
Hypofosfatázia – biochemické a klinické prejavy, molekulovo-genetická podstata
Hypophosphatasia – biochemical and clinical manifestations, molecular enetic principles
Hypophosphatasia is a rare hereditary metabolic disorder accompanying deficit of tissue nonspecific serum alkaline phosphatase. The incidence of overt forms is estimated about 1 : 100000 live births. In the prenatal manifestation the disease may cause severe damage to the foetus with intrauterine death. In hildren here is efect of mineralization ith ickets signs and the subsequent hypercalcaemia a ypercalciuria may lead to death. In adults the main manifestation is osteomalacia, skeletal eformities and ractures, arly rthritis. In evere orms the heredity is autosomal ecessive ype. In mild orms the heredity may e ominant r ecessive. In two case reports we present clinical ourse of he disease n wo dult isters, where diagnosis of hypophosphatasia was first time onfirmed n Slovak population using olecular enetic methods.
Key words:
hypophosphatasia, alkaline phosphatase, osteomalacia, amino acid changes – T83, E174K.
Autoři:
Ilja Chandoga 1; Ján Futas 2; RNDr. Robert Petrovič, Ph.D. 2; Ján Chandoga 2
Působiště autorů:
II. ortopedicko-traumatologická klinika LF UK a N, Bratislava
1; Ústav lekárskej biológie, genetiky a linickej genetiky LF UK a N, Bratislava
2
Vyšlo v časopise:
Čas. Lék. čes. 2011; 150: 541-545
Kategorie:
Kazuistika
Souhrn
Hypofosfatázia je zriedkavé dedičné metabolické ochorenie podmienené deficitom aktivity sérovej tkanivovo nešpecifickej alkalickej fosfatázy. Výskyt manifestných foriem sa odhaduje na 1 : 100 000 živonarodených detí. Pri prenatálnej manifestácii ochorenia môže dôjsť k ťažkému intrauterínnemu poškodeniu plodu s erinatálnym úmrtím. U detí dochádza k oruchám mineralizácie s rachitickými príznakmi, následné zmeny charakteru hyperkalcémie a hyperkalciúrie môžu viesť až k úmrtiu. U dospelých je hlavným prejavom osteomalácia, deformity a raktúry skeletu, včasná artróza. U ťažkých foriem je dedičnosť autozómovorecesívne hotypu, u ľahších foriem môže byť dedičnosť dominantná aj recesívna.
V kazuistike uvádzame klinický obraz a priebeh ochorenia u dvoch dospelých sestier, u ktorých bola prvý krát v slovenskej populácii potvrdená hypofosfatázia pomocou molekulárno-genetických metód.
Kľúčové slová:
hypofosfatázia, alkalická fosfatáza, osteomalácia, zámeny aminokyselín – T83, E174K.
Úvod
Alkalická fosfatáza (ALP-ortofosformonoesterfosfohydroláza, EC3.1.3.1) je enzým kódovaný minimálne štvoricou génov. Tri izoenzýmy sú tkanivovo-špecifické: intestinálny, placentárny a enzým zárodočných buniek. Štvrtý enzým – tkanivovo nešpecifická alkalická fosfatáza – je prítomný takmer vo všetkých tkanivách, zvlášť v pečeni, kostnom tkanive a obličkách.
Funkcia enzýmu spočíva v hydrolýze fosfoesterov, prirodzenými substrátmi pre enzým sú fosfoetanolamín, pyridoxal-5-fosfát, pyrofosfát. Enzým prispieva k ineralizácii skeletu okrem zvýšenia lokálneho výskytu fosfátov hydrolýzou nukleotidfosfátov aj hydrolýzou inhibítoramineralizácie – pyrofosfátu (1).
Gén pre ALPL je lokalizovaný na prvom chromozóme v blasti 1p36.1-34. Je dlhý viac ako 50kb, má 12 exónov a óduje proteín s ĺžkou 507 aminokyselín. Doposiaľ bolo popísaných 204 mutácií génu (http://www.sesep.uvsq.fr/Database.html).
Korelácia medzi fenotypom a enotypom sa nedá dobre určiť, pretože väčšina pacientov sú zložení heterozygoti pre mutácie meniace zmysel. Medzi kaukazoidnými pacientmi je rozšírená mutácia E174K, ktorá má pôvod v everozápadnej Európe a utácia A23V, pravdepodobne germánskeho pôvodu.
Hypofosfatázia (deficit tkanivovo nešpecifickej alkalickej fosfatázy – ALP, TNSALP) je vrodené ochorenia spojivového tkaniva prejavujúce sa deformitami a oškodením pohybového aparátu. Výskyt manifestných foriem je približne 1 : 100 000 živonarodených detí (2).
Kazuistiky
Na našich pracoviskách boli diagnostikované dve pacientky v ríbuzenskom vzťahu (sestry) so zníženými aktivitami tkanivovo nešpecifickej alkalickej fosfatázy.
Kazuistika 1
U taršej zo sestier sa objavili prvé známky ochorenia vo veku 32 mesiacov (zmeny na dlhých kostiach a rebrách, predčasná strata mliečneho chrupu, nízka aktivita ALP). Opakovane boli namerané nízke hodnoty ALP (posledné vyšetrenie 0,06 μkat/l), bez zmien v odnotách vápnika a osfátov v ére a moči. V voch rokoch bola u acientky vykonaná kostná biopsia, histologicky bol preukázaný deficit ALP.
Od detstva boli u acientky prítomné kostné zmeny a mierne rastové zaostávanie. V účasnosti vo veku 42 rokov sú pacientky prítomné ťažké artrotické zmeny pravého členkového kĺbu, obe chodidlá sú valgózne deformované. Pacientka má chronické vertebrogénne ťažkosti s kalcifikátmi v igamentum longitudinale anterius, polytópne artralgie. V röntgenovom obraze kolien dominujú skoré artrotické zmeny III. stupňa s hypopláziou laterálnych kondylov femuru. Realizované bolo denzitometrické vyšetrenie s normálnym nálezom.
Kazuistika 2
Mladšej zo sestier bola diagnostikovaná hypofosfatázia vo veku štyroch mesiacov na základe rodinnej anamnézy, biochemických parametrov a klinických prejavov (obojstranná lobulárna pneumónia, celková hypotónia, mäkké záhlavie, naznačená Harrisonova ryha, hrubnuté osteochondrálne spojenia rebier). Opakovane boli namerané nízke aktivity ALP (posledné vyšetrenie 0,08 μkat/l). Vo veku 10 mesiacov bola u acientky vykonaná kraniotómia pre intrakraniálnu hypertenziu z ôvodu predčasnej kraniosynostózy.
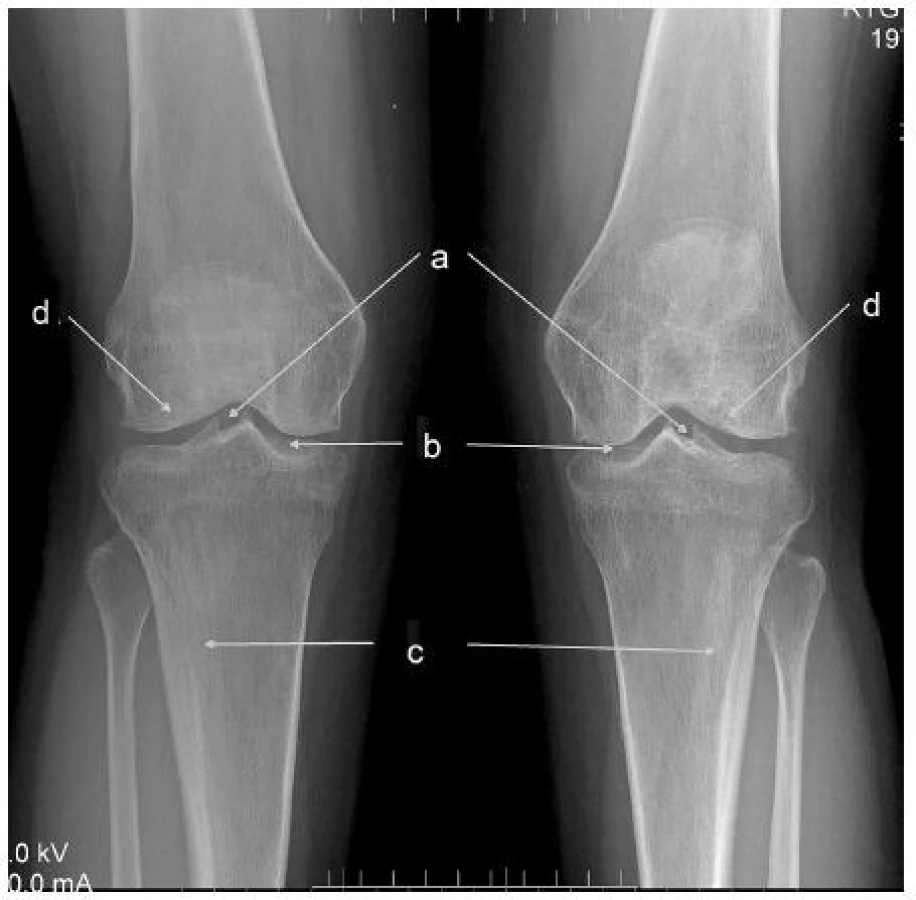
V účasnosti vo veku 32 rokov má mladšia pacientka vertebrogénne ťažkosti, bolesti oboch kolien a členkov s eformitami a artrotickými zmenami. Na röntgenovom vyšetrení aj na magnetickej rezonancii bola verifikovaná závažná osteochondrálna lézia laterálneho kondylu ľavého kolena (obr. 1). Pre výrazné klinické ťažkosti bola na tomto kolene vykonaná artroskopická operácia s dstránením deštruovaných a uvoľnených častí chrupavky. Prognóza je nepriaznivá.

V öntgenovom náleze sú zmeny typické pre hypofosfatáziu ešte markantnejšie ako u taršej sestry. V oblasti kolien je badateľné zahrotenie eminentia intercondylaris (depozity vápnika), typické kyjakovité rozšírenie metafýz tíbie. Obojstranne je prítomná hypoplázia laterálnych kondylov stehennej kosti, pokročilé artrotické zmeny. Vľavo je prítomný stav po nezvyčajnej únavovej zlomenine proximálnej fibuly (obr. 2). Oba členkové kĺby sú dysplastické, valgózne deformované (obr. 3).

Porovnanie
U oboch pacientiek bolo indikované molekulárno-genetické vyšetrenie. Genomická DNA bola izolovaná kolónkovou metódou zo vzorky periférnej krvi. Vzhľadom na veľký počet mutácii v géne ALPL, často prítomných v heterozygotnom stave, molekulárno-genetické vyšetrenie pacientov s podozrením na hypofosfatáziu vyžaduje sekvenčnú analýzu celej kódujúcej oblastí tohto génu. Pomocou polymerázovej reťazovej reakcie (PCR) sme amplifikovali všetkých 12 exónov génu ALPL, ktoré sme podrobili sekvenčnej analýze pomocou ABI PRISM Dye Terminatorkitu a kapilárnej elektroforézy na prístroji ABI 310. S výnimkou exónu 8 a 12 sekvencie primérov a podmienky amplifikácie boli rovnaké, ako sú uvedené v práci Morneta et al. (3).
U oboch vyšetrených sestier boli zistené totožné genetické zmeny – identifikované boli dve kauzálne mutácie (zložení heterozygoti). Prvá mutácia bola zistená v 5. exóne (c.299 C>T) a spôsobuje zámenu treonínu za metionín v pozícii 83 proteínového reťazca (T83M). Druhá mutácia sa nachádzala v 6. exóne (c.571 G>A) a spôsobuje zámenu glutámovej kyseliny za lyzín v pozícii 174 reťazca (E174K). Výsledok vyšetrenia je znázornený na obrázku 4. Mutácia v 5. exóne bola verifikovaná aj pomocou restrikčnej analýzy a výsledok je dokumentovaný na obrázku 5.


Liečba je u boch pacientiek symptomatická. Vychádza z ríležitostného podávania analgetík, užívania chondroprotektív, prechodný efekt mala aj viskosuplementačná intraartikulárna liečba kyselinou hyalurónovou. Vertebrogénne bolesti majú dobrú odozvu na rehabilitačnú liečbu a procedúry. Deformity členkov a ôh vyžadujú korekciu individuálnymi ortopedickými vložkami.
Diskusia
U oboch pacientiek boli zistené mutácie, ktoré podľa literárnych údajov majú kauzálny vzťah k hypofosfatázii. Mutáciu E174K ako prví popísali Henthorn a Whyte u troch pacientov v kombinácii s mutáciou D277A (4). V dvoch prípadoch (súrodenci) ochorenie sa manifestovalo ako detská forma. Rodinní príslušníci, ktorí mali iba jednu mutáciu, nemali žiadne príznaky ochorenia, išlo teda o autozómovorecesívny typ dedičností. U tretieho pacienta sa ochorenie manifestovalo ako adultná forma s fraktúrami dlhých kostí, hoci v anamnéze sa predsa objavili údaje o rachitíde v detstve. Hérasse et al. uviedli, že táto mutácia bola prítomná až u 1 % pacientov hodnoteného súboru kaukazoidnej populácie s miernou formou hypofosfatázie (5). Ide o mutáciu, ktorá vznikla pomerne dávno a rozšírila sa pomocou predkov zo severozápadnej Európy. Výskyt tejto mutácie v stredoeurópskom regióne nie je známy. Mutácia sa nachádza v oblastí proteínu (active site valley), v ktorej zmeny nemajú výrazný efekt na funkciu enzýmu a aminokyselinové substitúcie sú lepšie tolerované (mierny fenotyp). Mutácia T83M je zriedkavou a je asociovaná s ťažkým fenotypom, čo možno vysvetliť tým, že postihuje aktívne miesto enzýmu (6).
Klinický obraz tohto raritného ochorenia je veľmi variabilný, výrazne závislý od veku, v torom sa ochorenie manifestuje. V raxi pozorujeme, že pri zriedkavých metabolických ochoreniach dochádza k výšenému záchytu pacientov po rozšírení diagnostiky a výšení edukácie odbornej verejnosti. Aj v rípade hypofosfatázie očakávame, že skutočný počet pacientov v ašej populácii bude niekoľko desiatok. Posledné údaje odhadujú výskyt miernych foriem vrátane odontohypofosfatázie až 1 : 6000 (7).
Obvykle sa ochorenie člení do štyroch základných klinických foriem podľa vekovej manifestácie (perinatálna, infantilná, detská a dultná forma).
Aj napriek tomu, že najťažšou formou je perinatálna hypofosfatázia, nestretli sme sa zo strany pediatrov s podozrením požiadavkami na potvrdenie ochorenia. V omto veku ú prítomné ťažké deformity skeletu a vysoká letalita pre respiračné komplikácie z dôvodu rachitických deformít hrudníka a hypoplastických pľúc (2). Obdobne pri infantilnej forme, ktorá je definovaná manifestáciou do šiestich mesiacov, sa doposiaľ diagnostika nepožadovala. Ochorenia dávame do pozornosti neurológom, vzhľadom k omu, že u acientov dochádza k evnému zrastu lebečných švov a unkčnej kraniosynostóze. Hyperkalcémia s yperkalciúriou vedie k nefrokalcinóze a obličkovému zlyhaniu. Práve v omto veku sú markantne zvýšené hodnoty sérového fosfátu a alcia, ktoré môžu byť pri iných formách normálne. Pri diagnostike je užitočné vyšetrenie hladiny fosfoetanolamínu, jeho hodnoty sú v ére aj moči zvýšené (8). Toto vyšetrenie však nepatrí v ašich laboratóriach medzi rutínne, je nutné použiť chromatografické metódy.
Pri podozrení na hypofosfatáziu boli laboratórne vyšetrenia v inulosti dopĺňané invazívnou kostnou biopsiou. V dnešnej dobe sú k ispozícii molekulárno-genetické metódy, umožňujúce stanoviť diagnózu hypofosfatázie s rčením konkrétnej mutácie.
Na neskoršiu, detskú formu, by sa mala sústrediť hlavne pozornosť ortopédov, stomatológov. Prejavuje sa totiž predčasným vypadávaním mliečnych zubov pri zníženej tvorbe dentálneho cementu. Rachitída spôsobuje nízky vzrast, poruchy chôdze. Práve v omto veku môže byť klinický nález diskrétny a hodnoty enzýmu môžu byť mierne znížené až normálne. Ku skresleniu hodnôt a ch zvýšeniu dochádza najmä počas rastovej akcelerácie v puberte, ktorá nastupuje skôr u dievčat ako u chlapcov. Bežne sú akceptované referenčné hodnoty u ospelých 0,5–1,7 μkat/l a etí 1,3–5,8 μkat/l. Pri hypofosfatázii platí, že čím nižšie sú sérové aktivity ALP, tým je klinická forma ťažšia. V etskom veku je pri diferenciálnej diagnostike potrebné odlíšiť osteogenesis imperfecta, hypochondropláziu a achitídu (2). Do pozornosti dávame rádiologický nález rozšírenia metafýz v blasti kolien, zápästí a členkov – „kyjakovité metafýzy“. Typický je aj rádiologický nález jazykovitých zón prejasnenia od kĺbových štrbín smerom do metafýz (9).
Hypofosfatáziu môžu imitovať aj niektoré ojedinelé skeletálne dysplázie. Pri kleidokraniálnej dysplázii môže byť rítomný rádiologický a iochemický nález podobný hypofosfatázii (zvýšenie hladín fosfoetanolamínu v ére a moči). Znížené hodnoty ALP sú pravdepodobne následok zníženej expresie génu pre alkalickú fosfatázu u ýchto pacientov (10).
Dospelá forma sa obvykle sa prejaví v strednom veku a pacienti často ako jediný anamnestický údaj uvádzajú predčasnú stratu zubov. Táto forma môže prebiehať pod obrazom osteomalácie s bolesťami dolných končatín, chrbtice, proximálnou svalovou slabosťou. Často dochádza ku stresovým pseudofraktúram, najčastejšie metatarzálnym a emorálnym, s typickým nálezom tzv. Looserových zón na laterálnej ploche femuru. Artralgie sú spôsobené kryštálmi kalcium pyrofosfátu. Na odlíšenie je dôležitý nález zvýšených hodnôt alkalickej fosfatázy a sekundárny hyperparatyreoidizmus pri rachitídide a steomalácii iného pôvodu (11). Hladiny fosfoetanolamínu bývajú pri iných osteomaláciach v orme (12). Hlavne u dospelých pacientov s iskrétnym priebehom a mierne zníženými až hraničnými hodnotami alkalickej fosfatázy je možné sa stretnúť s epovšimnutím si tohto nálezu. Klinický rutinný prístup sa vyznačuje tendenciou nepovažovať nízke hodnoty enzýmov za patologické.
Navyše ú ešte definované dve osobitné formy. Pri odontohypofosfatázii sú prítomné len dentálne prejavy. Pseudohypofosfatázia je osobitná a xtrémne zriedkavá forma s ríznakmi infantilného typu avšak s ormálnym laboratórnym nálezom.
Možnosti liečby hypofosfatácie sú závislé od vekovej manifestácie, závažnosti ochorenia a onkrétnych klinických prejavov. Liečba zinkom a agnéziom (katalyzujúce ióny enzýmu) ako aj pyridoxal-5-fosfátom neprináša žiadaný efekt. Zmiernenie prejavov ochorenia je možné dosiahnuť nízkofosfátovou diétou (2). Výraznú opatrnosť je nutné venovať snahám o iečbu vitamínmi skupiny D a ápnikom. Tá je odporúčaná len pri ich dokázanom nedostatku pre zvýšené riziko tvorby obličkových kameňov. U ospelých pacientov nie je vhodná inak štandardná liečba osteoporózy bisfosfonátmi, ktoré enzýmu ešte znižujú. V osledných rokoch bol popisovaný prínos liečby teriparatidom v rípade výskytu patologických fraktúr u ospelých (13). Táto liečba u našich acientiek nebola osteológom indikovaná, vzhľadom k ch aktuálnej neprítomnosti. Otázne je, či by ich aplikácia neviedla aspoň k pomaleniu progresie závažného nálezu na ľavom kolene.
V ahraničí bolo popísané zlepšenie priebehu ochorenia transplantáciou kostnej drene u etskej formy hypofosfatázie (14, 15). Vo zvieracom experimente bolo úspešné použitie génovej a nzýmovej terapie (16, 17).
Z linického a raktického hľadiska je dôležitý komplexný prístup k iečbe týchto pacientov. Základom je dentálna starostlivosť, fyzioterapia, protetická starostlivosť a ravidelné sledovanie. V ndikovaných prípadoch je nutná chirurgická liečba. Pri výskyte diafyzárnych zlomenín je indikovaná vnútrodreňová osteosyntéza klincami („rodding“). V rípade kostných a kĺbových deformít predstavujú možnosti chirurgickej liečby artroskopické operácie, osteotómie, artrodézy a endoprotézy.
Záver
Deficiencia ALP je zriedkavé genetické ochorenie spôsobené širokou škálou mutácii génu ALPL, o výskyte ktorého v lovenskej republike ako aj v blízkych krajinách (Česká republika, Poľsko) chýbajú informácie. Príčinou môže byť stav diagnostiky, ktorý sa doposiaľ opieral o biochemický nález znížených aktivít enzýmu v sére a klinický nález zmien skeletu. Samotné vyšetrenie hodnôt ALP však nemusí odhaliť podstatne zníženie aktivít alebo nízke aktivity môžu byť prehliadnuté. Aj pozitívny nález enzýmového vyšetrenia by vyžadoval dôkaz zmien ďalších metabolitov (fosfoetanolamínu, pyridoxálfosfátu), čomu tak v rutinnej diagnostike nie je. Zavedenie molekulárno-genetických metód a identifikácia mutácii v géne ALPL poskytuje možnosť kauzálnej diagnostiky u pacientov s vysloveným podozrením na hypofosfatáziu, má význam pri určení prognózy ochorenia a pre genetickú konzultáciu vo vzťahu k potomstvu. V kazuistike prezentujeme dva prípady tohto ochorenia, u ktorých sme identifikovali mutácie v heterozygotnom stave (mutáciu T83M a E174K) a popisujeme klinický a diagnostický nález. Nakoľko ochorenie sa prejavuje variabilným fenotypom, pozornosť by mu mali venovať pediatri, neurológovia, nefrológovia, ortopédi ako aj stomatológovia. Hlavne vo vzťahu k dospelým pacientom je nutné upozorniť na možnosť diskrétneho nálezu a potrebu povšimnúť si aj mierne znížené hodnoty aktivít ALP.
Skratky
- DNA – deoxyribonukleová kyselina
- MUT – mutovaná alela
- PCR – polymerázová reťazová reakcia
- TNSALP, ALP – tkanivovo nešpecifická alkalická fosfatáza, alkalická fosfatáza
- UN – Univerzitná nemocnica
- WT – Wild type mutácia
ADRESA PRO KORESPONDENCI:
MUDr. Ilja Chandoga
II. ortopedicko-traumatologická klinika LF UK a N
Antolská 11, 851 07 Bratislava, Slovenská republika
e-mail: chandoga@gmail.com
Zdroje
1. Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, et al. The Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill 2001.
2. Mornet E. Hypophosphatasia. http://www.OJRD.com/content/ 2/1/40
3. Mornet E, Taillandier A, Peyramaure S, Kaper F, Muller F, Brenner R, BussiŹre P, Freisinger P, Godard J, LeMerrer M, Oury JF, Plauchu H, Puddu R, Rival JM, Superti-Furga A, Touraine RL, Serre JL, Simon-Bouy B. Identification of fifteennovel mutations in the tissue-nonspecifical kalinephosphatase (TNSALP) gene in European patients with severe hypophosphatasia. Eur J HumGenet 1998; 6 : 308–314.
4. Henthorn PS, Whyte MP. Missense mutations of the tissue-nonspecifical kalinephosphatase gene in hypophosphatasia. Clin Chem 1992; 38 : 2501–2505.
5. Hérasse M, Spentchian M, Taillandier A, Mornet E. Evidence of a founder effect for the tissue-nonspecifical kalinephosphatase (TNSALP) gene E174K mutation in hypophosphatasia patients. Eur J Hum Genet 2002; 10 : 666-668.
6. Mornet E, Stura E, Lia-Baldini AS, Stigbrand T, Ménez A, LeDu MH. Structural evidence for a functional role of human tissue nonspecifical kalinephosphatase in bone mineralization. J BiolChem 2001; 276 : 31171–31178.
7. Mornet E, Yvard A, Taillandier A, Fauvert D, Simon-Bouy B. A molecular-base destimation of the prevalence of hypophosphatasia in the European population. Ann Hum Genet 2011; 75 : 439–445.
8. Davies AM, Johnson K, Whitehouse RW. Imaging of the Hip & Bony Pelvis. Berlin, Heidelberg: Springer-Verlag 2006.
9. Castriota-Scanderbeg A, Dallapiccola B. Abnormal Skeletal Phenotypes: From Simple Signs to Complex Diagnoses. Berlin, Heidelberg: Springer-Verlag 2005.
10. Unger S, Mornet E, Mundlos S, Blaser S, Cole D. Severe cleidocranial dysplasia can mimic hypophosphatasia. Eur J Pediatr 2002; 161 : 623–626.
11. Barvencik F, Gebauer M, Schinke T, Amling M. Saethre-Chotzen: Multiple Fractures in a Patient with Mutations of TWIST1 and TNSALP. Clin Orthop Relat Res 2008; 466 : 990–996.
12. Maricic M. Osteomalacia. Curr Osteoporos Rep 2008; 6 : 130–133.
13. Whyte MP, Mumm S, Deal C. Adult Hypophosphatasia Treated with Teriparatide. J Clin Endocrinol Metab 2007; 92 : 1203–1208.
14. Whyte MP, Kurtzberg J, Mcalister WH, Mumm S, Podgornik MN, Coburn SP, Ryan RM, Miller CR, Gottesma GS, Smith AK, Douville J, Waters-Pick B, Armstrong RD, Martin PL. Marrow cell transplantation for infantile hypophosphatasia. J Bone Miner Res 2003; 18 : 624–636.
15. Cahill RA, Wenkert D, Perlman SA, Steele A, Coburn SP, Mcalister WH, Mumm S, Whyte MP. Infantile Hypophosphatasia: Transplantation Therapy Trial Using Bone Fragments and Cultured Osteoblasts. J Clin Endocrinol Metab 2007; 92 : 2923–2930.
16. Hessle L, Johnson KA, Anderson HC, Narisawa S, Sali A, Goding JW, Terkeltaub R, Millán JL. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci USA 2002; 99 : 9445–9449.
17. Millán JL, Narisawa S, Lemire I, Loisel TP, Boileau G, Leonard P, Gramatikov S, Terkeltaub R, Camacho NP, Mckee MD, Crine P, Whyte MP. Enzyme replacement therapy for murine hypophosphatasia. J Bone Miner Res 2008; 23 : 777–778.
Štítky
Adiktologie Alergologie a imunologie Angiologie Audiologie a foniatrie Biochemie Dermatologie Dětská gastroenterologie Dětská chirurgie Dětská kardiologie Dětská neurologie Dětská otorinolaryngologie Dětská psychiatrie Dětská revmatologie Diabetologie Farmacie Chirurgie cévní Algeziologie Dentální hygienistkaČlánek vyšel v časopise
Časopis lékařů českých

- Jaké zdravotní benefity může mít popíjení kávy nebo čaje?
- Jak a kdy u celiakie začíná reakce na lepek? Možnou odpověď poodkryla čerstvá kanadská studie
- Efektivita kartáčku Sonicare For Kids u dětí předškolního věku
- Přerušovaný půst může mít významná zdravotní rizika
- Ingenol-mebutát rozšiřuje možnosti léčby aktinické keratózy
Nejčtenější v tomto čísle
- Biochemické mechanismy účinků antidepresiv
- Hypofosfatázia – biochemické a klinické prejavy, molekulovo-genetická podstata
- Zemřel prof. MUDr. Vladimír Pacovský, DrSc.
- Hygiena ženy IV - 19. století
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy