
Trombocyty v sepsi
Thrombocytes in sepsis
Sepsis and septic shock are the most frequent causes of death in intensive care units. Thrombocytopenia and/or platelet function impairment are common parts of the multiple organ dysfunction syndrome. However, thrombocytes are not mere bystanders as documented in the present literature. Platelets, primary actors in haemostasis and thrombin generation during endothelial integrity disruption, similarly to leukocytes, maintain important functions of innate defense mechanisms of the host. These cellular fragments express and secrete numbers of adhesive and pro-inflammatory molecules that serve to initiate and modulate the primary immune response. Such haemostatic and especially non-haemostatic functions of platelets and their role in the pathogenesis of systemic inflammation will be discussed in this review.
Keywords:
platelets – sepsis – innate immunity – intercellular interaction
Autoři:
Karvunidis Thomas 1; Chvojka Jiří 1; Lysák Daniel 2; Kroužecký Aleš 1; Raděj Jaroslav 1; Novák Ivan 1; Matějovič Martin 1
Působiště autorů:
I. interní klinika, Jednotka intenzivní péče, Univerzita Karlova v Praze, Lékařská fakulta v Plzni a Fakultní nemocnice Plzeň
1; Hematologicko-onkologické oddělení, Fakultní nemocnice Plzeň
2
Vyšlo v časopise:
Anest. intenziv. Med., 21, 2010, č. 6, s. 342-350
Kategorie:
Intenzivní medicína - Přehledový článek
Souhrn
Sepse a septický šok jsou jednou z nejčastějších příčin smrti na jednotkách intenzivní péče. Trombocytopenie a/nebo porucha funkce krevních destiček jsou poměrně častou součástí syndromu multiorgánové dysfunkce, přičemž trombocyty nemusí hrát v patofyziologii těchto stavů pouze pasivní roli. Krevní destičky plnící primárně hemostatickou funkci při porušení integrity endotelu, zastávají, obdobně jako leukocyty, důležitou funkci v primárních obranných mechanismech hostitele. Tyto buněčné fragmenty na svém povrchu exprimují a secernují řadu adhezivních a prozánětlivých molekul, které slouží k iniciaci a modulaci primární imunitní odpovědi. Tyto hemostatické a zejména nehemostatické funkce trombocytů a jejich úloha v patogenetických mechanismech při systémovém zánětu budou autory shrnuty a autoři o nich budou diskutovat v tomto přehledovém článku.
Klíčová slova:
trombocyty – sepse – přirozená imunita – intercelulární interakce
Úvod
Sepse a septický šok jsou desátou nejčastější příčinou smrti všeobecně a nejčastější příčinou smrti na nekoronárních jednotkách intenzivní péče. Incidence sepse je 50–95 případů na 100 000 obyvatel a zvyšuje se cca o 9 % ročně. I přes značné pokroky v diagnostice a léčbě je mortalita těchto stavů 30–90% a koreluje s počtem dysfunkčních orgánů/systémů a s délkou trvání těchto stavů [1, 2].
Trombocyty jsou bezjaderné a nejmenší krevní elementy (3,6 x 7,0 μm; povrch 8 μμm2; objem 7 fl) vzniklé odštěpením z megakaryocytů v kostní dřeni. Proliferace a diferenciace megakaryocytů a následná produkce krevních destiček je pod kontrolou trombopoetinu (a v menší míře i jiných cytokinů: IL-3, IL-6, IL-11, G-CSF atd.). Trombopoetin (TPO) je produkován konstantní rychlostí v játrech a ledvinách a následně z cirkulace vychytáván vazbou se specifickým receptorem (c-Mpl) na povrchu trombocytů a megakaryocytů. Destičky slouží i jako zásobárna TPO a po jejich aktivaci dochází v procesu degranulace k jeho zpětnému uvolnění do cirkulace [3, 4]. Trombopoetin je eliminován/metabolizován výhradně cirkulujícími trombocyty. Pokles počtu cirkulujících destiček tudíž vede ke zvýšení hladiny trombopoetinu a naopak [5]. Trombocyty sehrávají centrální úlohu v procesu hemostázy a trombózy. Navíc jsou i specializovanými buňkami přirozené imunity, modulátory zánětlivé odpovědi hostitele; jsou zavzaty do procesů hojení ran a hematogenních metastáz [6]. Jsou významným zdrojem velkého množství bioaktivních mediátorů, které produkují, skladují a secernují [7]. Za normálních okolností, průměrně 1 x 1012 destiček, cirkuluje v krevním řečišti podél endotelu cév o ploše až 1000 m2, aniž by docházelo k jejich aktivaci, adhezi, agregaci a degranulaci. Při jakémkoliv porušení integrity cévní stěny dochází k nastartování procesu vzájemných interakcí cirkulujících trombocytů, endoteliálních buněk a subendoteliálních struktur v procesu hemostázy [8].
Trombocytopenie v sepsi
Trombocytopenie, arbitrárně definovaná jako počet trombocytů < 150 x 109/l, je častým jevem v sepsi či septickém šoku. Její incidence je přibližně 35–44 %. U 20–25 % těchto nemocných zjišťujeme počet destiček < 100 x 109/l a 12–15 % pacientů má těžkou trombocytopenii (< 50 x 109/l) [9, 10, 11, 12]. Počet destiček typicky klesá v prvních čtyřech dnech pobytu na JIP [13]. Mechanismus vzniku trombocytopenie v sepsi/septickém šoku není v současnosti zcela jasný (tab. 1). Vysvětlení sníženou produkcí trombocytů v kostní dřeni je v logickém rozporu s vysokými hladinami produkce destiček stimulujícími prozánětlivými cytokiny (TNF-α, IL-6 atd.) a trombopoetinu u nemocných v sepsi [3, 4]. Jednou z možných příčin může být nekontrolovaná fagocytóza krevních elementů a jejich prekurzorů v procesu systémového zánětu aktivovanými makrofágy. Tyto tzv. hemofagocytární syndromy (HPS) tvoří vzácnou skupinu vrozených a získaných onemocnění, pro která je společná neadekvátní buněčná imunitní odpověď u nemocných s defektem cytotoxické aktivity T-lymfocytů a NK-buněk. Hemofagocytóza se může podílet na jinak nevysvětlené trombocytopenii u některých pacientů v sepsi/septickém šoku. Za těchto klinických situací může být tento proces relativně častý [14, 15]. Kromě trombocytů jsou častým cílem hemofagocytózy i erytrocyty a jejich jaderné prekurzory v kostní dřeni. Právě erytrofagocytóza je potentním stimulem pro zvýšení exprese hemoxygenázy-1 (HO-1), proteinu, jenž má svůj podíl v protektivních mechanismech v sepsi/septickém šoku [16]. HO-1 je enzym účastnící se degradace hemu. Jeho zvýšená aktivita je spojená s protizánětlivými, antiapoptotickými a antioxdativními efekty. Tyto nejsou zprostřekovány enzymem samotným, ale právě degradačními produkty hemu – bilirubinem, oxidem uhelnatým (CO) a ferritinem [17]. Jako řadu jiných patofyziologických mechanismů v sepsi nelze ani hemofagocytózu, i přes její dopad na počty cirkulujících krevních elementů, z výše uvedených důvodů považovat za jednoznačně negativní proces.

Imunitní (autoprotilátkami zprostředkovaná) trombocytopenie může být příčinou sníženého počtu krevních destiček u kriticky nemocných v daleko větší míře, než jak je na ni pomýšleno. Nespecifické autoprotilátky proti trombocytům mohou být dekekovány až u 30 % těchto jedinců [18]. Většina IgG autoprotilátek se váže samostatně na bakteriální komponenty nacházející se na povrchu aktivovaných destiček nebo ve formě imunokomplexů. Pouze u menší části nemocných (30 % [18]) nacházíme specifické autoprotilátky namířené na membránové glykoproteiny (GPIIb/IIIa, GPIb/IX). Takto vyvolaná trombocytopenie tak pravděpodobně sdílí řadu patofyziologických mechanismů popsaných u imunitní (idiopatické) trombocytopenické purpury (ITP). Pacienti s trombocytopenií a detekovatelnými autoprotilátkami mají menší pravděpodobnost normalizace počtu destiček než jedinci bez těchto protilátek, nicméně nevyžadují větší substituci krevními deriváty (trombokoncentráty) [18]. Dalším patologickým stavem charakterizovaným často těžkou trombocytopenií s přítomností autoprotilátek je heparinem-indukovaná trombocytopenie (HIT). Tyto protilátky třídy IgG jsou namířeny proti komplexu destičkový faktor 4 a heparin (PF4/heparin, HIT-IgG) [19]. Incidence je odhadována na 1–3 % všech nemocných léčených nefrakcionovaným heparinem (UFH), až 0,8 % při aplikaci nízkomolekulárních heparinů (LMWH) [20, 21] a typicky se rozvíjí po více než 5 dnech léčby. I přes často závažnou trombocytopenii je HIT stavem jednoznačně prokoagulačním, díky uvolnění velkého množství prokoagulačních substancí (serotonin, histamin, ADP) při degranulaci trombocytů stimulované navázáním multimerů PF4/HIT/HIT-IgG na jejich FcγRIIa receptory [19]. Při suspekci na HIT je nezbytné neprodleně ukončit léčbu heparinem, stejně jako odstranit všechny heparin obsahující intravaskulární katétry a jiné zařízení, přerušit extrakorporální očišťovací (CRRT) a podpůrné (ECMO) metody užívající jako antikoagulans heparin. Pouhé vysazení heparinu však není adekvátní léčbou HIT. U 30–50 % nemocných, kteří v době HIT neměli HIT-asociovanou trombózu, se do jednoho měsíce rozvine trombembolická příhoda, pokud nejsou léčeni alternativním antikoagulanciem (přímé inhibitory trombinu) [22]. Mnohem častější než výše popsaná autoprotilátkami zprostředkovaná HIT je neimunitní HIT (nonimmune-HIT, N-HIT, dříve HIT type I). Mechanismem je nejspíše přímá vazba heparinu na trombocyty [23]. Taková trombocytopenie je většinou pouze mírná a rezultuje i bez přerušení léčby heparinem.
Relativně vzácnější příčinou trombocytopenie u kriticky nemocných včetně těch v sepsi/septickém šoku může být tzv. etylendiamintetraacetát-dependentní pseudotrombocytopenie (EDTA-PTCP). Literatura udává incidenci 0,1% [24, 25]. Jedná se o in vitro fenomén agregace destiček v přítomnosti EDTA a autoprotilátek proti trombocytům (tříd IgG, IgM i IgA; ligand: GPIIb/IIIa) [26]. Automatické analyzátory nebývají schopny přítomnost shluků krevních destiček vyhodnotit a výsledkem bývá nesprávně udaný nízký počet trombocytů. Tento fenomén by měl být vždy zvážen v diferenciální diagnostice trombocytopenie (nejen kriticky nemocných) a skutečný počet destiček ověřen analýzou z krevního vzorku odebraného do odběrových souprav s Na-citrátem jako antikoagulans.
Konzumpce trombocytů hraje také důležitou roli v rozvoji trombocytopenie v sepsi, a to v důsledku vysoké produkce trombinu, který je nejvíce potentním aktivátorem destiček in vivo. Trombocyty mohou být také aktivovány přímo endotoxinem a dalšími prozánětlivými mediátory (PAF atd.) [15, 16].
Primárním důsledkem trombocytopenie u kriticky nemocných je zvýšení rizika krvácení. U pacientů s těžkou trombocytopenií (< 50 x 109/l) je toto riziko čtyřikrát až pětkrát vyšší [11, 12] a obecně se zvyšuje při systémovém zánětu [17]. Riziko intracerebrálního krvácení nemocných přijatých na JIP je relativně nízké (0,3–0,5 %), ale až 88 % těchto nemocných má současně počet krevních destiček < 100 x 109/l [18]. Bez ohledu na příčinu je trombocytopenie nezávislým prediktorem mortality na JIP s relativním rizikem 1,9–4,2 podle multivariantních analýz [11, 12]. Trombocytopenie perzistující déle než 4 dny po přijetí na JIP či pokles počtu krevních destiček o > 50 % výchozích hodnot v průběhu pobytu na JIP jsou spojeny se čtyřnásobným až šestinásobným zvýšením mortality [11, 13]. Počet trombocytů se ukazuje být silnějším nezávislým ukazatelem mortality na JIP než některé skórovací systémy (APACHE II, MODS). Sepse je jasným nezávislým rizikovým faktorem pro rozvoj trombocytopenie a současně tíže sepse/septického šoku koreluje i s její závažností [11, 12, 19]. Trombocytopenie může být důsledkem i jiných stavů nesouvisejících se sepsí/septickým šokem (tab. 2).

Funkce trombocytů v sepsi
Trombocyty jsou vysoce diferencované buňky sehrávající esenciální úlohu v procesu primární zástavy krvácení a současně i v přirozených obranných mechanismech hostitele. Tvorbou trombu zajišťují hemostázu a brání krevním ztrátám při porušení cévní stěny, produkcí cytokinů, expresí adhezivních molekul s následnou přímou intercelulární interakcí a signalizací (trombocyt-endoteliální buňka, trombocyt-neutrofil) iniciují a modulují imunitní odpověď [7, 10, 20, 21, 22, 23, 24]. Nelze pominout ani vliv vazoaktivních látek secernovaných trombocyty na tonus cév při systémovém zánětu (tab. 3). Funkce destiček může být primárně rozdělena do čtyř fází: adheze, aktivace, agregace a sekrece.

Všechny zmíněné funkce a interakce trombocytů jsou podmíněny intaktní kaskádou změn, kterými procházejí v průběhu jejich aktivace. Při porušení integrity cévní stěny dochází k odhalení subendoteliálního kolagenu. Na ten se vážou cirkulující multimery von Willebrandova faktoru (vWF), které se dále vážou na membránový receptorový komplex trombocytů – glykoprotein Ib/V/IX (GP Ib/V/IX). Dochází k aktivaci a změně tvaru destiček. Diskoidní tvar s homogenní distribucí granul trombocytů v klidovém stavu se mění s charakteristickou excesivní tvorbou pseudopodií za aktivní účasti cytoskeletu [25], fuzí granul s membránou v procesu exocytózy a sekrecí jejich obsahu (tab. 4). Adheze destiček je možná i přímou interakcí membránového komplexu GP Ia/IIa (eventuálně i GP VI či CD36) s kolagenem jako jeho ligandem. Aktivace je provázena expresí a konfirmační změnou integrinu αIIbβ3 – receptorového komplexu GPIIb/IIIa (CD41/CD61). Následuje vzájemná agregace trombocytů pomocí tohoto receptoru a cirkulujícího fibrinogenu jako ligandu pro tento receptor. Lipoproteiny s nízkou denzitou (low-density lipoproteins, LDL) jsou rovněž ligandem GPIIb/IIIa a modulátorem destičkových funkcí. Uvádí se vztah vysoké hladiny LDL a hyperreaktivity trombocytů [26]. Možný vztah mezi LDL a protrombotickým stavem může vysvětlovat vyšší výskyt kardiovaskulárních komplikací po vysazení statinů (inhibitorů HMG-CoA reduktázy). Další konsekvencí aktivace destiček je sekrece obsahu granul – degranulace. Dochází k uvolnění několika agonistů destiček, koagulačních faktorů, heparin-binding proteinů, růstových faktorů, vazoaktivních látek a k expresi membránových receptorů zprostředkovávajících interakce s leukocyty či endoteliálními buňkami. Řada z takto uvolněných molekul (ADP, serotonin atd.) potencují stimulaci a aktivaci dalších trombocytů a atrahují je do místa poškození cévní stěny či infekce (tab. 4).

Jen několik málo kvalitních studií se dosud zabývalo problematikou alterace funkce destiček v sepsi. Práce Yaguchiho et al. za použití flowcytometrie, agregometrie a imunoanalýzy prokazuje snížení agregability kontrastující se zachováním exprese adhezivních molekul, intaktní sekrecí obsahu α-granul a zvýšení uvolňování některých růstových faktorů (VEGF) trombocytů jedinců v sepsi v porovnání se zdravými kontrolami [27].
Produkce bioaktivních mediátorů trombocyty
Krevní destičky nejsou jen pasivní zásobárnou bioaktivních látek. V průběhu jejich aktivace je řadou enzymatických pochodů produkováno množství lipidových derivátů – eikosanoidů, jako tromboxan A2 (TXA2) a prostaglandiny. Současně v transcelulárním metabolismu produkují prozánětlivé i protizánětlivé cytokiny. Trombocyty mají navíc několik jedinečných extranukleárních cest translace mRNA na proteiny tzv. signál-dependentním mechanismem, na jejichž konci je sekrece mimo jiné i IL-1β či TF a tedy i propojení hemostázy a zánětu [28].
Vascular endothelial growth factor (VEGF) je klíčová molekula v kontrole cévní permeability. Její působení je zprostředkováno vazbou s povrchovým VEGF-receptor-2 v membráně endoteliálních buněk. Kromě toho má významnou roli v angiogenezi a hojení ran. VEGF je produkován řadou buněk, mimo jiné monocyty periferní krve a trombocyty. Množství stimulů asociovaných se zánětem/sepsí (jako LPS, TNF-α) a hypoxií (cestou hypoxia-inducible factor 1a transcription factor) zvyšuje produkci a sekreci VEGF [29]. Řada studií prokazuje asociaci mezi zvýšenou plasmatickou hladinou VEGF a sepsí [29, 30]. Navíc některá data ukazují na možnou souvislost mezi plasmatickou hladinou VEGF a tíží sepse/orgánové dysfunkce a mortalitou [29]. Platelet-derived growth factor (PDGF) lze použít jako jeden z laboratorních markerů degranulace trombocytů [27].
Trombocyty a přirozená imunita
Přirozená imunita je tvořena fyzikálními, chemickými a buněčnými komponentami, které dohromady tvoří první obrannou linii organismu před invazí mikroorganismů. Zapojení těchto antigen-nespecifických mechanismů je bezprostřední (v několika sekundách) po rozpoznání infekčního agens. Kaskáda imunitní odpovědi organismu a propojení přirozené a adaptivní imunity cestou rozpoznání specifických molekulárních charakteristik konkrétních patogenů (pathogen-associated molecular patterns, PAMPs), jejich zpracování a prezentace antigen-prezentujícími buňkami (APCs) v asociaci s HLA molekulami s následnou aktivací (efektorových) buněk a mechanismů specifické/získané imunity jsou popsány v řadě studií a přehledných článků [31, 32, 33].
Trombocyty jsou schopny aktivně a účinně propojit primární imunitní reakce se specifickými získanými mechanismy. Na svém povrchu exprimují imunostimulační molekuly, jako např. CD40L (CD154), a touto cestou mohou např. stimulovat specifické antivirové CD8+ T-lymfocyty [34].
Invadující patogeny jsou rozpoznávány Toll-like receptory (TLRs) na profesionálních fagocytech. TLRs vážou řadu molekulárních struktur a jsou klíčové v procesu stimulace imunitního systému [35, 36, 37]. TLRs jsou exprimovány na nejrůznějších imunitních buňkách a recentní literatura dokumentuje přítomnost některých subtypů TLRs (TLR 1, 2, 4 a 9) i na neaktivovaných trombocytech [38, 39]. Jejich aktivace vazbou specifických PAMPs spouští kaskádu dobře popsaných dějů, které vedou k aktivaci destiček, produkci pro - i protizánětlivých mediátorů, sekvestraci trombocytů v mikrocirkulaci, zejména plic a jater (TLR-dependentní trombocytopenie), a aktivaci imunitního systému cestou prezentace PAMPs klasickým buňkám imunitního systému [23,40–43]. TLRs jsou přítomny rovněž na megakaryocytech v kostní dřeni [4] a existuje řada důkazů experimentálních studií o jejich roli v trombopoéze [44]. Nicméně, logicky odvozeno, aktivované destičky (včetně aktivace cestou TLRs) secernují TPO a další cytokiny stimulující jejich produkci, jak již bylo zmíněno výše [3, 4].
Jedním z mnoha mechanismů přirozené imunity je i fagocytóza zprostředkovaná granulocyty a monocyty/ makrofágy, podílející se na izolaci, destrukci a prezentaci antigenů invadujících agens. Trombocyty jsou rovněž schopny internalizace mikroorganismů či jejich částí v procesu pseudofagocytózy („engulfment“) [24]. Ačkoliv byla dokumentována parciální digesce bakterií v destičkových lysozomech [45], trombocyty nejsou v těchto kompartmentech schopny generovat baktericidní pH vzhledem ke komunikaci těchto intracelulárních prostor s okolním prostředím kanalikulárním systémem [46].
Intercelulární interakce trombocytů
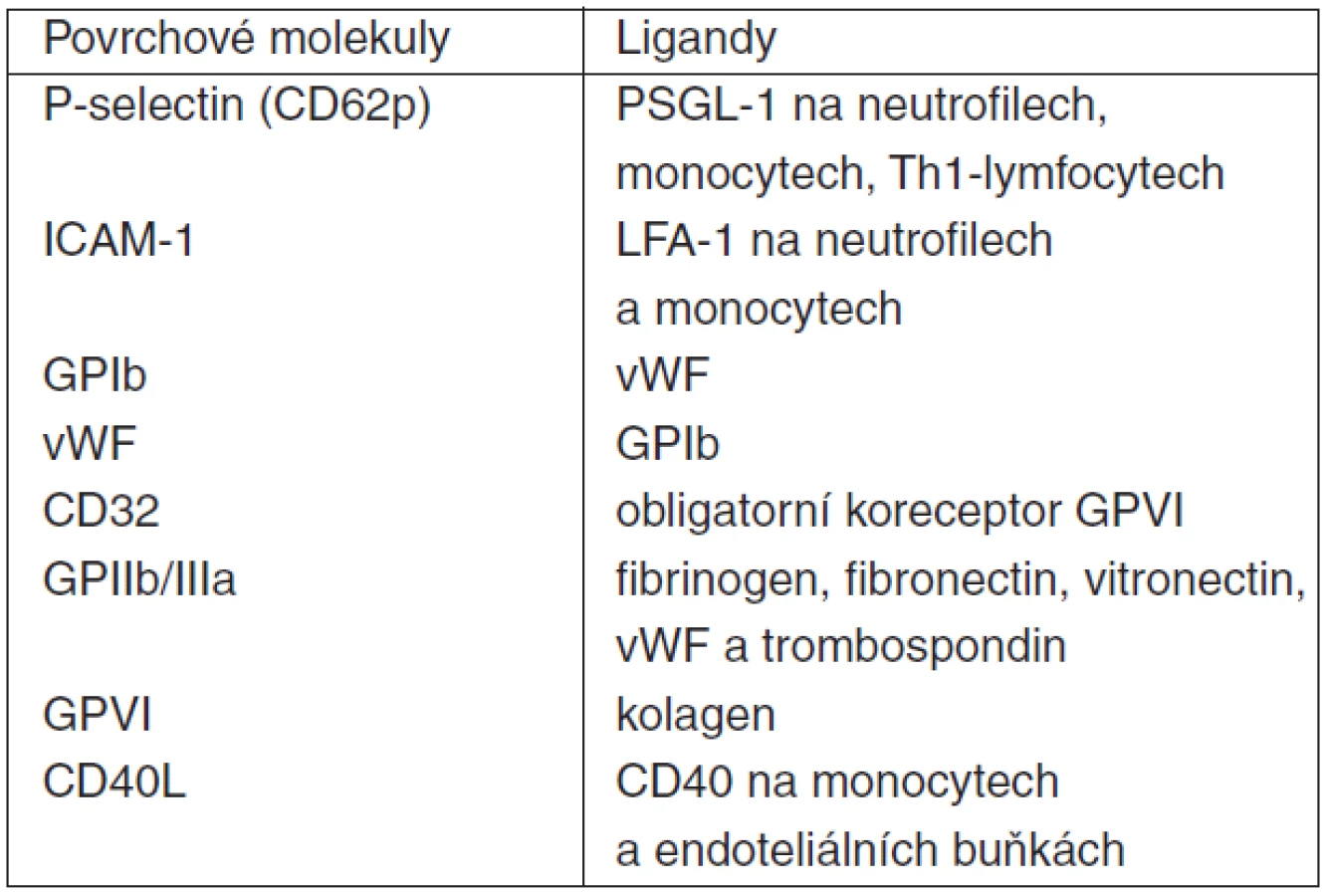
Aktivované trombocyty exprimují na svém povrchu řadu adhezivních molekul sloužících k mezibuněčnému kontaktu a signalizaci (tab. 5 a 6). Jednou z nich je i P-selektin (CD62p), integrální membránový glykoprotein patřící do rodiny selektinových adhezivních receptorů. Nachází se na povrchu nejen destiček, ale i makrofágů/monocytů a aktivovaných endoteliálních buněk. Dalšími selektiny jsou E-selektin exprimovaný na membráně aktivovaných endoteliálních buněk a L-selektin na povrchu leukocytů. P-selektin hraje esenciální roli v interakci trombocytů s leukocyty. Jeho vazebným protějškem s velkou afinitou je tzv. P-selektin glycoprotein ligand-1 (PSGL-1), který se nachází na povrchu leukocytů i samotných destiček. Vazba P-selektin – PSGL-1 (eventuálně současně GP Ib/V/IX – vWF či GP Ia/IIa – kolagen při porušení integrity endotelu) a následná silná vazba komplexu GPIIb/IIIa v membráně trombocytů přes fibrinogenový můstek na intercelular adhesion molekule-1 (ICAM-1) na povrchu endotelu umožňuje adhezi destiček v místě invaze patogenů či porušení cévní stěny. Současně dochází obdobnými mechanismy (selektin, ICAM-1) na stejném místě i k adhezi leukocytů (polymorfonukleárů, monocytů/makrofágů). Nicméně, k zachycení, adhezi a aktivaci leukocytů může dojít i mechanismem tzv. second capture, kdy k endotelu pevně adherující trombocyty zachytávají cirkulující („resting“) leukocyty, dochází k interakci trombocyt-leukocyt (polymorfonukleáry, PMN), aktivaci PMN a následně k „rollingu“ těchto PMN po vrstvě destiček a postupnému průniku přes cévní stěnu [22]. Stabilní kontakt trombocytu s PMN může být zprostředkován i vazbou ICAM-2 (trombocyt) s lymphocyte function-associated antigen-1 (LFA-1, CD11a/CD18, PMN) [47].

Sepse je provázena signifikantní aktivací neutrofilů. PAMPs jako lipopolysacharid (LPS) spouští kaskádu změn vedoucí k strukturálním změnám neutrofilů, zvýšení jejich rigidity, expresi adhezivních molekul, jejich cílenou migraci („homing“) do mikrocirkulace plic a jater, což může být jedním z mechanismů následné dysfunkce těchto orgánů. Na druhou stranu může být migrace neutrofilů do plic i aktivním mechanismem imunitního systému v průběhu systémového zánětu. Sekvestraci PMN do těchto orgánů záhy následuje i akumulace trombocytů v takto postižené mikrocirkulaci. Úloha neutrofilů se v tomto procesu jeví jako esenciální, jak dokumentují některé experimentální práce [39]. Patofyziologické pochody tohoto fenoménu nejsou zcela objasněny. Existuje několik možností: přímá interakce trombocytů s imobilizovanými PMN a aktivovanými ECs v cílové oblasti, atrakce destiček jejich agonisty (ADP, trombin) secernovanými v těchto cílových tkáních aktivovanými PMN a ECs [22, 48], zvýšená vazba trombocytů exponovaných LPS na imobilizované neutrofily [49]. Aktivované neutrofily navázané na trombocyty uvolňují do cirkulace řadu biologicky aktivních molekul a navíc i DNA a dohromady vytvářejí tzv. neutrophil extracellular traps (NETs), které hrají roli v zachytávání a likvidaci bakterií v cévním řečišti [50, 51].
Další cestou intercelulární komunikace trombocytů s buňkami přirozené imunity je interakce receptoru CD40 a jeho ligandu CD40L. Exprese transmembránového proteinu CD40L (CD154) na povrchu destiček se zvyšuje po jejich aktivaci [52]. Vazba na již zmíněný CD40 na povrchu endoteliálních buněk, monocytů, dendritických buněk, B-lymfocytů a trombocytů vede ke zvýšení exprese adhezivních molekul (ICAM-1, VCAM-1), produkci cytokinů a tím další aktivaci imunitního systému [52, 53, 54, 55]. Trombocyty interakcí CD40L-CD40 indukují maturaci dendritických buněk, což je centrálním mechanismem v rozvoji získané imunity proti invadujícím patogenům [56]. Dendritické buňky jsou rovněž velmi potentními APCs, a mohou tak sehrát roli i v imunitně podmíněné trombocytopenii. Stejným mechanismem mohou destičky indukovat změnu izotypů B-lymfocytů a augmentaci odpovědi CD8+ T-lymfocytů, což vede k lepší obraně proti virovým infekcím [56]. CD40L se z membrány trombocytů uvolňuje do cirkulace jako 18-kDa solubilní CD40L (sCD40L). Tento již není schopen aktivovat endoteliální buňky [52, 57], ale stále si zachovává potenciál stimulovat destičky přes jejich receptor CD40 s již výše diskutovanou konsekvencí. Několik studií poukazuje na možnost významného podílu vyplavení sCD40L z trombocytů (eventuálně po jejich aktivaci via TLRs) na patogenezi transfuzí indukovaného postižení plic (transfusion-related acute lung injury, TRALI). T lymfocyty s exprimovanými CD40L jsou schopny aktivovat trombocyty k uvolnění RANTES, které zpětnovazebně stimulují a aktivují další T lymfocyty, a potencují tak specifickou imunitní odpověď organismu hostitele [55].
Trombocyty a infekční agens
Interakce bakterií s krevními destičkami je možná třemi mechanismy. Tím prvním je nepřímé působení cestou aktivace trombocytů zvýšením hladiny prozánětlivých cytokinů v odpovědi imunitního systému na přítomnost invadujících bakterií. Dále mohou destičky reagovat na látky secernované (toxiny; toxiny syndromu toxického šoku, enterotoxiny aj.) či uvolněné (komponenty buněčných stěn bakterií; LPS E. coli O157) ze samotných mikroorganismů. V neposlední řadě může dojít k vazbě samotných bakterií s trombocyty. Tato vazba může být přímá či zprostředkovaná společným ligandem (většinou plasmatický protein). Všechny zmíněné interakce mohou při dostatečné intenzitě spouštět již výše diskutovanou kaskádu změn vedoucí k aktivaci destiček [58, 59, 60]. Rovněž bakteriální fenotyp hraje významnou roli v intenzitě a rychlosti odpovědi trombocytů na tuto stimulaci [58]. I přes značnou variabilitu mechanismů přímé interakce bakterií s trombocyty vede zablokování destičkového receptoru pro Fc fragment IgG (FcγRIIa) k prevenci aktivace a agregace trombocytů v přítomnosti bakterií [61].
Trombocytopenie je častým jevem při systémových virových infekcích. Podle okolností a závažnosti může vyústit až do formy idiopatické trombocytopenické purpury (ITP) [58]. Trombocyty hrají důležitou roli v obraně organismu proti invazi a/nebo progresi virových infekcí. Detailní mechanismy však nejsou dosud známy.
Testování funkce trombocytů
Kromě obligatorního stanovení počtu krevních destiček je k testování jejich stavu a funkcí, eventuálně i k funkci hemostatického systému jako celku, možno využít řadu etablovaných metod, jejichž podrobný popis je nad rámec tohoto sdělení. Jejich stručný seznam a popis je uveden v tabulce 7.

Naopak relativně novou a výkonnou metodou s potencionálním širokým uplatněním je proteomická analýza. Proteomika analyzuje proteiny, jako komplementární složky ke genům, které je kódují. Umožňuje tedy identifikaci, kvantifikaci a analýzu funkcí proteinů [62]. Zejména v oblasti intenzivní péče může být – a také již je – proteomika metodou, jež detailně odhaluje mechanismy patologických stavů, protože právě proteiny a peptidy, jako konečné produkty transkripce a translace genů aktivovaných v kaskádě systémové zánětlivé odpovědi a multiorgánového selhání, jsou efektorovými molekulami. Jejich identifikace, znalost funkce a vzájemných interakcí může vést k nalezení nových biomarkerů a terapeutických cílů [63].
Vzhledem k absenci buněčného jádra a jen malému obsahu mRNA, jsou trombocyty výborným cílem pro proteomické studie. Existuje řada prací popisujících proteom destiček v různých stavech aktivace. Množství proteomických technik (2-DE či chromatografie následovaná různými variantami hmotnostní spektrometrie) umožňujících charakterizaci různých skupin proteinů trombocytů – proteiny cytoskeletu, proteiny uplatňující se v přenosu signálů, proteiny syntetizované destičkami v neaktivním stavu a proteiny syntetizovanými a secernovanými aktivovanými trombocyty. Technologie a metodika proteomické analýzy trombocytů je shrnuta v přehledné práci Angela Garcíi [64].
Trombocyty jako cíl léčebných intervencí v sepsi
Ačkoliv je normální počet krevních destiček 150 až 350 . 109/l, pouze malá část z tohoto počtu (< 10 . 109/l) je za normálních okolností skutečně třeba k prevenci běžných krvácení. K hemostáze při větších poraněních a běžných chirurgických výkonech je zpravidla trombocytů potřeba více (40–50 . 109/l), podle závažnosti stavu, rozsahu poranění a výkonu [58, 65]. Také od tohoto poznání by se měla odvíjet i strategie v jejich substituci. Excesivní převody trombocytárních koncentrátů mohou zvyšovat „klasická“ rizika spojená s převodem krevních derivátů/trombocytů, ale při znalostech výše diskutovaných nehemostatických funkcí destiček mohou vést i k potenciaci a zhoršení systémové inflamace a MODS.
Recentní literatura dokumetuje minimálně asociaci ALI/ARDS s transfuzemi krevních produktů bohatých na plasmu (plasma-rich blood derivates; čerstvá zmrazená plasma, trombokoncentráty), a to zejména u kriticky nemocných v sepsi/septickém šoku a/nebo s pneumonií či po aspiraci žaludečního obsahu [78]. Trombocyty včetně těch transfundovaných se také mohou podílet na poškození, ale na druhé straně i na regeneraci jater po jejich primárním postižení [79].
Spolu s rostoucí incidencí závažných onemocnění vyvolaných polyrezistentními mikroorganismy se stává léčba těchto infekcí čím dál tím náročnější a komplikovanější. Nahlížíme-li na trombocyty jako na velmi potentní iniciátory a modulátory imunitní odpovědi hostitele, jeví se cílení našich léčebných či případně preventivních snah na tyto elementy jako racionální. Například antagonizace/blokáda destičkového receptoru FcγRIIa může zabránit či zmírnit aktivaci trombocytů a tím neadekvátní odpovědi imunitního systému, jež může vést k septickému šoku a MODS [61]. Inhibice FcγRIIa neovlivňuje reakce trombocytů na jiné agonisty, a tedy ani např. jejich hemostatické funkce [61].
V neposlední řadě může být rezoluce trombocytopenie v sepsi/septickém šoku považována za jednu ze známek správně zvolené léčebné strategie a příznivého ovlivnění základního onemocnění [11, 12, 13].
Závěr
Trombocyty tvoří nedílnou součást hemostatického systému nezbytného k udržení homeostázy organismu. Nicméně, jak ukazuje současný výzkum a literatura, jejich funkce zasahují daleko a intenzivně do vrozené i adaptivní imunity a velkou měrou se podílejí na imunitní odpovědi organismu hostitele při invazi patogenů. I když je řada patofyziologických mechanismů, kterými mohou trombocyty iniciovat a modulovat zánětlivou odpověď, známa, je třeba dalšího výzkumu k podrobnému odhalení jejich funkcí a případnému nalezení nových biomarkerů a terapeutických cílů.
Použité zkratky:
JIP – jednotka intenzivní péče
APACHE II – acute physiology and chronic health evaluation score
MODS – multiple organ dysfunction syndrome/score
EC(s) – endoteliální buňka (buňky)
DC(s) – dendritická buňka (buňky)
PMN – polymorfonukleáry
APC(s) – antigen prezentující buňka (buňky)
NET(s) – neutrophil extracellular trap(s)
PAMP(s) – pathogen-associated molecular pattern(s)
TLR – Toll-like receptor
TPO – trombopoetin
EPO – erytropoetin
G-CSF – granulocyte-colony stimulating factor
M-CSF – macrophage/monocyte-colony stimulating factor
HLA – human leukocyte antigen
IL-1β – interleukin-1β
IL-3 – interleukin-3
IL-6 – interleukin-6
IL-11 – interleukin-11
TNF-α – tumor necrosis factor α
PAF – platelet activating factor
VEGF – vascular endothelial growth factor
PDGF – platelet-derived growth factor
GP – glykoprotein
PSGL-1 – P-selectin glycoprotein ligand-1
RANTES – regulated on activation normal T-cell expressed and secreted
vWF – von Willebrandův faktor
LDL – low-density lipoprotein
ADP – adenindifosfát
TXA2 – tromboxan A2
TF – tissue factor
LPS – lipopolysacharid
ICAM-1 – intercellular adhesion molekule-1
ICAM-2 – intercellular adhesion molekule-2
VCAM-1 – vascular-cell adhesion molekule-1
LFA-1 – lymfocyte function-associated antigen-1
2-DE – dvojdimenzionální elektroforéza
Práce byla podpořena výzkumným záměrem “MSM 0021620819 Náhrada a podpora funkce některých životně důležitých orgánů“.
Došlo dne 3. 8. 2010
Přijato dne 21. 9. 2010.
Adresy pro korespondenci:
Prof. MUDr. Martin Matějovič, PhD.
I. interní klinika FN Plzeň
Alej Svobody 80
304 60 Plzeň
e-mail: matejovic@fnplzen.cz
MUDr. Thomas Karvunidis
JIP, I. Interní klinika
Fakultní nemocnice Plzeň
Alej Svobody 80
304 60 Plzeň
e-mail: karvunidist@fnplzen.cz
Zdroje
1. Martin, G. S., Mannino, D. M., Eaton, S. et al. The epidemiology of sepsis in the United States from 1979 through 2000. N. Engl. J. Med., 2003, 348, p. 1546–1554.
2. Annane, D., Bellissant, E., Cavaillon, J. M. Septic shock. Lancet, 2005, 365, p. 63–78.
3. Folman, C. C., Linthorst, G. E., van, M. J. et al. Platelets release thrombopoietin (Tpo) upon activation: another regulatory loop in thrombocytopoiesis? Thromb. Haemost., 2000, 83, p. 923–930.
4. Stohlawetz, P., Folman, C. C., von dem Borne, A. E. et al. Effects of endotoxemia on thrombopoiesis in men. Thromb. Haemost., 1999, 81, p. 613–617.
5. Kaushansky, K. Thrombopoietin. N. Engl. J. Med., 1998, 339, p. 746–754.
6. Jurk, K., Kehrel, B. E. Platelets: physiology and biochemistry. Semin. Thromb. Hemost., 2005, 31, p. 381–392.
7. Smyth, S. S., McEver, R. P., Weyrich, A. S. et al. Platelet functions beyond hemostasis. J. Thromb. Haemost., 2009, 7, p. 1759–1766.
8. Levi, M. Platelets. Crit. Care Med., 2005, 33, p. S523–S525.
9. Levi, M. Platelets at a crossroad of pathogenic pathways in sepsis. J. Thromb. Haemost., 2004, 2, p. 2094–2095.
10. Vincent, J. L., Yagushi, A., Pradier, O. Platelet function in sepsis. Crit. Care Med., 2002, 30, p. S313–S317.
11. Vanderschueren, S., De, W. A., Malbrain, M. et al. Thrombocytopenia and prognosis in intensive care. Crit. Care Med., 2000, 28, p. 1871–1876.
12. Strauss, R., Wehler, M., Mehler, K. et al. Thrombocytopenia in patients in the medical intensive care unit: bleeding prevalence, transfusion requirements, and outcome. Crit. Care Med., 2002, 30, p. 1765–1771.
13. Akca, S., Haji-Michael, P., de, M. A. et al. Time course of platelet counts in critically ill patients. Crit. Care Med., 2002, 30, p. 753–756.
14. Francois, B., Trimoreau, F., Vignon, P. et al. Thrombocytopenia in the sepsis syndrome: role of hemophagocytosis and macrophage colony-stimulating factor. Am. J. Med., 1997, 103, p. 114–120.
15. Zimmerman, G. A., McIntyre, T. M., Prescott, S. M. et al. The platelet-activating factor signaling system and its regulators in syndromes of inflammation and thrombosis. Crit. Care Med., 2002, 30, p. S294–S301.
16. Weyrich, A. S., Zimmerman, G. A. Platelets: signaling cells in the immune continuum. Trends Immunol., 2004, 25, p. 489–495.
17. Levi, M., Lowenberg, E. C. Thrombocytopenia in critically ill patients. Semin. Thromb. Hemost., 2008, 34, p. 417–424.
18. Oppenheim-Eden, A., Glantz, L., Eidelman, L. A. et al. Spontaneous intracerebral hemorrhage in critically ill patients: incidence over six years and associated factors. Intensive Care Med., 1999, 25, p. 63–67.
19. Russwurm, S., Vickers, J., Meier-Hellmann, A. et al. Platelet and leukocyte activation correlate with the severity of septic organ dysfunction. Shock, 2002, 17, p. 263–268.
20. Levi, M. Platelets in sepsis. Hematology., 2005, 10 Suppl 1, p. 129–131.
21. Levi, M. Platelets. Crit. Care Med., 2005, 33, p. S523–S525.
22. Zarbock, A., Polanowska-Grabowska, R. K., Ley, K. Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood Rev., 2007, 21, p. 99–111.
23. von, H. P., Weber, C. Platelets as immune cells: bridging inflammation and cardiovascular disease. Circ. Res., 2007, 100, p. 27–40.
24. Semple, J. W., Freedman, J. Platelets and innate immunity. Cell Mol. Life Sci., 2010, 67, p. 499–511.
25. Fox, J. E. The platelet cytoskeleton. Thromb. Haemost., 1993, 70, p. 884–893.
26. Puccetti, L., Pasqui, A. L., Pastorelli, M. et al. Platelet hyperactivity after statin treatment discontinuation. Thromb. Haemost., 2003, 90, p. 476–482.
27. Yaguchi, A., Lobo, F. L., Vincent, J. L. et al. Platelet function in sepsis. J. Thromb. Haemost., 2004, 2, p. 2096–2102.
28. Weyrich, A. S., Schwertz, H., Kraiss, L. W. et al. Protein synthesis by platelets: historical and new perspectives. J. Thromb. Haemost., 2009, 7, p. 241–246.
29. van der, F. M., van Leeuwen, H. J., van Kessel, K. P. et al. Plasma vascular endothelial growth factor in severe sepsis. Shock, 2005, 23, p. 35–38.
30. Yano, K., Liaw, P. C., Mullington, J. M. et al. Vascular endothelial growth factor is an important determinant of sepsis morbidity and mortality. J. Exp. Med., 2006, 203, p. 1447–1458.
31. Annane, D., Bellissant, E., Cavaillon, J. M. Septic shock. Lancet, 2005, 365, p. 63–78.
32. Tsiotou, A. G., Sakorafas, G. H., Anagnostopoulos, G. et al. Septic shock; current pathogenetic concepts from a clinical perspective. Med. Sci. Monit., 2005, 11, p. RA76–RA85.
33. Brown, K. A., Brain, S. D., Pearson, J. D. et al. Neutrophils in development of multiple organ failure in sepsis. Lancet, 2006, 368, p. 157–169.
34. Elzey, B. D., Tian, J., Jensen, R. J. et al. Platelet-mediated modulation of adaptive immunity. A communication link between innate and adaptive immune compartments. Immunity, 2003, 19, p. 9–19.
35. Annane, D., Bellissant, E., Cavaillon, J. M. Septic shock. Lancet, 2005, 365, p. 63–78.
36. Matsuda, N., Hattori, Y. Systemic inflammatory response syndrome (SIRS): molecular pathophysiology and gene therapy. J. Pharmacol. Sci., 2006, 101, p. 189–198.
37. McGettrick, A. F., O’Neill, L. A. Toll-like receptors: key activators of leucocytes and regulator of haematopoiesis. Br. J. Haematol., 2007, 139, p. 185–193.
38. Shiraki, R., Inoue, N., Kawasaki, S. et al. Expression of Toll--like receptors on human platelets. Thromb. Res., 2004, 113, p. 379–385.
39. Andonegui, G., Kerfoot, S. M., McNagny, K. et al. Platelets express functional Toll-like receptor-4. Blood, 2005, 106, p. 2417–2423.
40. Beaulieu, L. M., Freedman, J. E. The role of inflammation in regulating platelet production and function: Toll-like receptors in platelets and megakaryocytes. Thromb. Res., 2010, 125, p. 205–209.
41. Ishii, K. J., Akira, S. Toll-like Receptors and Sepsis. Curr. Infect. Dis. Rep., 2004, 6, p. 361–366.
42. Aslam, R., Speck, E. R., Kim, M. et al. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood, 2006, 107, p. 637–641.
43. Alves-Filho, J. C. Toll-like receptors on platelets: the key for disseminated intravascular coagulation in sepsis? Thromb. Res., 2005, 115, p. 537–538.
44. Jayachandran, M., Brunn, G. J., Karnicki, K. et al. In vivo effects of lipopolysaccharide and TLR4 on platelet production and activity: implications for thrombotic risk. J. Appl. Physiol., 2007, 102, p. 429–433.
45. Lewis, J. C., Maldonado, J. E., Mann, K. G. Phagocytosis in human platelets: localization of acid phosphatase-positive phagosomes following latex uptake. Blood, 1976, 47, p. 833–840.
46. White, J. G. Platelets are covercytes, not phagocytes: uptake of bacteria involves channels of the open canalicular system. Platelets., 2005, 16, p. 121–131.
47. Diacovo, T. G., deFougerolles, A. R., Bainton, D. F. et al. A functional integrin ligand on the surface of platelets: intercellular adhesion molecule-2. J. Clin. Invest, 1994, 94, p. 1243–1251.
48. Peters, M. J., Heyderman, R. S., Hatch, D. J. et al. Investigation of platelet-neutrophil interactions in whole blood by flow cytometry. J. Immunol. Methods, 1997, 209, p. 125–135.
49. Clark, S. R., Ma, A. C., Tavener, S. A. et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med., 2007, 13, p. 463–469.
50. Brinkmann, V., Reichard, U., Goosmann, C. et al. Neutrophil extracellular traps kill bacteria. Science, 2004, 303, p. 1532–1535.
51. Ma, A. C., Kubes, P. Platelets, neutrophils, and neutrophil extracellular traps (NETs) in sepsis. J. Thromb. Haemost., 2008, 6, p. 415–420.
52. Inwald, D. P., McDowall, A., Peters, M. J. et al. CD40 is constitutively expressed on platelets and provides a novel mechanism for platelet activation. Circ. Res., 2003, 92, p. 1041–1048.
53. Henn, V., Slupsky, J. R., Grafe, M. et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature, 1998, 391, p. 591–594.
54. Pignatelli, P., Sanguigni, V., Lenti, L. et al. gp91phox-dependent expression of platelet CD40 ligand. Circulation, 2004, 110, p. 1326–1329.
55. Danese, S., de la, M. C., Reyes, B. M. et al. Cutting edge: T cells trigger CD40-dependent platelet activation and granular RANTES release: a novel pathway for immune response amplification. J. Immunol., 2004, 172, p. 2011–2015.
56. Elzey, B. D., Tian, J., Jensen, R. J. et al. Platelet-mediated modulation of adaptive immunity. A communication link between innate and adaptive immune compartments. Immunity., 2003, 19, p. 9–19.
57. Henn, V., Steinbach, S., Buchner, K. et al. The inflammatory action of CD40 ligand (CD154) expressed on activated human platelets is temporally limited by coexpressed CD40. Blood, 2001, 98, p. 1047–1054.
58. Clemetson, K. J. Platelets and pathogens. Cell Mol. Life Sci., 2010, 67, p. 495–498.
59. Yeaman, M. R. Platelets in defense against bacterial pathogens. Cell Mol. Life Sci., 2010, 67, p. 525–544.
60. Yeaman, M. R. Bacterial-platelet interactions: virulence meets host defense. Future. Microbiol., 2010, 5, p. 471–506.
61. Kerrigan, S. W., Cox, D. Platelet-bacterial interactions. Cell Mol. Life Sci., 2010, 67, p. 513–523.
62. Peng, J., Gygi, S. P. Proteomics: the move to mixtures. J. Mass Spectrom., 2001, 36, p. 1083–1091.
63. Karvunidis, T., Mares, J., Thongboonkerd, V. et al. Recent progress of proteomics in critical illness. Shock, 2009, 31, p. 545–552.
64. Garcia, A., Watson, S. P., Dwek, R. A. et al. Applying proteomics technology to platelet research. Mass Spectrom. Rev., 2005, 24, p. 918–930.
65. Kor, D. J., Gajic, O. Blood product transfusion in the critical care setting. Curr. Opin. Crit. Care, 2010, 16, p. 309–316.
Štítky
Anesteziologie a resuscitace Intenzivní medicínaČlánek vyšel v časopise
Anesteziologie a intenzivní medicína

2010 Číslo 6
- Neodolpasse účinně snižuje pooperační bolest
- Neodolpasse jako ideální volba tam, kde je bolest provázena spasmem kosterního svalstva
- Optimalizace léčby pooperační bolesti snižuje nároky na zdravotní péči
- Použití Neodolpasse v indikaci pooperační bolesti
- Neodolpasse je bezpečný přípravek v krátkodobé léčbě bolesti
Nejčtenější v tomto čísle
- Septický šok při fatálně probíhající stafylokokové pneumonii: význam Pantonova-Valentinova leukocidinu – kazuistika
- Srovnání spokojenosti pacientů po celkové a regionální anestezii u operačních náhrad kyčelních a kolenních kloubů
- Trombocyty v sepsi
- Katétrová vysokofrekvenčná ventilácia pľúc pri tracheostómii podľa Fantoniho
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy