
Klinický případ: Intertriginózní purpura
Autoři:
D. Kovandová 1; O. Kodet 1,2; J. Štork 1
Působiště autorů:
Dermatovenerologická klinika 1. LF UK a VFN
přednosta prof. MUDr. Jiří Štork, CSc.
1; Anatomický ústav 1. LF UK
přednosta prof. MUDr. Karel Smetana, DrSc.
2
Vyšlo v časopise:
Čes-slov Derm, 88, 2013, No. 2, p. 83-86
Kategorie:
Repetitorium
Pacientkou byla 72letá žena, v jejíž rodinné anamnéze matka zemřela na mnohočetný myelom a otec na ileus, jinak byla bez pozoruhodností. Z léků dlouhodobě užívala amiodarone hydrochlorid, spironolakton, furosemid, levothyroxin, diosmin s hesperidinem, gabapentin, omeprazol, rutosidum s ac. ascorbicum. Nemocná byla dlouhodobě léčena pro arteriální hypertenzi, ischemickou chorobu srdeční, hypotyreózu, polyneuoropatie dolních končetin, asi rok byla vyšetřována na revmatologii pro nediferencovanou polyartritidu. Před rokem byla provedena levostranná hemikolektomie pro enteroragii s histologickým nálezem segmentálních zánětlivých změn. Nález nebyl jednoznačně uzavřen, byla zvažována ischemická kolitida, divertikulóza či zánětlivý proces. Při vyšetření byla hospitalizována na interní klinice k celkovému přešetření pro hmotnostní úbytek 12 kg za poslední 3 měsíce, otoky dolních končetin, polykací obtíže, tvorbou erozí v dutině ústní a vznik makulózního exantému periorbitálně, pod prsy, v oblasti zevního genitálu a perianálně.
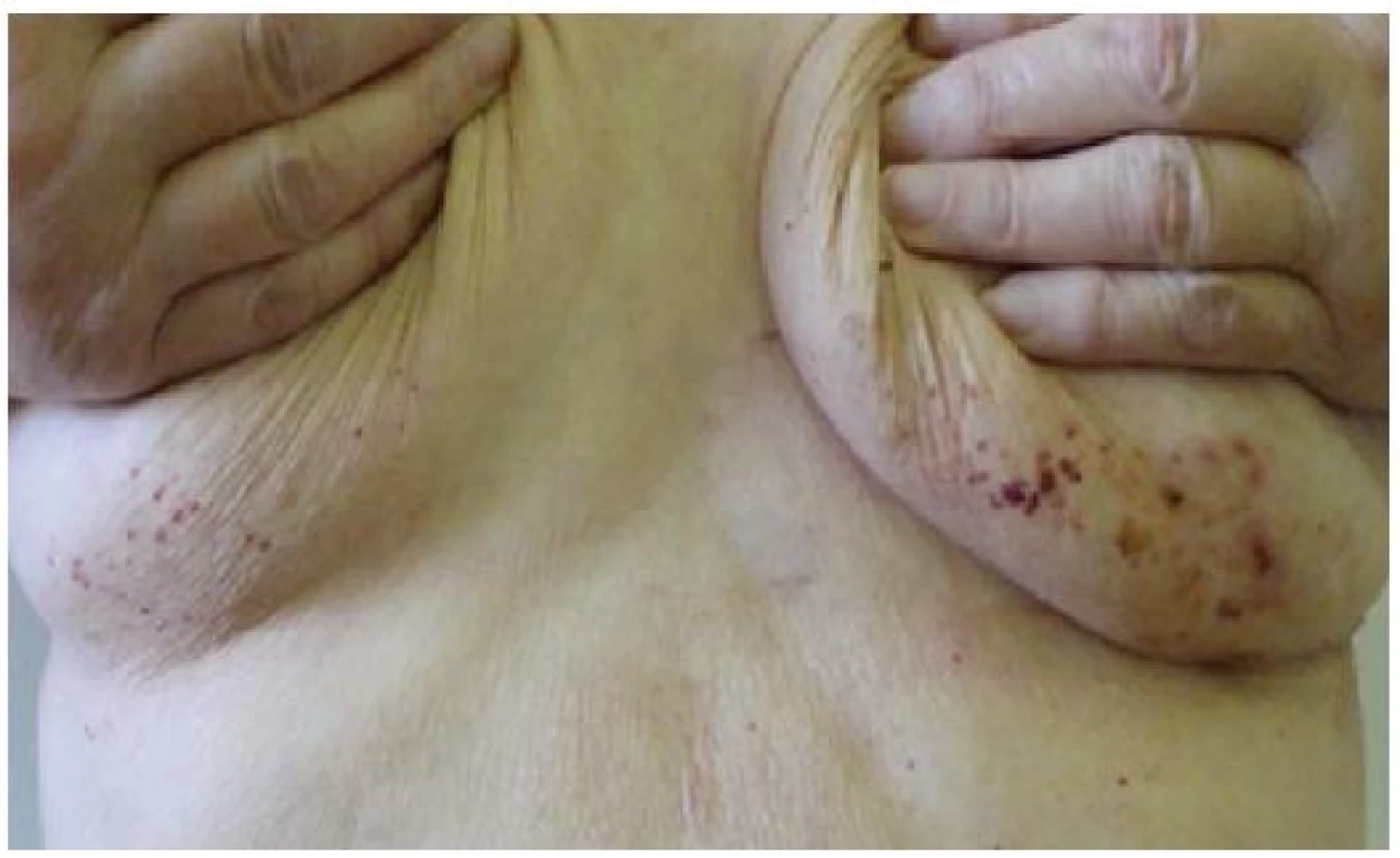
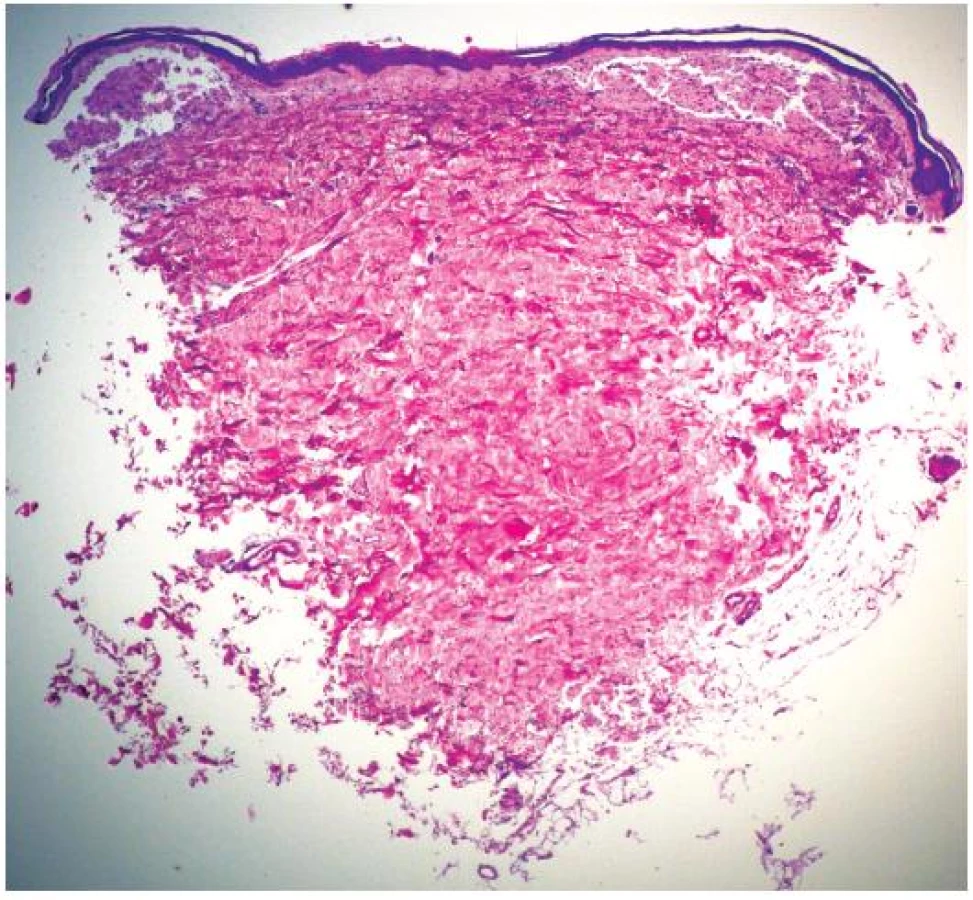
Při dermatologickém konziliu byl patrný nález purpury v oblasti pod prsy (obr. 1), v tříselné krajině, perianálně a v oblasti zevního genitálu. V oblasti víček byl patrný edém s purpurou (obr. 2). V rámci konziliárního vyšetření byla provedena biopsie kůže v oblasti pravého třísla (obr. 3, 4).


HISTOPATOLOGICKÝ NÁLEZ
Epidermis je mírně atrofická. Horní korium je místy vyplněno hrudkovitými eozinofilními hmotami s erytrocytárními extravazáty (obr. 3), které jsou přítomny i v části lalůčků podkožní tukové tkáně. Barvení metodou PAS a metodou Kongo červeně vykazuje pozitivitu stěn některých cév (obr. 4) a hrudkovitých hmot v horním koriu. Závěr: nález svědčí pro amyloidózu, nejspíše systémovou.
Provedené doporučené vyšetření krve imunoelektroforézou prokázalo monoklonální gamapatii IgG typ lambda v oblasti gama a Bence-Jonesovu bílkovinu typ lambda v oblasti beta1 globulinů.
ZÁVĚR
Amyloidóza kůže v rámci sekundární systémové amyloidózy při mnohočetném myelomu IgG typ lambda postihující též gastrointestinální trakt.
PRŮBĚH
Pacientka byla předána do péče hematoonkologie. Další vyšetření ozřejmila amyloidózu gastrointestinálního traktu, trepanobiopsie prokázala infiltraci kostní dřeně nezralým plazmocelulárním myelomem typu lambda (60–70%).
DISKUSE
Amyloidóza je soubor relativně vzácných onemocnění charakterizovaných extracelulárním ukládáním vláknitých proteinů, amyloidu, v intersticiu různých orgánů. Jako první popsal amyloid německý patolog Rudolf Virchow v roce 1854, který si povšiml ukládání bílkovinných substancí ve tkáních, které bylo spojeno s orgánovou dysfunkcí. Etiopatogeneze amyloidózy není zcela jednoznačná, pravděpodobně se nejedná jen o pasivní ukládání proteinů, ale prekurzorový protein je před vazbou v tkáni orgánů upravován buňkami imunitního systému – makrofágy.
Nativní buňky mají dva mechanismy k tvorbě proteinů. V prvním případě dochází k tvorbě proteinu v jednom celku, ve druhém případě jsou syntetizovány proteinové fragmenty, které se následně skládají do celku. Tyto proteiny, typické pro amyloid, se posléze mohou rozpadat do původních proteinových fragmentů, a tvořit tak fibrilární proteinové hmoty obtížně podléhající proteolýze [5]. Podle struktury fibril amyloidu bylo popsáno již více jak 20 různých proteinů tvořících amyloid. Nejčastějším typem amyloidu je AL amyloid, který je tvořen lehkými řetězci imunoglobulinů a jejich fragmenty. Tento typ nacházíme u monoklonálních gamapatií nebo u mnohočetného myelomu, kde se vyskytuje až v 15 % případů. Postihuje nejčastěji srdce, ledviny, gastrointestinální trakt, nervový systém a kůži. Dalším častým typem je AA amyloid, typický pro chronická zánětlivá onemocnění s nadprodukcí IL-1, IL-6 a TNF-alfa a následnou aktivací sekrece sérového amyloidového proteinu, který je nedokonale degradován v makrofázích a ukládán v játrech, ledvinách a slezině [11]. U lokalizovaných kožních amyloidóz jsou proteiny tvořící amyloidové fibrily degradované části keratinu [6].
Amyloidové fibrily mají společné fyzikálněchemické vlastnosti, nezávislé na biochemickém složení. Jednotlivé fibrily jsou pevné, lineární, různé délky, nevětvící se a jsou navzájem propletené do husté sítě. Pomocí rentgenové a infračervené krystalografie bylo prokázáno, že tyto fibrily prostorově zaujímají konfiguraci beta-skládaného listu, což může být jeden z hlavních důvodů odolnosti k proteolytickému trávení [6, 7]. Vedle vláknitých fibril se amyloid skládá také z nefibrilárních proteinů, označovaných jako komponenta P. Sérová složka tohoto amyloidového proteinu P napomáhá také k obraně proti proteolytickým enzymům a je zajímavé, že je přítomna i u zdravých jedinců [7]. Amyloid obsahuje i komponenty extracelulární matrix, jako jsou proteoglykany a glykosaminoglykany.
Rozeznáváme primární a sekundární lokalizovanou amyloidózu a systémovou amyloidózu se sekundárním kožním ukládáním amyloidu. Primární lokalizovaná kožní amyloidóza kůže má jen kožní postižení bez postižení vnitřních orgánů. Etiologie je většinou neznámá, může být však součástí familiárního výskytu. Největší výskyt je v Číně a Jižní Americe, naproti tomu v Evropě a Severní Americe je spíše vzácnější. Tento typ kožní amyloidózy se dává do souvislosti s chronickým třením, a tedy možným epidermálním poškozením [10].
Lichen amyloidosus představuje mnohočetné, tuhé, ploché nahnědlé papuly s lichenoidním leskem doprovázené výrazným svěděním ložisek s typickou lokalizací na bércích [3]. Amyloidosis maculosa tvoří makulózní projevy, neostře ohraničené, šedohnědé až namodralé barvy. Tato ložiska nalézáme spíše v oblasti zad, lopatek, případně na hrudníku [8]. Amyloidosis cutis nodularis (atrophicans) je vzácněji variantou postihující především ženy středního věku. Lze zde nalézt plošné léze nodulárního charakteru až s tvorbou infiltrátů s naznačenou povrchovou atrofií kůže. Tato ložiska postihují typicky končetiny a mají červenohnědé až žlutavé zbarvení [9]. Nodulární typ amyloidózy je spojen s lokálními depozity AL amyloidu a je někdy také označován za formu extramedulárního plazmocytomu. Zdrojem AL amyloidu jsou v tomto případě shluky plazmatických buněk klonálního původu [4, 9].
Sekundární depozita lokalizovaného amyloidu nacházíme jako průvodní nález u řady kožních nádorů, jako např. u bazaliomů, adnexálních nádorů, seboroických veruk, dále u solárních keratóz, pilomatrixom, dermatofibrom nebo i u intradermálních melancytárních névů [9]. Podobné lokalizované sekundární ukládání amyloidu můžeme pozorovat i u pacientů podstupujících PUVA terapii [1]. Množství amyloidu je u těchto lézí většinou klinicky nevýznamné, ale při akumulaci může tvořit diagnostické obtíže.
Systémové amyloidózy mohou doprovázet monoklonální gamapatie, mnohočetný myelom, nebo být přidružené u chronických systémových onemocnění. Systémové amyloidózy jsou součástí i tzv. primárních systémových amyloidóz a hereditárních amyloidóz [9].
Kožní postižení však nejčastěji nalézáme u sekundárních AL amyloidóz doprovázejících monoklonální gamapatie a mnohočetný myelom. Zde se kožní postižení popisuje asi v 15 % případů. AL amyloid je tvořen imunoglobuliny z lehkých řetězců typu lambda. Tento abnormální imunoglobulin z lehkých řetězců nalézáme rovněž v séru a moči postižených pacientů a obecně je označován jako tzv. Bence-Jonesova bílkovina. Jen část tohoto paraproteinu může tvořit po proteolýze fibrilární struktury, což vysvětluje relativně omezený výskyt amyloidu u pacientů s mnohočetným myelomem. Při klinickém postižení nejčastěji nalézáme nažloutlé lesklé, tuhé, hladké papuly, někdy až noduly, které jsou bez subjektivních příznaků. Křehkost cév vede ke krvácení do projevů a ke vzniku purpury, petechií a ekchymóz, výjimečně se mohou tvořit až hemoragické buly. Postižení nalézáme zpravidla na víčkách, periorbitálně, na krku, v oblasti axil, pod prsy, v tříselné krajině, v oblasti zevního genitálu a perianálně [9]. Na prstech rukou se může tvořit tvrdá kůže připomínající sklerodermii. V dutině ústní můžeme nalézat podobné projevy jako na kůži s nejčastějším postižením jazyka (makroglosie) a na bukální sliznici. Ve kštici můžeme nacházet lokalizovanou i difuzní alopecii. Nehty jsou postiženy spíše vzácněji, ale při postižení nehtové matrix se objevuje podélné proužkování, drolení a nadměrná křehkost. Z celkových příznaků dominuje únava, ubývání na hmotnosti, otoky končetin, dušnost či synkopy. Při postižení faryngu může být prvotním příznakem chrapot. Celkové příznaky jsou ale do značné míry ovlivněny případným orgánovým postižením. Jen z klinických příznaků je možné na systémovou amyloidózu pomyslet při klinické trias u pacientů, kde nalézáme současně syndrom karpálního tunelu, makroglosii a nález popsaných kožních či slizničních lézí.
Diagnostika amyloidózy se opírá především o histopatologické vyšetření, při kterém nacházíme eozinofilní amorfní hmoty uložené v dermálních papilách, ložiska amyloidu v retikulární dermis pak odpovídají papulózním až nodulárním projevům. Dále můžeme nacházet depozita ve stěně cév, což vysvětluje klinicky patrnou purpuru.
Stanovení amyloidu v tkáni se standardně provádí pomocí kongo červeně s indukcí dvojlomu a dichroismu v polarizovaném světle. Amyloid lze také znázornit pomocí metody PAS, nebo metylenové violeti. Při nižším zastoupení amyloidu ve tkáni mohou být tyto metody na klasických parafinových řezech nepříliš průkazné a lze použít i kryostatické řezy, kde je detekce amyloidu citlivější. Další možností je použití thioflavinu T při fluorescenční mikroskopii, kde však můžeme nalézat řadu falešně pozitivních výsledků, např. u onemocnění jako lichen ruber planus. Velmi obtížná je v praxi identifikace typu amyloidu pomocí imunohistochemické detekce za použití specifických protilátek. U primárně lokalizované kožní amyloidózy lze použít i monoklonální protilátky proti pancytokeratinu, a detekovat tak depozita degradovaného keratinu [2]. Optimálně by měl být amyloid vyšetřen biochemicky po extrakci nefixovaného vzorku, např. kryostatového řezu (ideálně po selekci od ostatních komponent mikrodisekcí) a podroben sekvenační nebo hmotnostní analýze. Při systémových amyloidózách lze detekovat prekurzorový paraprotein amyloidu v cirkulující krvi nebo v moči pomocí elektroforézy.
Léčba lokalizované formy amyloidózy je poměrně omezená a málo účinná. Lokálně aplikujeme kortikoidy v okluzi nebo intralezionálně, z dalších preparátů lze zmínit lokální aplikaci dimetylsulfoxidu či kalcipotriolu. Při subjektivních obtížích lze zvolit i chirurgickou excizi ložisek či ošetření pomocí laseru. Z celkových léků je popsán příznivý efekt acitretinu [3]. U systémových amyloidóz je nutná léčba základního onemocnění, po kompletním přešetření pacienta. Nejčastěji u monoklonálních gamapatií a mnohočetného myelomu se podává chemoterapie melfalan v kombinaci s kortikoidy nebo cyklofosfamidem, případně se provádí transplantace kostní dřeně. Při postižení srdce a ledvin je možné přistoupit k transplantaci těchto orgánů, což významně prodlužuje délku přežívání. Systémová amyloidóza není absolutní kontraindikací k transplantaci orgánů, i když postupně dochází k opětovnému poškození transplantovaného orgánu amyloidem. Dlouhodobá prognóza pacientů se systémovou amyloidózou je však vysoce nepříznivá [12].
Podpořeno projektem UK: SVV 264 510.
Do redakce došlo dne 1. 3. 2013.
Kontaktní adresa:
MUDr. Ondřej Kodet
Dermatovenerologická klinika 1. LF UK
U Nemocnice 2
128 08 Praha 2
e-mail: ondrej.kodet@fl1.cuni.cz
Zdroje
1. HASHIMOTO, K., KUMAKIRI, M. Colloid-amyloid bodies in PUVA-treated human psoriatic patients. J. Invest. Dermatol., 1979, 72, p. 70–80.
2. CHANG, Y. T., LIU, H. N., WANG, W. et al. A study of cytokeratin profiles in localized cutaneous amyloids. Arch. Dermatol. Res., 2004, 296, s. 83–88.
3. KOJANOVÁ, M., BÄUMELTOVÁ, I., ŠTORK, J. Lichen amyloidosus: příznivý účinek acitretinu. Čes-slov Derm, 2003, 78, 6, s. 239–243.
4. LOVE, W. E., MIEDLER, J. D., SMITH, M. K. et al. The spectrum of primary cutaneous nodular amyloidosis: two illustrative cases. J. Am. Acad. Dermatol., 2008, 58, s. 33–35.
5. MERLINI, G., SELDIN, D. C., GERTZ, M. A. Amyloidosis: pathogenesis and new therapeutic options. J. Clin. Oncol., 2011, 29, 14, s. 1924–1933.
6. MORRIS, K. L., RODGER, A., HICKS, M. R. et al. Exploring the sequence-structure relationship for amyloid peptides. ., 2013, 450, 2, s. 275–283.
7. PEPYS, M. B. Amyloidosis. Ann. Rev. Med., 2006, 57, s. 223–241.
8. POCK, L., DRLÍK, L., MIŇOVSKÁ, V. et al. Makulózní amyloidóza. Čes-slov Derm., 1999, 74, 5, s. 212–215.
9. POCK, L. Kožní amyloidózy. Čes-slov Derm, 82, 2007, 82, 5, s. 245–253.
10. SUMITRA, S., YESUDIAN, D. Friction amyloidosis a variant or an etiologic factor in amyloidosis cutis? Int. J. Dermatol., 1993, 32, s. 422–423.
11. TAMURA, H., HASEGAEA, K. A review for recent advances in AA amyloid research and therapeutic approach to AA amyloidosis complicating rheumatoid arthritis]. Nihon Rinsho Menęeki Gakki kaishi, 2009, 32, 1, s. 35–42.
12. WECHALEKAR, A. D., HAWKINS, P. N., GILLMORE, J. D. Perspectives in treatment of AL amyloidosis. Br. J. Haematol., 2008, 140, p. 365–377.
Štítky
Dermatologie Dětská dermatologieČlánek vyšel v časopise
Česko-slovenská dermatologie

2013 Číslo 2
- Nástroje k hodnocení závažnosti psoriázy v klinických studiích
- Systémová léčba atopické dermatitidy konečně i u dětí
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
Nejčtenější v tomto čísle
- Psoriáza u dětí
- Súčasné možnosti liečby infantilných hemangiómov
- Dermatoskopie usnadňuje diferenciální diagnózu lupus erythematodes chronicus discoides a keratosis actinica
-
Významné životní jubileum
Doc. MUDr. Marie Viktorinové, CSc.
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy