičné neurokutánní onemocnění způsobené mutací tumor supresorových genů se vznikem mnohočetných hamartomů, benigních a maligních nádorů. Onemocnění se v současné době dělí na 3 samostatné jednotky s odlišným klinickým obrazem, podmíněným mutací různých genů – neurofibromatózu typu 1 (NF1), neurofibromatózu typu 2 (NF2) a schwannomatózu. Nejčastějším typem je NF1. Projevy onemocnění jsou věkově vázané, závažnost klinického obrazu je variabilní. Kožní projevy se vyskytují zejména u NF1, méně u ostatních typů. Jedná se o mnohočetné skvrny charakteru „café-au-lait“ a drobné skvrnité hyperpigmentace („freckling“) v axillách a tříslech. U NF1 se nachází neurofibromy a plexiformní neurofibromy, gliomy optiku, patognomický je nález Lischových nodulů na duhovce. U NF2 se nachází vestibulární schwannomy, u schwannomatózy mnohočetné schwannomy periferních nervů. Prognóza je individuální podle klinického obrazu a komplikací. K diagnostice jednotlivých forem neurofibromatóz slouží diagnostická kritéria, klíčovou roli hraje vyšetření dermatologem. Pacienti by měli být koncentrováni ve specializovaných centrech. Léčba neurofibromatózy je symptomatická.
Klíčová slova:
neurofibromatóza – café-au-lait – freckling – neurofibrom – schwannom – gliom – specializovaná centra odborné péče
ÚVOD
Neurofibromatóza patří do skupiny neurokutánních syndromů, dříve označovaných jako fakomatózy. Jedná se o multisystémová onemocnění, která postihují především kůži a nervový systém. Onemocnění vznikají mutací genů, jejichž produkty mají vliv na regulaci buněčného růstu a proliferaci, s možným vlivem na zvýšený výskyt nádorů. Dědičnost neurokutánních syndromů je převážně autozomálně dominantní, častý je také sporadický výskyt. Klinické projevy jsou značně variabilní, objevují se i v kojeneckém věku a s věkem jejich výskyt narůstá.
Neurofibromatóza je nejčastější neurokutánní onemocnění. První zmínky se objevují v literatuře již ve 13. století. V roce 1882 Friedrich Daniel von Recklinghausen popsal mnohočetný výskyt neurofibromů na kůži ve spisu „Űber die multiplen Fibrome der Haut und ihre Beziehung zu den multiplen Neuromen“ [17]. Toto onemocnění bylo později pojmenováno von Recklinghausenova choroba. V 70. letech 20. století byly popsány případy nemocných s oboustrannými vestibulárními schwannomy. Diagnóza neurofibromatózy tak byla rozdělena podle klinického obrazu na 2 typy. Na počátku 90. let 20. století byly objeveny podmiňující geny obou typů neurofibromatózy, a byly tak rozlišeny neurofibromatóza typu 1 a typu 2 jako dvě samostatné klinické jednotky [6, 18].
KLASIFIKACE NEUROFIBROMATÓZY
Klasifikace neurofibromatózy se v průběhu posledních třiceti let neustále vyvíjí. V roce 1982 byla Riccardim publikována klasifikace 8 typů neurofibromatózy podle klinického obrazu. Toto dělení nebylo zcela akceptováno, bylo mnohokrát přepracováno a s ohledem na pozdější molekulárně-genetické nálezy není v současné době považováno za platné. Aktuální celosvětově uznávaná klasifikace neurofibromatózy vychází z National Institute of Health Consensus Conference on Neurofibromatosis z roku 1987, která rozlišuje 3 typy neurofibromatózy – neurofibromatózu typu 1, typu 2 a schwannomatózu. V minulosti užívané označení pro NF1 periferní a pro NF2 centrální neurofibromatóza se pro nepřesnost těchto termínů již nepoužívá [17, 18]. Později popsané vzácné varianty neurofibromatózy označované jako gastrointestinální a familiární spinální neurofibromatóza, neurofibromatosis – Noonanové syndrom a Watsonův syndrom jsou podle nejnovějších molekulárně-genetických poznatků považovány za varianty NF1.
NEUROFIBROMATÓZA TYPU 1
Nejčastějším typem neurofibromatózy je neurofibromatóza typu 1, označovaná také jako neurofibromatosis von Recklinghausen typ 1 (NF1) nebo morbus von Recklinghausen typ 1, dříve také periferní neurofibromatóza. Tímto typem je postiženo více než 90 % všech pacientů s neurofibromatózou [1]. Celosvětově se vyskytuje s frekvencí 1 : 2 500–4 000 obyvatel. Onemocnění postihuje všechny rasy a obě pohlaví stejně často. Asi polovina nemocných má pozitivní rodinnou anamnézu [1].
ETIOPATOGENEZE
NF1 je onemocnění s autozomálně dominantním typem dědičnosti, variabilní expresivitou a téměř úplnou penetrancí. Gen pro NF1 je lokalizován na dlouhém raménku chromozomu 17 (17q11.2). Představuje 280 kb genomové DNA, obsahuje 61 exonů. Produktem NF1 genu je neurofibromin, cytoplazmatický protein o velikosti 2 818 aminokyselin s molekulovou hmotností 320 kD. Neurofibromin je exprimován v neuronech, Schwannových buňkách, oligodendrocytech, astrocytech a leukocytech. Působí jako negativní růstový regulátor tím, že aktivací GTPázy inhibuje funkci proteinů Ras. Během GTP hydrolýzy funguje jako akcelerátor konverze aktivovaného GTP-vázaného proteinu Ras na inaktivní GDP-vázaný protein Ras. Ztráta funkce NF1 genu mutací a tím chybění či porucha funkce neurofibrominu vede např. ve Schwannově buňce ke zvýšení hladiny aktivovaného GTP-vázaného proteinu Ras a tím k urychlení růstu a proliferace této buňky. Z toho pak vychází hypotéza vzniku neurofibromu či plexiformního neurofibromu.
Četnost mutací v genu NF1 je ve srovnání s jinými lidskými geny jedna z nejvyšších, což je považováno za klinicky významné (až u 50 % případů vzniká onemocnění v důsledku nové mutace). V genu NF1 bylo dosud popsáno více než 1 300 mutací. Nejzávažnější klinický obraz byl pozorován u delečních mutací, naopak nejnižší riziko závažných zdravotních komplikací je u mozaiky [26]. Častější výskyt nových mutací je popisován na paternálních chromozomech. Příčina tohoto jevu zatím není známa, předpokládá se vliv vyššího věku otce v době početí [17].
KLINICKÝ OBRAZ
Variabilita klinického obrazu je značná i v rámci jedné rodiny. Klinické příznaky NF1 se manifestují postupně v závislosti na věku. V prvních letech života, výjimečně při narození, se objevují hyperpigmentace charakteru skvrn café-au-lait a vzácně plexiformní neurofibromy. Gliomy optiku mohou být kongenitální nebo se vyvíjí obvykle do 10 let věku, později jsou velmi vzácné. V předškolním a mladším školním věku vznikají drobné skvrnité hyperpigmentace v axillách a tříslech (freckling), před pubertou, během puberty a v těhotenství neurofibromy. Lischovy noduly (viz dále) přibývají během celého života. Kostní změny bývají kongenitální. Kromě jmenovaných klinických příznaků se také vyvíjí benigní a maligní nádory. Typickou věkovou manifestaci projevů NF1 znázorňuje graf 1.
![Přehled obvyklé počáteční manifestace projevů neurofibromatózy typu 1 v závislosti na věku Modifikováno podle [26].](https://pl-master.mdcdn.cz/media/image/19145a3a82e048667fd510906da0d87a.jpg?version=1537792676)
A. Kožní projevy
Dermatolog může při fyzikálním vyšetření pacienta zaznamenat typické kožní projevy neurofibromatózy. Tabulka 1 uvádí přehled kožních projevů NF1 a chorob sdružených s NF1.

- Skvrny café-au-lait
Kávové skvrny (café-au-lait macules – CALM) jsou ostře ohraničené makuly charakteristického vzhledu (obr. 1). Vyskytují se kdekoliv na kůži, typicky na trupu a končetinách. Na dlaních, ploskách, ve kštici a na sliznicích se nevyskytují. Skvrny café-au-lait jsou obvykle oválného tvaru, s vlnitými ostře ohraničenými okraji, hladkého povrchu bez zvýšeného ochlupení. Barva skvrn je rovnoměrná, připomíná barvu bílé kávy, odstín však může být velmi variabilní, a to od světle hnědé až po tmavě hnědou v závislosti na fototypu. Po expozici UV záření se barva nemění. Velikost skvrn je u malých dětí obvykle 0,5–3 cm, skvrny se zvětšují proporcionálně s růstem těla, u dospělých mohou dosáhnout velikosti i více než 20 cm. CALM mohou být přítomny na kůži již při narození nebo se objevují v raném dětství. Během života většinou přibývají, nejvíce ve věku do 10 let. Skvrny café-au-lait nemají tendenci k malignímu zvratu [22].

Solitární kávová skvrna se nachází až u 36 % obyvatel [2], nález mnohočetných kávových skvrn je vzácný, objevuje se u řady syndromů. Přítomnost skvrn café-au-lait je jedním ze 7 diagnostických kritérií NF1 (tab. 2) a vyskytují se zhruba u 95 % pacientů [1]. Pro NF1 je významný nález 6 a více skvrn o průměru u prepubertálních osob 0,5 cm a více, u postpubertálních 1,5 cm a více, přičemž není přímá úměra mezi jejich počtem a závažností klinického průběhu [17, 22]. Kávové skvrny jsou obvykle prvním a často několik let jediným klinickým příznakem neurofibromatózy.

Dermatoskopický obraz se liší podle lokalizace [10]. Na obličeji nacházíme homogenní hnědé skvrny s perifolikulární hypopigmentací, na ostatních částech těla pouze nenápadnou hnědou pigmentovou síť (obr. 2).

Histologický nález je nepříznačný, neodlišitelný od pihy, pigmentace v axillách u neurofibromatózy (frecklingu) a chloazmatu. Epidermis má normální konfiguraci odpovídající věku, není akantotická ani atrofická. Množství melaninového pigmentu v keratinocytech je zvýšeno, zejména ve spodních vrstvách epidermis, někdy s přítomností makromelanozomů (obr. 3). Zřídka jsou nacházeny melanofágy v horních částech koria. Hyperpigmentace nebývá patrna v místech kožních adnex [19].

Elektronmikroskopicky může být oproti normální kůži počet melanocytů větší, bývají patrny velké melanozomy v melanocytech a keratinocytech, podobně jako u jiných melanocytových projevů [19].
Diferenciální diagnóza CALM zpravidla pro velmi typický vzhled nečiní potíže. Vzácně může být obtížné odlišení od pihy, lentiga, kongenitálního melanocytárního névu, Beckerova névu, névu spilu, pozánětlivé hyperpigmentace, lézí při urticaria pigmentosa či solitárního mastocytomu [22].
- Drobné skvrnité pigmentace v axillách a tříslech
Skvrnitá hyperpigmentace v axillách a tříslech, označovaná jako Croweho znamení či jako „freckling“, je specifickým diagnostickým znakem NF1 (obr. 4). Jedná se o makuly světle až středně hnědé barvy, velikosti 1–3 mm. Skvrny se obvykle vyskytují v místech minimální expozice UV záření, zejména v intertriginózní lokalizaci. Příčina jejich vzniku je nejasná, předpokládá se vliv zvýšeného tření, teploty a vlhkosti. Objevují se zpravidla ve věku 3–5 let v návaznosti na výsev skvrn café-au-lait (obvykle předchází vývoji neurofibromů). Freckling v axillách se nachází až u 90 % dospělých pacientů s NF1. Freckling může být lokalizován i na krku, okolo úst, na trupu a na perineu, v těchto lokalitách však není diagnostickým znakem [2].


- Generalizovaná difuzní hyperpigmentace
U pacientů s NF1 byla pozorována v pubertě a vyšším věku zvýšená bazální pigmentace na kůži při srovnání s jejich nepostiženými příbuznými. U segmentální neurofibromatózy jsou hyperpigmentované okrsky kůže ostře ohraničeny od nepostižené kůže.
- Pruritus
Dalším běžným příznakem NF1 je generalizovaný pruritus. Patogeneze je nejasná, pruritus by pravděpodobně mohl být způsoben zvýšeným počtem mastocytů v neurofibromech. Tyto mastocyty uvolňují histamin způsobující pocit svědění [2].
Kožní choroby sdružené s NF1
Juvenilní xantogranulom (JXG) je benigní histiocytóza kůže, sliznic a očí u dětí, jejíž etiologie a patogeneze dosud nejsou objasněny. U nemocných s NF1 byl popsán současný výskyt JXG a velmi vzácné juvenilní myelomonocytární leukémie (JMML) [2].
Glomus tumor je vzácný benigní nádor glomových buněk, které se vyskytují v glomových tělíscích v retikulární vrstvě dermis, zejména v oblasti nehtových lůžek. Jedná se o hojně inervované buňky hladkého svalu zodpovědné za regulaci krevního průtoku. Klinicky nacházíme modročervené makuly či papuly pod nehtovou ploténkou, projevují se záchvatovitou bolestivostí postiženého prstu, kterou lze vyprovokovat chladem a stlačením nehtové ploténky. Solitární glomus tumory se vyskytují obvykle po traumatu nehtového aparátu. Mnohočetné glomus tumory bez vazby na úraz byly popsány u řady dědičných onemocnění včetně neurofibromatózy.
Vztah melanomu a neurofibromatózy je kontroverzní, různé studie dokumentují přítomnost kožního a mimokožního melanomu u pacientů s NF1 s frekvencí výskytu 0,12–5,4 % [2]. U kožního melanomu pacientů s NF1 oproti melanomům v kontrolní skupině převládaly ženy, melanom se vyskytl v nižších věkových skupinách a u nemocných byly v době diagnózy stanoveny vyšší hodnoty indexu podle Breslowa [2].
Naevus anaemicus je vrozené ostře ohraničené bílé ložisko nepravidelného tvaru na trupu nebo končetinách. Bledost ložiska je způsobena zvýšenou citlivostí cévní stěny ke katecholaminům, po mechanickém podráždění nezčervená. Asociace NF1 a naevus anaemicus je sporná, vzhledem k nevýznamnosti dosud nebyly provedeny klinické studie.
B. Mimokožní projevy NF1
U NF1 se setkáváme se širokou škálou klinických příznaků, může být postižen takřka každý orgánový systém. Tabulka 3 uvádí přehled mimokožních projevů NF1.
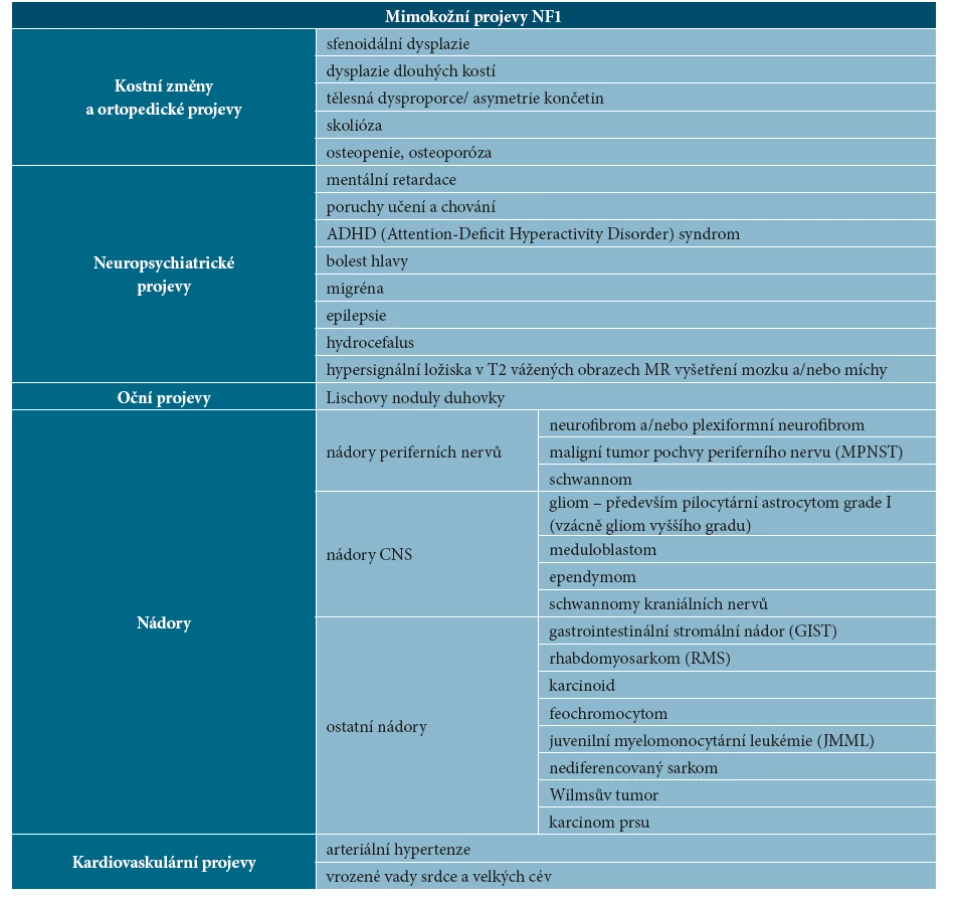
- Kostní změny a ortopedické projevy
Kostní abnormality u NF1 jsou různorodé. Byla popsána sfenoidální dysplazie, dysplazie dlouhých kostí, skolióza, osteopenie a osteoporóza. Patogenetické mechanismy nejsou zatím dostatečně vysvětleny. Předpokládá se nerovnováha způsobená deficitem osteoblastů, osteoklasty mají naopak prodloužené přežívání, což vede pravděpodobně ke zvýšené degradaci kostní tkáně [2].
Sfenoidální dysplazie je vrozený jednostranný defekt stěny orbity a sfenoidální kosti. Může být doprovázen herniací mozkové tkáně do orbity s následným exoftalmem [21, 26]. Je diagnostickým znakem neurofibromatózy, snadno odhalitelným na CT nebo MR hlavy.
Dalším častým nálezem je dysplazie dlouhých kostí, postihuje až 14 % nemocných [2]. První projevy jsou detekovatelné zobrazovacími metodami již v prvním roce života. Jedná se o kongenitální pseudoartrózy postihující nejčastěji tibii, jenž bývá často vychýlena anterolaterálně. Ztenčení kortikalis dlouhých kostí vede ke vzniku patologických fraktur s obtížným hojením.
Mezi další projevy NF1 patří tělesná disproporce – malá postava, nadměrný růst některých kostí [17], makrocefalie, prominující čelo a obočí, asymetrie končetin [13]. Tělesná asymetrie může být u nemocných s NF1 způsobena kromě kostních abnormalit také zmnožením měkkých tkání, zejména v souvislosti s přítomností neurofibromů (viz dále).
Skolióza je nejčastějším ortopedickým nálezem u NF1, vyskytuje se až u 10 % pacientů. Přes velmi častý výskyt skolióza nepatří mezi diagnostická kritéria. Obvykle se manifestuje ve věku kolem 10 let, patogeneze je nejasná. Častá je rychlá progrese s nutností rehabilitační či operační terapie [7, 13, 18]. Při zachycení skoliózy během fyzikálního vyšetření je třeba odeslat pacienta k ortopedickému vyšetření.
- Neuropsychiatrické projevy
Mentální retardace bývá obvykle lehká, postihuje méně než 10 % nemocných s NF1. Téměř u poloviny pacientů s NF1 byly pozorovány poruchy učení, zejména obtíže s matematikou a čtením. Byly také zaznamenány časté poruchy chování a vysoká četnost ADHD (Attention-Deficit Hyperactivity Disorder) syndromu [13]. Bolest hlavy je u dětí s NF1 častá a obvykle se rozvíjí bez organické příčiny (bývá způsobena blokem krční páteře). Migréna se objevuje až u 25 % dětí s NF1. Epilepsie se vyskytuje u 7 % dětí s NF1, hydrocefalus u 9 %, obě komplikace bývají projevem expanzivního procesu CNS, méně často bývá příčinou hydrocefalu stenóza distální části mokovodu [18].
Hypersignální ložiska CNS – až u 85 % dětí s NF1 byla zjištěna hypersignální ložiska v T2 vážených obrazech na MR zobrazení mozku a míchy. Ložiska jsou označována jako FASI (Foci of Abnormal Signal Intensity) nebo UBO (Unidentified Bright Object). Někdy jsou nesprávně označována jako hamartomy, protože podle současných znalostí se pravděpodobně jedná o vakuolární změny myelinu [18]. FASI/UBO se nejčastěji nachází v oblasti bílé hmoty mozkových hemisfér nebo bazálních ganglií (obr. 5). Obvykle mírně rostou a přibývají v prvních letech života, v dospělosti výrazně regredují. Z hlediska rozvoje ložiskového neurologického nálezu jsou asymptomatická, ale bývají dávána do souvislosti s častým výskytem poruch učení, soustředění a chování. Ložiska nesmí být zaměněna za nádory a neměla by být chirurgicky řešena.

- Oční projevy
Typickými očními projevy NF1 jsou Lischovy noduly duhovky, méně často se vyskytují glaukom, trofické změny papil optického nervu nebo jejich městnání, okohybné poruchy, ptóza a hypertelorismus.
Lischovy noduly jsou drobné polokulovité hamartomy na oční duhovce, dobře patrné při vyšetření štěrbinovou lampou. Zpočátku jsou světlé, s věkem tmavnou a přibývají. Objevují se v dětství (ale vyskytují se jen u malého procenta dětí do 6 let věku), u dospělých s neurofibromatózou typu 1 se vyskytují u 95 % nemocných (obr. 6a, 6b). Lischovy noduly nezpůsobují poruchy zraku, patří mezi diagnostická kritéria NF1. Pacienti s nejasnou diagnózou NF1 a rodinní příslušníci nemocných NF1 by měli být odesláni k očnímu vyšetření se zaměřením na vízus (někteří autoři doporučují i vyšetření barevného vidění), oční pozadí a k vyšetření předního segmentu oka štěrbinovou lampou na přítomnost Lischových nodulů [17].


- Nádory
Pacienti s NF1 mají zvýšené riziko vzniku benigních i maligních nádorů. Jednoznačně převažují benigní nádory periferních nervů, mezi které patří neurofibromy, plexiformní neurofibromy a schwannomy. Z benigních nádorů CNS se nejčastěji vyskytuje pilocytární astrocytom zrakové dráhy, především optického nervu a chiasmatu, méně často se vyskytují gliomy mozkového kmene a mozečku a vzácně ependymom. Ze zhoubných nádorů to jsou gliomy vyššího gradu a je uváděn také meduloblastom. Nejčastějšími zhoubnými nádory u NF1 jsou maligní nádory pochev periferních nervů (malignant peripheral nerve sheath tumor, MPNST). Dalšími malignitami popisovanými u NF1 jsou gastrointestinální stromální nádory (GIST), rhabdomyosarkom (RMS), karcinoid, feochromocytom, hematologické malignity (např. vzácná juvenilní myelomonocytární leukémie – JMML), nediferencovaný sarkom, Wilmsův tumor a karcinom prsu. Méně často se vyskytuje malobuněčný karcinom plic, kolorektální karcinom a karcinom rekta, ovariální karcinomy, myelodysplastický syndrom a anaplastický astrocytom [16]. U nemocných s NF1 je 2,7krát vyšší riziko zhoubných nádorů oproti zdravé populaci, zhoubné nádory se vyskytují v nižších věkových skupinách a někdy mají horší prognózu. Zhoubný nádor je nejčastější příčina úmrtí u nemocných s NF1, zkracuje délku života o 10–15 let. Rozdílná mortalita ve věkových skupinách je podmíněna rozdílnou incidencí malignit – mortalita je mnohem vyšší u nemocných do 40 let než ve skupině nad 40 let [16].
Nádory periferních nervů
Neurofibrom je benigní nádor pochvy periferního nervu vycházející ze Schwannových buněk, fibroblastů a mastocytů. Rozlišují se 4 typy neurofibromů.
Kožní neurofibromy postihují většinu nemocných s NF1. Vyvíjí se obvykle před pubertou, během puberty a v těhotenství. Jsou měkké, postupně se vyklenují nad úroveň kůže, až se stávají pendulujícími. Jejich počet značně kolísá od několika až po desítky až stovky (obr. 7). Nemají tendenci k malignímu zvratu, často však zadrhávají o oděv, způsobují nemocným kosmetické potíže a někdy svědí.

Subkutánní neurofibromy jsou hlouběji uložené v dermis, hmatatelné, obrůstají podkožní nervy. Často bývají bolestivé, maligní zvrat je vzácný [22].
Nodulární neurofibrom může vyrůstat z periferních nervů v různých lokalitách. V závislosti na postiženém nervu se mohou v jeho inervační zóně vyskytovat bolesti, parestezie, vzácně i senzitivní či motorický deficit [26]. Vyskytují se i spinální neurofibromy vyrůstající z nervových kořenů, které mohou být solitární nebo mnohočetné (obr. 8).

Plexiformní neurofibrom vytváří rozsáhlé tumory postihující dlouhé úseky nervů. Jsou obvykle přítomny již při narození, jejich růst je nepředvídatelný, někdy dosahují značné velikosti. Mohou být klinicky němé až do dospělosti, někdy bývá na kožním povrchu přítomna hyperpigmentace (obr. 9) a hypertrichóza. Klinicky mohou být zaměněny za kongenitální melanocytární névus [2]. Hrozí u nich riziko maligního zvratu [22].

Neurofibromy se mohou vyskytovat také u osob bez diagnózy NF1. Mikroskopický obraz neurofibromu je charakteristický, průřez kožním neurofibromem znázorňují obrázky 10 a 11.


Nález dvou a více neurofibromů nebo alespoň jednoho plexiformního neurofibromu patří mezi diagnostická kritéria NF1.
Maligní tumor pochvy periferního nervu (MPNST), bývá někdy označován jako maligní schwannom, neurofibrosarkom, neurogenní sarkom nebo anaplastický neurofibrom. Vyskytuje se kdekoliv na těle, nejčastěji na proximálních částech končetin a trupu [16]. Celoživotní riziko vzniku MPNST u pacientů s NF1 je 8–13% (oproti 0,001% v ostatní populaci), nejčastěji k němu dochází ve věku 20 až 35 let [6]. Deset až padesát procent MPNST vzniká maligním zvratem plexiformního neurofibromu nebo hlouběji uloženého neurofibromu, nikdy ne z kožního neurofibromu, ostatní MPNST vznikají de novo [16]. U dětí se MPNST objevuje velmi vzácně, a to především v době dospívání [18]. Příznakem je klidová bolest a bolest narušující spánek, obvyklá je změna konzistence plexiformního neurofibromu z měkké na tvrdou a rychlý růst, může se nově objevit neurologický deficit. Odlišení MPNST od plexiformního neurofibromu může být problematické, někteří autoři doporučují použití pozitronové emisní tomografie s fluorodeoxyglukózou (FDG-PET) [16].
Schwannom nazývaný též neurilemom či neurinom je benigní nádor vycházející ze Schwannových buněk pochvy periferního nervu. Schwannomy rostou zpravidla pomalu, jen velmi vzácně se stávají maligními. Vyskytují se kromě NF1 zejména u NF2 a schwannomatózy, mohou však být i sporadické u pacientů bez neurofibromatózy.
Nádory CNS
Z nádorů CNS u NF1 jsou nejčastější gliomy, vyskytující se kromě zrakové dráhy a chiasmatu také v oblasti mozečku a mozkového kmene. Méně častými nádory popisovanými u NF1 jsou meduloblastom, ependymom a schwannomy kraniálních nervů. Neurologické vyšetření u pacienta s NF1 by mělo být provedeno jedenkrát ročně. Při neobvyklých neurologických příznacích ihned, zejména při poruchách čití, motorickém výpadku, poruchách koordinace a sfinkterových potížích, neboť mohou být projevem nitrolebního nebo míšního procesu. Bolesti hlavy, zvracení a poruchy vědomí mohou být způsobeny zvýšeným nitrolebním tlakem a musí být důvodem urgentního vyšetření neurologem [6].
Gliom optiku (optic pathway glioma, OPG) je charakteristickým nálezem pro NF1. Jedná se o benigní nádor zrakové dráhy, především optického nervu (obr. 12) či chiasmatu. OPG patří mezi pilocytární astrocytomy WHO grade 1. Výskyt gliomu optiku je nejčastěji udáván u 15–20 % nemocných NF1 [7, 13], ale někteří autoři udávají nález OPG až u 27 % pacientů s NF1 [5]. Oboustranné gliomy optiku jsou popisovány pouze u diagnózy NF1 [5, 18]. OPG mohou být dlouhodobě nebo i trvale stacionární nebo pomalu rostoucí. Jsou v polovině případů asymptomatické nebo se manifestují exoftalmem, poruchou vízu, perimetru, ptózou a okohybnými poruchami. Gliom chiasmatu tlakem na hypotalamus ovlivňuje hypotalamo-hypofyzárně-gonadální osu, což může být příčinou zrychleného růstu nebo předčasné puberty. Symptomatické gliomy optiku se objevují zpravidla do 6 let věku, nejvíce gliomů je diagnostikováno ve věku 3 let [2]. Gliom optiku je významným nálezem, pro který je třeba provádět u nemocných s NF1 oční vyšetření v prvních šesti letech života alespoň jednou za 6 měsíců a později minimálně jedenkrát za rok [6, 18]. Rutinní screening magnetickou rezonancí k vyhledávání gliomů u všech asymptomatických pacientů s NF1 se podle některých autorů neukázal být přínosný [6].

Juvenilní myelomonocytární leukémie (JMML) je vzácným typem leukémie, postihuje především kojence a malé děti. Řadí se mezi myelodysplasticko-myeloproliferativní onemocnění (MDS/MPD) se závažnou prognózou. Jedinou léčbou JMML je alogenní transplantace kmenových buněk, bez transplantace je medián přežití asi 1 rok. U dětí s NF 1 bylo zjištěno zvýšené riziko juvenilní myelomonocytární leukémie 200–500krát oproti běžné populaci [2]. Je proto doporučována zvýšená pozornost při sledování možných příznaků JMML (náchylnost k infekcím, hepatosplenomegalie, lymfadenopatie, bledost, petechie, leukocytóza v periferní krvi obvykle 20–30 × 109/l s monocytózou), avšak rutinní screening krevního obrazu a diferenciálního počtu leukocytů u všech pacientů s NF1 se jeví podle provedených studií nepřínosný [2].
Kardiovaskulární projevy
U pacientů s NF1 je častá arteriální hypertenze. Obvykle vzniká na podkladě fibromuskulární dysplazie, která způsobuje pomalý rozvoj stenózy některých tepen, u NF1 nejčastěji renálních tepen. Arteriální hypertenze může být i projevem feochromocytomu, který se vyskytuje u 1–4 % dospělých s NF1 [17]. Popsány byly i vrozené vady srdce a velkých cév, zejména stenóza plicnice a aorty. Velmi vzácně se objevuje postižení mozkových tepen charakteru moya-moya syndromu, jehož rozvoj je u NF1 vázán na předchozí radioterapii nádoru mozku (především gliomu chiasmatu).
NEUROFIBROMATÓZA TYPU 1 V TĚHOTENSTVÍ
Během těhotenství může dojít k růstu a zvýšení počtu neurofibromů [7, 13]. Mezi rizikové patří spinální neurofibromy, které mohou svým růstem do páteřního kanálu způsobit míšní kompresi. Pánevní neurofibromy mohou komplikovat vaginální porod [6].
DIAGNOSTIKA
Diagnóza NF1 je stanovena především na základě hodnocení klinického obrazu. Diagnostická kritéria byla vytvořena na National Institute of Health Consensus Development Conference v r. 1987 [15], pro stanovení diagnózy NF1 je nutná přítomnost 2 nebo více diagnostických kritérií ze 7 možných (viz tab. 2).
Pro časné stanovení diagnózy NF1 u dětí do 3 let věku a pro pacienty s málo vyjádřenými příznaky je cenným diagnostickým znakem výskyt NF1 u příbuzného 1. stupně [6, 17, 18]. Oba rodiče by proto měli být vyšetřeni dermatologem se zhodnocením výskytu skvrn café-au-lait, frecklingu a kožních neurofibromů, dále očním lékařem k vyšetření předního segmentu oka na přítomnost Lischových nodulů. Biopsie skvrn café-au-lait a asymptomatických kožních neurofibromů se běžně neprovádí. Děti se šesti skvrnami café-au-lait bez dalších klinických projevů a bez rodinné anamnézy by měly být dále sledovány, 95 % z nich bude mít neurofibromatózu typu 1 [6].
DIFERENCIÁLNÍ DIAGNÓZA NÁLEZU MNOHOČETNÝCH SKVRN CAFÉ-AU-LAIT
Diagnostické příznaky NF1 se vyskytují v populaci celkem často. Například skvrny café-au-lait se na kůži nachází zhruba u 3–36 % obyvatel [2]. Je proto velmi důležité držet se striktně diagnostických kritérií, neboť nesprávně stanovená nebo nedostatečně podložená diagnóza může způsobit pacientovi a potažmo i jeho rodině značné potíže [18].
Mnohočetné skvrny café-au-lait na kůži mohou být příznakem řady onemocnění. Vyskytují se kromě neurofibromatózy u McCune-Albrightova syndromu, neurofibromatosis type 1-like syndromu (Legiova syndromu), tuberózní sklerózy, LEOPARD syndromu, Cowdenovy choroby, ataxia-teleangiectasia, Bloomova syndromu, mnohočetné endokrinní neoplazie 1 a 2B. Všechny tyto choroby mají ještě další kožní a mimokožní projevy [12]. Přehled nejčastějších onemocnění s výskytem skvrn café-au-lait zobrazuje tabulka 4.

VZÁCNÉ FORMY NF1
Segmentální neurofibromatóza
Jedná se o vzácnou formu neurofibromatózy s lokalizovanými projevy a negativní rodinnou anamnézou [2], dříve v Riccardiho klasifikaci označovaná jako NF5, dnes je považována za podtyp NF1. Vzniká postzygotickou mutací genu NF1, a dochází tak k somatickému mozaicismu. Závažnost klinického obrazu záleží na době vzniku mutace – časná mutace může vést k obrazu generalizované neurofibromatózy. Klinický obraz zpravidla postihuje pouze jeden segment, obvykle dermatom na trupu, končetinách nebo v obličeji, projevy však mohou být i oboustranné nebo ve více segmentech [8]. Projevy jsou velmi variabilní, vyvíjí se s věkem, v dětství se vyvíjí pigmentové změny a plexiformní neurofibromy, v dospělosti neurofibromy. Přenos onemocnění na potomky je možný, v případě gonadální mozaiky by mohl být potomek postižen i systémovou formou neurofibromatózy [8].
Gastrointestinální neurofibromatóza
Vzácná forma NF1, charakteristická nálezem mnohočetných neurofibromů v gastrointestinálním traktu, skvrn café-au-lait, frecklingu a Lischových nodulů. Objevuje se v dospělosti.
Familiární spinální neurofibromatóza (FSNF)
Je vzácnou formou NF1, charakteristickou vznikem mnohočetných spinálních neurofibromů. U nemocných se z diagnostických příznaků NF1 mohou nacházet skvrny café-au-lait, freckling a Lischovy noduly [20].
Mnohočetné skvrny café-au-lait
Bývají označovány jako multiple café-au-lait syndrome, familial café-au-lait spots (FCALS), multiple café-au-lait spots, dříve podle Riccardiho dělení byly označovány jako neurofibromatóza typu 6 (NF6). Jedná se o vzácnou formu neurofibromatózy, u které jsou přítomny pouze mnohočetné skvrny café-au-lait, není přítomen freckling, neurofibromy ani Lischovy noduly [12]. Tato diagnóza může být stanovena pouze v případě, pokud starší dítě nebo dospělý nemá přítomny další příznaky NF1 a v rodině je jasná anamnéza mnohočetných skvrn café-au-lait bez dalších příznaků NF1 [22]. Existence této varianty neurofibromatózy jako samostané formy je sporná [12). Rozhodující je výsledek molekulárně-genetického vyšetření NF1 genu.
Neurofibromatosis – Noonanové syndrom (NFNS)
O této jednotce se diskutovalo v 70.–90. letech 20. století jako o autozomálně dominantním onemocnění, jehož projevy zahrnují mnohočetné pigmentové névy a skvrny café-au-lait, nemocní mají světlé vlnité vlasy, hypertelorismus, krátkou postavu, pectus excavatum, vrozené srdeční vady, mentální retardaci, kryptorchismus, hypogonadismus a lymfedém dolních končetin [23]. Za příčinu „klasického“ syndromu Noonanové je t. č. považována mutace PTPN11 genu na 12. chromozomu (12q24.1). NFNS je nyní hodnocen jako alelický s NF1 a je u něj nalézána především mutace v NF1 genu nebo vzácně v PTPN11 genu [4]. Zajímavé je, že byli popsáni pacienti s NFNS, u nichž byla nalezena mutace jak v NF1 genu, tak v PTPN11 genu [25].
Watsonův syndrom
Watsonův syndrom byl v 80. letech 20. století popsán jako autozomálně dominantně dědičné onemocnění s přítomností skvrn café-au-lait, s nečetnými kožními neurofibromy, mentální retardací a pulmonální stenózou [6, 17]. Nyní je hodnocen jako alelický s NF1, tedy s mutací v NF1 genu.
ÚLOHA DERMATOLOGA, DOPORUČENÁ VYŠETŘENÍ A DALŠÍ SLEDOVÁNÍ PACIENTŮ S NF1
Dermatolog hraje zásadní roli v prvotním rozpoznání příznaků neurofibromatózy na základě pečlivého odebrání anamnézy a vyšetření kožních projevů pacienta. Při podezření na NF1 odesílá pacienta v místě bydliště k vyšetření dalších projevů (k očnímu a neurologickému vyšetření, eventuálně RTG dlouhých kostí).
Všichni pacienti s NF1 by měli být v ČR dispenzarizováni na specializovaném pracovišti pro neurokutánní onemocnění. V České republice zatím nebylo Ministerstvem zdravotnictví uznáno žádné centrum vysoce specializované péče o pacienty s neurokutánními syndromy. Česká dermatovenerologická společnost ČLS JEP, z. s., navrhla 2 dermatologická specializovaná centra pro vzácná onemocnění (VO), kam NF patří, ve FN Motol a FN Brno.
Rozvrh a rozsah potřebných kontrol u specialistů vyplývá z přítomnosti klinických projevů NF1 a rizika vzniku nádorů podle věkové skupiny. Pacienti by měli být sledováni v místě bydliště pediatrem či obvodním lékařem, oftalmologem, neurologem, dermatologem, podle potřeby dalšími specialisty (neurochirurgem, ortopedem). V případě potřeby se na doporučení specialistů provádí zobrazení mozku, míchy, vnitřních orgánů a měkkých tkání, metodou volby je magnetická rezonance (MR), často je používán i ultrazvuk. Úkolem spádového dermatologa je pravidelné sledování kožních projevů NF1. Rozpis doporučených vyšetření uvádí tabulka 5.

GENETICKÉ VYŠETŘENÍ A PORADENSTVÍ
Ověření klinické diagnózy na molekulárně-genetické úrovni by mělo být v současné době standardní součástí diagnostického procesu u všech pacientů s klinickými projevy NF1. Genetické vyšetření je indikováno také u případů, kdy potomek klinicky zdravých rodičů splňuje pouze 1 kritérium pro NF1. DNA analýza u NF1 prošla v posledních letech prudkým rozvojem. Molekulárně-genetická vyšetření se zrychlují a zpřesňují, mají možnost odhalit stále větší procento kauzálních mutací. Tabulka 6 shrnuje indikace vyšetření genu NF1.

Přímá DNA analýza slouží k identifikaci kauzální mutace genu NF1, k testování příbuzných nebo je využívána pro potřeby prenatální nebo preimplantační genetické diagnostiky (DNA analýza embrya, buněk CVS nebo amniocytů v plodové vodě). Na základě nalezené mutace však není možné předpovídat průběh onemocnění, klinické manifestace stejné mutace jsou rozdílné i mezi členy jedné rodiny.
Nepřímá DNA analýza genu NF1 se používá v rodinách se dvěma a více členy s NF1, u kterých nebyla nalezena kauzální mutace genu (neumožňuje detekci mutací de novo). Používá se k určení rizika dalších členů rodiny, u kterých se zatím neprojevily klinické příznaky, a v preimplantační diagnostice.
Rodinám s výskytem neurofibromatózy by mělo být nabídnuto genetické poradenství zejména v prekoncepční přípravě. Analýzou genu NF1 se v ČR v současné době zabývají 2 pracoviště – Ústav biologie a lékařské genetiky UK 2. LF a FN Motol Praha a Oddělení lékařské genetiky FN Brno [18].
TERAPIE
Léčba neurofibromatózy je symptomatická.
Pigmentové projevy obvykle nevyžadují léčbu, doporučuje se dodržování fotoprotekce. Skvrny café-au-lait mohou být odstraněny laserem, po ošetření jsou však časté recidivy. Lokální aplikace analogů vitaminu D3 podle některých klinických studií vedla k znatelnému vyblednutí frecklingu a skvrn café-au-lait [2], běžně se však nepoužívá.
Kožní neurofibromy se odstraňují, pokud činí potíže – jsou kosmeticky rušivé, zadrhávají o oděv, jsou bolestivé, rychle rostou nebo mají negativní vliv na okolní struktury. Menší léze mohou být odstraněny laserem či diatermokoagulací, větší neurofibromy se odstraňují chirurgicky.
Podkožní neurofibromy se obvykle neodstraňují.
Kožní a podkožní plexiformní neurofibromy by měly být široce excidovány plastickým chirurgem. Riziko hojení hypertrofickou jizvou a recidivy plexiformního neurofibromu je vysoké, u dětí s rozsáhlým plexiformním neurofibromem je riziko recidivy až 75% [18]. Nodulární a hlouběji uložené plexiformní neurofibromy postihující periferní nervy patří do rukou neurochirurga.
Léčba maligního nádoru pochvy periferního nervu (MPNST) je chirurgická a onkologická. Je nutná rozsáhlá excize nádoru do zdravé tkáně, často doprovázená vznikem neurologického deficitu z vytětí příslušného nervu s jeho širokým okolím, bývá nutná i amputace končetiny. Metastázy jsou časté, mnoho případů je navíc rezistentní vůči chemoterapii [22]. Prognóza MPNST je špatná, pětileté přežití od stanovení diagnózy MPNST je 21% u pacientů s NF1 oproti 42% u sporadických případů MPNST bez diagnózy NF1 [5]. MPNST je nejčastější příčinou úmrtí dospělých s diagnózou NF1 [8, 17]. Také léčba ostatních zhoubných nádorů patří do rukou onkologa a příslušného specialisty, obvykle zahrnuje chirurgické odstranění, méně častá je chemo - či radioterapie.
Od 60. let 20. století probíhá řada klinických studií s mnoha farmakologickými přípravky k prevenci růstu a k léčbě neurofibromů a plexiformních neurofibromů, avšak studie dosud nebyly úspěšné. Sirolimus (rapamycin) a jeho derivát everolimus, inhibitory proteinkinázy mTOR (mammalian target of rapamycin) byly slibné, ale u NF1 nebyly úspěšné, léky se jeví účinné pouze u tuberózní sklerózy [2]. Imatinib mesylát, tipifarnib a pirfenidon měly nežádoucí účinky a neměly u NF1 žádaný efekt. Kauzální terapie neurofibromů tedy zatím nebyla nalezena a jejich terapie nadále zůstává symptomatická. Riccardiho návrh na podávání ketotifenu k ovlivnění mastocytů a pruritu v plexiformním neurofibromu může u některých pacientů vést k subjektivní úlevě.
PROGNÓZA
Jen relativně malé procento nemocných neurofibromatózou typu 1 má obtíže, které snižují kvalitu života, vedou k invaliditě nebo jsou příčinou úmrtí. U pacientů s NF1 je zvýšená mortalita ve srovnání s celkovou populací, zejména ve věku 10–40 let, více u žen. Nejčastější příčinou úmrtí je přítomnost maligního nádoru, která vede ke zkrácení života o 10–15 let oproti běžné populaci [16].
NEUROFIBROMATÓZA TYPU 2
Bývá označována jako neurofibromatosis von Recklinghausen typ 2, NF2, dříve také centrální neurofibromatóza. NF2 je geneticky podmíněné onemocnění odlišné od NF1, zahrnuje neurologické a oční příznaky s minimálním kožním nálezem. Incidence je udávána 1 : 33 000–40 000 obyvatel. Dědičnost onemocnění je autozomálně dominantní, avšak asi 50 % případů vzniká mutací de novo. Gen pro NF2 je lokalizován na dlouhých raménkách chromozomu 22 (22q12.2), genovým produktem je schwannomin (méně často označován jako merlin či neurofibromin-2). Schwannomin působí obdobně jako neurofibromin jako nádorový supresor potlačením buněčného růstu.
Klinický obraz NF2 se významně liší od NF1, projevy onemocnění se zpravidla objevují později, až během druhé dekády života. Klinicky nejvýznamnějším symptomem je oboustranný vestibulární schwannom (také označovaný jako neurinom akustiku), který se vyskytuje takřka u všech nemocných a při oboustranném výskytu je pro NF2 diagnostickým příznakem. Projevuje se tinnitem a poruchou až ztrátou sluchu, v pokročilých stadiích příznaky hydrocefalu či útlakem mozkového kmene. Pro NF2 je charakteristický a současně diagnostický mnohočetný výskyt dalších benigních nádorů centrálního nervového systému – mozkových a míšních gliomů, ependymomů, meningeomů, schwannomů a neurofibromů periferních nervů. Aby nález těchto nádorů byl pro NF2 diagnostický, musejí být nalezeny vždy v počtu 2 a více (např. 2 meningeomy a více). Klinicky se tyto benigní nádory projevují podle lokalizace bolestmi hlavy, křečemi nebo ložiskovým neurologickým nálezem (mozečkovou symptomatologií, syndromem mosto-mozečkového koutu, rozvojem paraparézy atd.). Skvrny café-au-lait se vyskytují u 43 % pacientů s NF2, přičemž 6 a více skvrn má pouze 1 % nemocných, freckling zcela chybí. Kožní neurofibromy jsou přítomny pouze u zhruba 20 % nemocných. Lischovy noduly se u NF2 nevyskytují. Dalším diagnostickým příznakem NF2 je juvenilní subkapsulární katarakta. Při očním vyšetření lze dále nalézt retinální hamartomy a epiretinální membrány [18].
Diagnostická kritéria pro neurofibromatózu typu 2 byla stanovena na National Institute of Health Consensus Development Conference v roce 1990 (tab. 7). Ke stanovení diagnózy NF2 je třeba splnit jedno z kritérií, Evans et al. v roce 1999 rozšířili diagnostická kritéria o kritérium kalcifikace mozkové tkáně. Metodu volby ke stanovení diagnózy NF2 představuje MR mozku a/nebo míchy. Pro úspěšné zobrazení vestibulárních schwannomů na MR vyšetření je nutné podat kontrastní látku.
![Diagnostická kritéria neurofibromatózy typu 2 podle National Institute of Health Consensus Development Conference (1990) [14]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/f00d1fc3d0f806997abd48ba2a9e1a30.png)
Léčba NF2 je symptomatická, spočívá v chirurgickém odstranění nádorů, chemoterapii či radioterapii, včetně ozáření nádorů na Leksellově gama noži.
SCHWANNOMATÓZA
Podle Riccardiho klasifikace neurofibromatózy by odpovídala NF7, tento název se však v současnosti nepoužívá. Jedná se o autozomálně dominantně dědičné onemocnění s vysokým výskytem mutací de novo – až v 80–90 % případů. Zodpovědný gen je lokalizován na 22. chromozomu v těsné blízkosti genu pro NF2, genový produkt však dosud není znám.
V klinickém obraze dominuje nález mnohočetných schwannomů, narozdíl od NF2 u schwannomatózy nejsou přítomny vestibulární schwannomy ani další mnohočetné benigní nádory centrálního a periferního nervového systému. Schwannomatóza má až u jedné třetiny případů segmentální charakter, tj. schwannomy jsou lokalizovány pouze na jedné části těla.
Diagnostická kritéria schwannomatózy byla stanovena Children’s Tumor Foundation International Consensus Conference on Schwannomatosis v roce 2005. Věkový limit pro hodnocení kritérií je udáván v literatuře rozdílně, zde je uveden pro osoby starší než 30 let věku (tab. 8). Segmentální schwannomatóza je diagnostikována při splnění výše uvedených kritérií na jedné z končetin nebo paravertebrálně v oblasti 5 a méně na sebe navazujících obratlů páteře. V případě podezření na schwannomatózu je třeba provést genetické vyšetření k vyloučení mutace v NF2 genu. Onemocnění nemusí výrazněji ovlivňovat kvalitu života.
![Diagnostická kritéria schwannomatózy podle Children’s Tumor Foundation International Consensus Conference on Schwannomatosis (2005) [11]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/6b6f0f51e2ae59529703d777149fdef4.png)
Léčba patří na specializované neurochirurgické pracoviště a na pracoviště zaměřená na léčbu bolesti.
ZÁVĚR
Kožní projevy neurofibromatózy jsou klinicky snadno zjistitelné. K časnému stanovení diagnózy může významně přispět vyšetření dermatologem, proto je znalost klinického obrazu velmi potřebná. Při podezření na neurofibromatózu na základě zhodnocení kožního nálezu dermatolog odesílá pacienta do specializovaných ambulancí ke komplexnímu vyšetření a v případě potvrzení neurofibromatózy k následné dispenzarizaci. Časná diagnostika neurofibromatózy významně přispívá ke zvýšení kvality nebo dokonce k záchraně života pacienta. Navzdory současným znalostem neurofibromatózy jsou léčebné možnosti omezené. Určitou naději pro nemocné představují studie nových léků.
Poděkování
Děkujeme Patologickému oddělení Masarykovy nemocnice v Ústí nad Labem, o. z., za poskytnutí snímků histologických preparátů, dále Radiodiagnostickému oddělení Masarykovy nemocnice v Ústí nad Labem, o. z., a Klinice zobrazovacích metod 2. LF UK a FN Motol za poskytnutí MR snímků.
Do redakce došlo dne 15. 4. 2015.
Adresa pro korespondenci:
MUDr. Daniela Humhejová
Kožní oddělení
Masarykova nemocnice v Ústí nad Labem, o. z.
Krajská zdravotní, a. s.
Sociální péče 3316/12A
401 13 Ústí nad Labem
e-mail: daniela.humhejova@kzcr.eu
Zdroje
1. ANTONIO, J. R., GOLONI, B., TRIDICO, L. A. Neurofibromatosis: chronological history and current issues. An. Bras. Dermatol., 2013, 88 (3), p. 329–343.
2. BOYD, K. P., KORF, B. R., THEOS, A. Neurofibromatosis type 1. J. Am. Acad. Dermatol., 2009, 61 (1), p. 1–16.
3. Brems, H., Pasmant, E., Van Minkelen, R., Wimmer, K., Upadhyaya, M., Legius, E., Messiaen, L. Review and Update of SPRED1 Mutations Causing Legius Syndrome. Hum Mutat, 2012, 33, p. 1538–1546.
4. De Luca, A., Bottillo, I., Sarkozy, A., Carta, C., Neri, C., Bellacchio, E., Schirinzi, A., Conti, E., Zampino, G., Battaglia, A., Majore, S., Rinaldi, M. M., Carella, M., Marino, B., Pizzuti, A., Digilio, M. C., Tartaglia, M., Dallapiccola, B. NF1 gene mutations represent the major molecular event underlying neurofibromatosis-Noonan syndrome. Am. J. Hum. Genet., 2005, 77, p. 1092–1101.
5. Evans, D. G. R., Baser, M. E., McGaughran, J., Sharif, S., Howard, E., Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med. Genet., 2002, 39, p. 311–314.
6. FERNER, R., HUSON, S., THOMAS, N. et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J. Med. Genet., 2007, 44, p. 81–88.
7. Goldstein, J., Gutmann, D. Neurofibromatosis type 1. In Roach, E. S., Miller, V. S., ed. Neurocutaneous Disorders. Cambridge University Press, U. K., 2004, p. 42–49.
8. JEDLIČKOVÁ, H. Segmentální neurofibromatóza. Čes-slov Derm, 2009, 84, No. 3, s. 145–147.
9. Leisti, E. L. Radiologic findings of the head and spine in neurofibromatosis 1 (NF1) in Northern Finland. Academic Dissertation. Oulu, Finland, University of Oulu and Oulu Universtity Hospital, 2003.
10. LUK, D. C., LAM, S. Y., CHEUNG, P. C., CHAN, B. H. Dermoscopy for common skin problems in Chinese children using a novel Hong Kong-made dermoscope. Hong Kong Med. J., 2014, 20 (6), p. 495–503.
11. MacCollin, M., Chiocca, E. A., Evans, D. G. R. et al. Diagnostic criteria for schwannomatosis. Neurology, 2005, 64, p. 1838–1845.
12. MADSON, J. Multiple or familial café-au-lait spots is neurofibromatosis type 6: Clarification of a diagnosis. Dermatol. Online J., 18 (5), 4.
13. Maria, B. L., Menkes, J. H. Neurocutaneous Syndromes. In Menkes, J. H., Sarnat, H. B., Maria, B. L. eds. Child Neurology, Philadelphia, Lippincott Williams Wilkins, 2005, p. 803–828.
14. Mulvihill, J. J., Parry, D. M., Sherman, J. L., Pikus, A., Kaiser-Kupfer, M. I., Eldridge, R. N. I. H Conference. Neurofibromatosis 1 (Recklinghausen disease) and neurofibromatosis 2 (bilateral acoustic neurofibromatosis). An update. Ann. Intern. Med., 1990, 113, p. 39–52.
15. National Institute of Health Consensus Development Conference. Neurofibromatosis: Conference Statement. Arch. Neurol. Chicago, 1988, 45, p. 575–578.
16. PATIL, S., CHAMBERLAIN, R. S. Neoplasms Associated with Germline and Somatic NF1 Gene Mutations. The Oncologist, 2012, 17, p. 101–116.
17. PETRÁK, B., BENDOVÁ, Š., LISÝ, J., KRAUS, J., ZATRAPA, T., GLOMBOVÁ, M., ZÁMEČNÍK, J. Neurofibromatosis von Recklinghausen typ 1 (NF1) – klinický obraz a molekulárně-genetická diagnostika. Cesk. Patol., 2015, 51 (1), s. 34–40.
18. PETRÁK, B., PLEVOVÁ, P., NOVOTNÝ, J., FORETOVÁ, L. Neurofibromatotis von Recklinghausen. Klin. Onkol., 2009, 22 (Suppl), s. 38–44.
19. PIZINGER, K. Kožní pigmentové projevy. 1. vyd. Praha: Grada Publishing, 2003, s. 31–32, ISBN 80-247-0616-4.
20. RUGGIERI, M. The different forms of neurofibromatosis. Child’s Nerv. Syst., 1999, 15, p. 295–308.
21. SEIDL, Z., VANĚČKOVÁ, M. Neuroradiologické vyšetřovací metody v diagnostice a jejich přínos pro další klinické vedení nemocných s neurokutánním onemocněním (fakomatózy, neurovývojová onemocnění). Neurol. Praxi, 2004, 2, s. 85–90.
22. SHAH, K. N. The Diagnostic and Clinical Significance of Café-au-lait Macules. Pediatr. Clin. North. Am., 2010, 57 (5), p. 1131–1153.
23. SPITZ, J. Genodermatoses. A Clinical Guide to Genetic Skin Disorders, second edition: Philadelphia, Lippincott Williams Wilkins, 2004. p. 80–87, ISBN-13 : 978-0781740883.
24. ŠTORK., J. et al. Dermatovenerologie.1. vyd. Praha: Galén, 2008, s. 341, 364–365, 408, ISBN 978-80-7262-371-6.
25. Thiel, C., Wilken, M., Zenker, M., Sticht, H., Fahsold, R., Gusek-Schneider, G. C., Rauch, A. Independent NF1 and PTPN11 mutations in a family with neurofibromatosis-Noonan syndrome. Am. J. Med. Genet., 2009, 149A, p. 1263–1267.
26. WILLIAMS, V., LUCAS, J., BABCOCK, M., GUTMANN, D., KORF, B., MARIA, B. Neurofibromatosis Type 1 Revisited. Pediatrics, 2009, 123 (1), p. 124–133.
Štítky
Dermatologie Dětská dermatologieČlánek vyšel v časopise
Česko-slovenská dermatologie

2015 Číslo 3
- Ingenol-mebutát rozšiřuje možnosti léčby aktinické keratózy
- Nástroje k hodnocení závažnosti psoriázy v klinických studiích
- Proces hojení ran krok za krokem a co ho může zkomplikovat
Nejčtenější v tomto čísle
- Neurofibromatóza z pohledu dermatologa
- Klinický případ: erytém glans penis
- Latentná tuberkulózna infekcia a biologická liečba
- Agminátní histiocytomy
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy