
Familiární plicní fibróza – doporučení pro diagnostiku a léčbu
Familial pulmonary fibrosis – guidelines for diagnostics and treatment
Familial pulmonary fibrosis (FPF) is defined as interstitial lung involvement in at least two members of the same biological family. Pathogenesis of FPF involves background of genetic risk factors further modified by environmental exposures and aging. Manifesta ‑ tion of FPF mirrors manifestation of interstitial lung diseases generally. Patients may present also with involvement of other organs, as seen usually in those affected by complex syndromes or telomeropaties. Described mutations concern telomeres homeostasis genes (TERT, TERC, RTEL1, PARN, DKC1, TINF, NAF1), surfactant genes (SFTPC, ABCA3, NFKX2‑1) or genes associated with complex syndromes (COPA, TMEM173, HPS‑1‑8, NF1, FAM111B, NDUFAF6, GATA 2). Genetic tests are indicated by specialist in clinical genetics, optimaly after consultation with respiratory specialist involved in interstitial lung diseases. Treatment of FPF is currently unknown. In patients with multiorgan involvement growing number of organs may be affected in time and sometimes dysfunction of mostly severe affected organ may manifest before interstitial lung involvement.
Keywords:
Genetics – treatment – familial pulmonary fibrosis – telomer – surfaktant
Autoři:
Martina Šterclová 1,2; Michael Doubek 3,4; Martina Doubková 5
Působiště autorů:
Pneumologická klinika 2. LF a FN Motol, Praha
1; Interní oddělení Nemocnice na Homolce, Praha
2; Interní hematologická a onkologická klinika FN Brno a LF Brno 4Středoevropský technologický institut, Masarykova univerzita, Brno
3; Klinika nemocí plicních a tuberkulózy FN Brno a LF Masarykovy univerzity Brno
5
Vyšlo v časopise:
Vnitř Lék 2020; 66(6): 365-370
Kategorie:
Přehledové články
Souhrn
Za familiární plicní fibrózu (FPF) je považován výskyt intersticiálního plicního procesu u dvou a více osob s intersticiálním plicním procesem, které jsou přímými příbuznými. Patogeneze onemocnění je pravděpodobně vícestupňový proces, na jehož počátku stojí patologie v germinálním genomu. Projevy FPF se neliší od projevů intersticiálních plicních procesů obecně. U části pacientů se můžeme setkat s izolovaným postižením v oblasti plicního parenchymu, možné je ale i postižení mimoplicní, zejména u komplexních syndromů a telomeropatií. Dosud nejčastěji popsané mutace se týkají genů uplatňujících se udržení homeostázy telomer (TERT, TERC, RTEL1, PARN, DKC1, TINF2, NAF1), surfaktantu (SFTPC, ABCA3, NFKX2‑1) nebo asociovaných s komplexními syndromy (COPA, TMEM173, HPS‑1‑8, NF1, FAM111B, NDUFAF6, GATA 2). Genetické vyšetření indikuje genetik nejlépe po konzultaci s pneumologem věnujícím se intersticiálním plicním procesům. Specifická léčba FPF není známa. U pacientů s multiorgánovým postižením se nezřídka setkáme s postupnou manifestací obtíží, která může vést k selhání funkce postiženého orgánu před rozvojem intersticiálního plicního procesu.
Klíčová slova:
familiární plicní fibróza – genetika – telomera – surfaktant – léčba
Úvod
Za familiární plicní fibrózu (FPF) je považován výskyt intersticiálního plicního procesu u dvou a více osob s intersticiálním plicním procesem, které jsou přímými příbuznými. FPF není jednotným plicním onemocnění, ale skupinou nemocí, které jsou podmíněné patologiemi v germinálním genomu a vedou k postižení plicního intersticia. Typ dědičnosti bývá různý, penetrance obvykle neúplná (1).
Původně byly FPF považovány za vzácné entity, postupem času se ale ukazuje, že minimálně u 10 % „sporadických“ idiopatických plicních fibróz (IPF) se jedná o hereditární stavy. FPF tak mohou tvořit 10–19,5 % všech případů intersticiálních plicních procesů. Nejčastější formou FPF je takzvaný syndrom krátkých telomer (definováno jako délka telomery < 10 percentil), který je nalézán až u 15 % případů FPF.
Patogeneze FPF
Patogeneze FPF je několikastupňový proces, který shrnuje obrázek 1. Mutace konkrétních genů se pak mohou podílet na rozvoji fibrotického plicního postižení na podkladě různých mechanismů.

Některé mutace genů pro surfaktantové proteiny (genů SFTBA1, SFTPA2 a SFTPC) vedou k nahromadění špatně sbalených proteinů v endoplazmatickém retikulu. Spouští se tzv. „unfolded protein response“, reakce buňky na stres endoplazmatického retikula, jejímž výsledkem může být v některých případech i apoptóza, nebo jsou aktivovány kaskády buněčné diferenciace a postižená buňka získá fenotypové znaky buňky mezenchymální. Dojde k epitelo-mezenchymové tranzici se všemi jejími známými důsledky.
Telomeropatie jsou spojeny úzce s předčasným stárnutím a snížením schopnosti buněk vyrovnat se s opakovaným poškozením. Podobné mechanismy se mohou uplatňovat i u pacientů s mukociliární dysfunkcí v důsledku vysoké exprese genu pro mucin (MUC5B) nebo při poruše integrity epitelu v dýchacích cestách při aberantní expresi desmoplakinu. Aktivace remodelace plicní tkáně může vyústit až v její fibrotickou přestavbu (2).
Klinické projevy FPF
Projevy FPF se neliší od projevů intersticiálních plicních procesů obecně. U části pacientů se můžeme setkat s izolovaným postižením v oblasti plicního parenchymu, možné je ale i postižení mimoplicní (viz tabulky 1 a 2). Mimoplicní postižení je typické především pro patogenní varianty genů, které vedou k rozvoji plicního postižení již v dětství, pro komplexní syndromy, jako je syndrom Heřmanského‑Pudláka, neurofibromatózu typu I nebo pro telomeropatie (3).
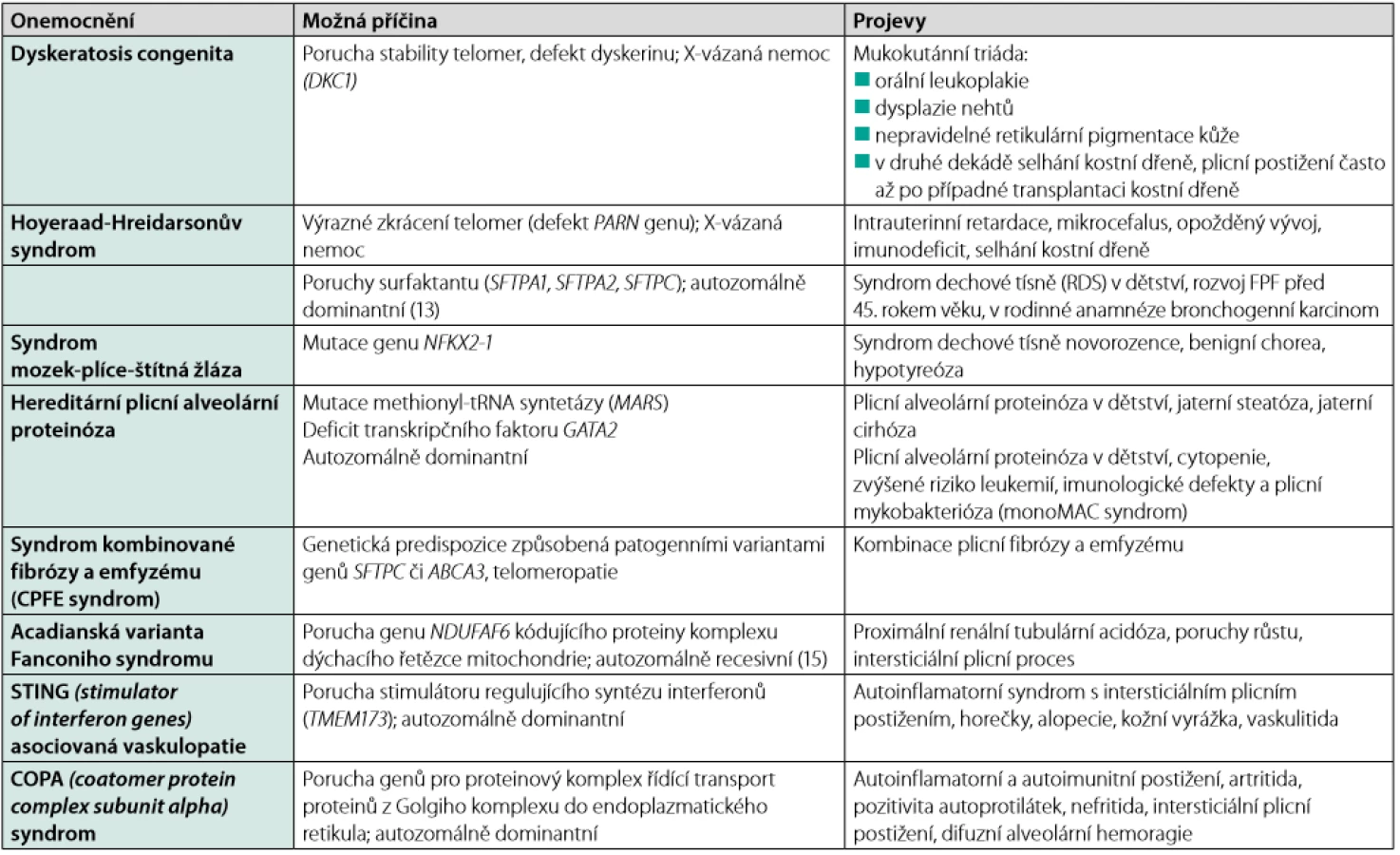

Fenotyp plicního postižení může být jakýkoliv a nemusí být u všech postižených v rámci jedné rodiny stejný. U dospělých se ale nejčastěji se setkáváme s fenotypem odpovídajícím neklasifikovatelné plicní fibróze (12–50 %) a obvyklé intersticiální pneumonii (80–22 %). Kombinace intersticiálního plicního postižení a bronchogenního karcinomu v mladším věku (32–50 let) budí podezření na patogenní variantu některého z genů pro surfaktant. U telomeropatií jsou častěji popisovány hematologické abnormality, a to včetně anémie (17–27 %), makrocytózy (24–41 %) a trombocytopenie (8–54 %), nebo jaterní onemocnění (4, 5).
Jak vyšetřit pacienta s podezřením na FPF?
Jak definice naznačuje, klíčovou pro vyslovení suspekce na FPF je kvalitně odebraná rodinná anamnéza. U většiny FPF se setkáváme s tzv. anticipačním fenoménem, tedy s narůstající tíží choroby a/nebo
častější manifestací choroby s každou další postiženou generací. Protože projevy některých FPF nezahrnují jen plicní tkáň, je doporučeno ptát se při odběru rodinné anamnézy nejen na přítomnost plicního onemocnění, ale i na výskyt jaterní cirhózy, aplastické anémie, předčasného šedivění vlasů, neurologických abnormalit a onemocnění štítné žlázy (6).
Při určení fenotypu postižení postupujeme stejně jako u jiných intersticiálních plicních procesů (anamnéza, fyzikální vyšetření, scree ‑ ning autoprotilátek, počítačová tomografue s vysokou rozlišovací schopností (HRCT) hrudníku, vyšetření plicních funkcí, bronchoskopie s bronchoalveolární laváží, transbronchiální biopsie případně kryobiopsie či chirurgická plicní biopsie). Důležité je identifikovat faktory, které mohou průběh onemocnění modifikovat nebo zhoršovat (kouření, environmentální a pracovní expozice, medikace, komorbidity). Vzhledem k tomu, že ne u všech nositelů genové mutace dojde k manifestaci onemocnění, hrají tyto zevní faktory v patogenezi též důležitou úlohu.
Genetické dispozice
V souvislosti s FPF byly popsány patogenní varianty řady genů. U FPF nalézáme varianty genů, které jsou v populaci vzácné (alelické frekvence alel < 0,1 % u tzv. genů velkého dopadu – major). Dědičnost FPF se řídí Mendelovými zákony (především autozomálně dominantní). Jak už ale bylo uvedeno výše, penetrance onemocnění je velice variabilní. Na rozdíl od vzácných variant genů s velkým dopadem mají běžné varianty menší účinek, ale jsou přítomny v populaci s vyšší frekvencí a celkově mohou přispět k většímu riziku rozvoje onemocnění – tyto varianty lze nalézt především u sporadických forem nemoci.
Genetické analýzy pacientů s FPF odhalily, že nemoc může být způsobena geny telomerázové katalytické aktivity (TERT ‑telomerase reverse transcriptase; TERC‑telomerase RNA component); geny ovlivňující biogenezi telomeráz (DKC1‑dyskerin; PARN ‑polyadenylation‑specific ribonuclease deadenylation nuclease; NAF1‑nuclear assembly factor 1 ribonucleoprotein) a geny, které mění telomery (TINF2‑telomere ‑interacting factor 2; RTEL1‑regulator of telomere‑elongation helicase‑1). Mutace spojené s FPF na počátku dospělosti se méně často nalézají v genech, které kódují surfaktantové proteiny (SFTPA1, SFTPA2 a SFTPC). Byly také popsány vzácné bialelické varianty v genech kódujících surfaktant protein B (SFTPB) a v genu pro ATP ‑vázající kazetový protein (ABCA3 – ATP ‑binding cassette subfamily A member). Celkem mutace v SFTPC, SFTPA2, TERT a TERC objasňují maximálně 20 % všech případů FPF. Byly však pozorovány i běžné varianty (frekvence alel v populaci nad 5 %) v genech spojených s rizikem FPF, nejčastěji byl pozorován jednonukleotidový polymorfismus (SNP) rs35705950 genu MUC5B (mucin 5 B) (7–10).
U koho indikovat genetické vyšetření pro podezření na FPF
- intersticiální plicní proces u dvou a více příbuzných prvního nebo druhého stupně
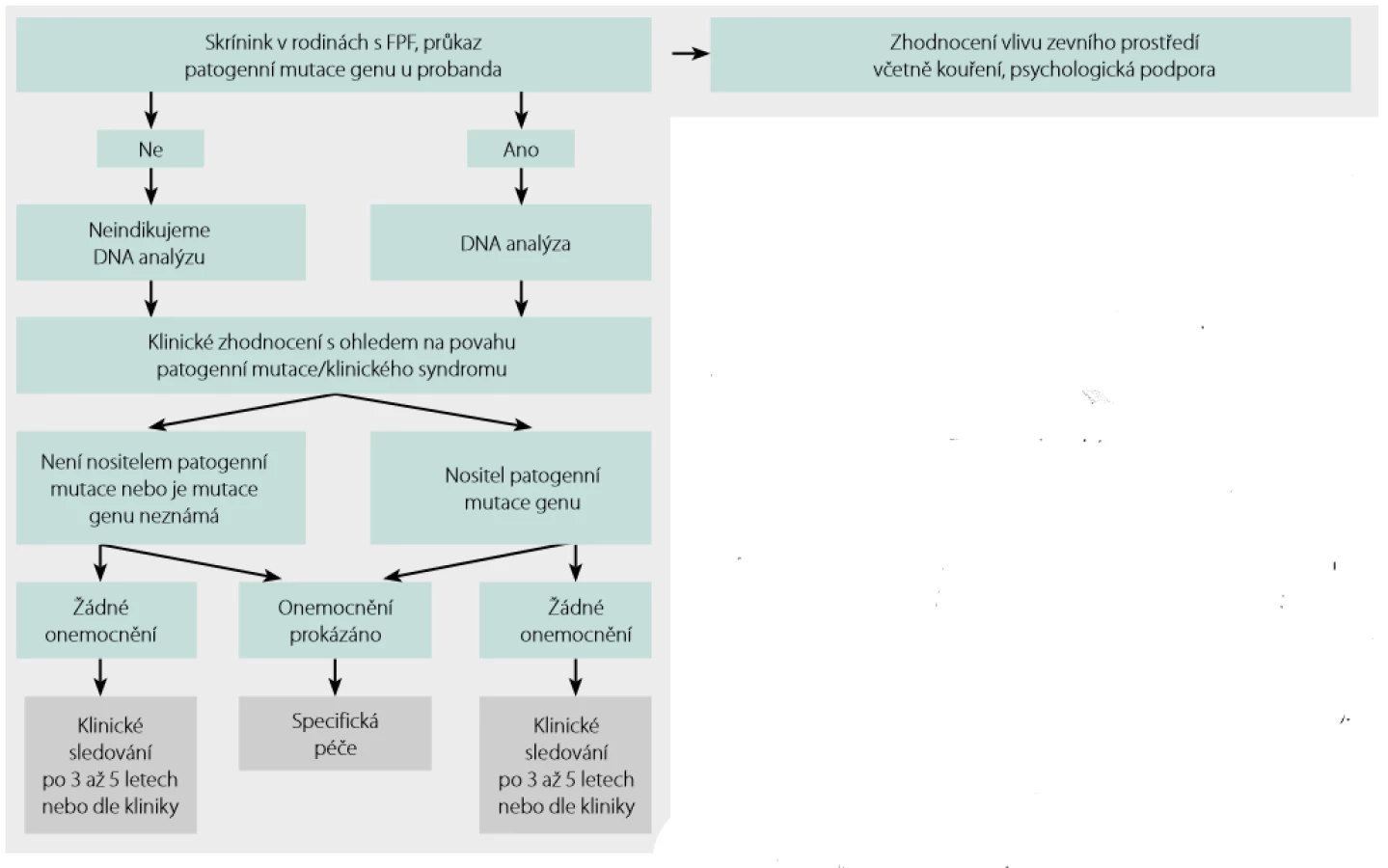
- intersticiální plicní postižení před 40. rokem věku, pokud se u příbuzných prvního nebo druhého stupně vyskytuje krevní onemocnění, jaterní onemocnění, neurologické symptomy nebo poruchy funkce štítné žlázy (tabulka 2), případně malignity v mladším věku Indikace a postup genetického testování u pacientů s podezřením na FPF shrnuje obrázek 2.

Vyhledávání a sledování rodin s FPF
Bohužel jednotná doporučení k provedení genetického testování neexistují. Odrazit se ale lze od bodů uvedených v předchozím odstavci. Genetické vyšetření indikuje genetik nejlépe po konzultaci s pneumologem věnujícím se intersticiálním plicním procesům. Na webu Společnosti lékařské genetiky a genomiky (http://www.slg.cz) lze nalézt seznam genetických ambulancí a databázi laboratoří molekulární genetiky (12). Příslušnou laboratoř pro analýzu konkrétního případu lze filtrovat podle genu, který bychom chtěli testovat. V případě nejasných nálezů a dosud neanotovaných variant genů je možné kontaktovat autory tohoto sdělení.
Je ‑li genetikem indikováno vyšetření patogenních variant genů spojených s FPF, lze se nejdříve zaměřit na vyšetření variant genů, které již byly s FPF asociovány dle literatury. Pokud nejsme úspěšní, pak lze použít metody exomového sekvenování a hledání variant genů, které dosud nebyly popsány. V tomto případě je nutné sekvenovat nejen probanda, ale rovněž jeho příbuzné, kteří dané onemocnění mají, i ty, kteří ho nemají. Srovnáním nalezených variant s fenotypem lze vytipovat geny, které hrají v rozvoji nemoci roli. Tyto nálezy je ale dále nutné ověřit analýzami in silico a případně i funkčním testováním. Postup takového vyšetření je uveden například v práci Doubková a kol. (13).
Uvedený postup schematicky shrnuje obrázek 3.

Skrínink a sledování asymptomatických členů rodiny probanda s FPF shrnuje obrázek 4.
Léčba FPF
Specifická léčba FPF není známa. U pacientů s multiorgánovým postižením se nezřídka setkáme s postupnou manifestací obtíží, která může vést k selhání funkce postiženého orgánu před rozvojem intersticiálního plicního procesu. Např. u pacientů s defektem GATA2 se může jednat o myelodysplastický syndrom nebo akutní leukemii, jejímž řešením může být transplantace krvetvorných buněk; u pacientů s Acadianskou variantou Fanconiho syndromu může jít selhání ledvin s nutností jejich transplantace (15, 16). V těchto případech dochází k rozvoji intersticiálního plicního postižení po transplantaci, a to pak může být příčinou smrti postiženého jedince.
U COPA (coatomer protein complex subunit alpha) syndromu nebo STING (stimulator of interferon genes) syndromu je léčba symptomatická.
Poruchy surfaktantu, které vedou k rozvoji syndromu dechové tísně (RDS) v dětském věku, mohou odpovídat na léčbu systémovými kortikosteroidy, azitromycinem nebo hydroxychlorochinem (17).
U dospělých pacientů je nezbytné vyloučit vliv faktorů, které mo ‑ hou průběh manifestace onemocnění zhoršovat – samozřejmostí by mělo být zanechání kouření, práce s fibrogenními prachy a vysazení potenciálně pneumotoxické medikace.
Pokud má FPF manifestaci familiární hypersenzitivní pneumonie a dominuje zánětlivý fenotyp s opacitami mléčného skla, může být kromě zamezení expozice vhodným léčebným opatřením systémová kortikoterapie.
Retrospektivní práce zahrnující pacienty s poruchami telomer ukazují, že podání azathioprinu a mykofenolátu může vést u této skupiny nemocných k akcentaci hematologických abnormalit a je dobré se jim vyhnout jak v posttransplantačních režimech (po transplantaci plic), tak nejspíše i v léčbě plicního postižení (připadalo by v úvahu u fenotypu nespecifické intersticiální pneumonie (NSIP) nebo hypersenzitivní pneumonie) (18).
U nemocných s fenotypem plicního postižení charakteru obvyklé intersticiální pneumonie (UIP), anebo s progresivní plicní fibrózou mohou být lékem volby antifibrotika, i když studie naznačují, že jejich efekt je ve srovnání se skupinou pacientů bez prokázané genetické abnormality horší. Studie srovnávající vývoj plicního postižení u nemocných s FPF léčených a neléčených antifibrotiky k dispozici není, a jelikož minimálně pirfenidon je touto skupinou nemocných tolerován dobře, u nemocných s FPF a fenotypem plicního postižení UIP a u pacientů s progresivní plicní fibrózou léčbu lze léčbu antifibrotiky doporučit (19).
U nemocných s telomeropatiemi je předmětem klinických studií použití danazolu – data ukazují, že léčba danazolem vede ke stabilizaci plicních funkcí, prodloužení telomer a zlepšení hematologických abnormalit. Lék měl ve studii ale výrazné vedlejší účinky, zejména v oblasti jater, a vysoké riziko tromboembolické nemoci. Někteří autoři vidí potenciální využití danazolu zejména v předtransplantační přípravě pacientů s prokázaným syndromem krátkých telomer, a to kvůli výše zmíněnému vysokému riziku selhání funkce kostní dřeně při imunosupresivní léčbě po transplantaci plic (20).
KORESPONDENČNÍ ADRESA AUTORA:
MUDr. Martina Šterclová, Ph.D.,
Pneumologická klinika 2. LF UK a FN Motol,
V Úvalu 84,
Praha 5, 150 06
Cit. zkr: Vnitř Lék 2020; 66(6): 365–370
Článek přijat redakcí: 14. 3. 2020
Článek přijat po recenzích k publikaci: 29. 7. 2020
Zdroje
1. Borie R, Crestani B. Familial pulmonary fibrosis: a world without frontiers. J Bras Pneumol 2019; 45: e20190303.
2. Kaur A, Mathai S, Schwartz DA. Genetics in idiopathic pulmonary fibrosis pathogenesis, prognosis and treatment. Front Med 2017; 4 : 154.
3. Doubková M, Trizuljak J, Vzralová Z, et al. Novel genetic variant of HPS1 gene in Hermansky‑Pudlak syndrome with fulminant progression of pulmonary fibrosis: a case report. BMC Pulm Med 2019; 19 : 178.
4. Krauss E, Gehrken G, Drakopanagiotakis F, et al. Clinical characteristics of patients with familial idiopathic pulmonary fibrosis. BMC Pulmonay Medicine 2019; 19 : 130.
5. Hortense AB, Santos MK, Wada D, et al. Familial pulmonary fibrosis: a heterogenous spectrum of presentations. J Bras Pneumol 2019; 45: e20180079.
6. Borie R, Kannengiesser C, Fontbrune FS, et al. Management of suspected monogenic lung fibrosis in a specialised center. Eur Respir Rev 2017; 26 : 160122.
7. Armanious MY, Chen JJ ‑L, Cogan JD, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med2007; 356 : 1317–1326.
8. Tsakiri KD, Cronkhite JT, Kuan PJ, et al. Adult – onset pulmonary fibrosis cause by mutations in telomerase. Proc Natl Acad Sci USA 2007; 104 : 7552–7557.
9. Fingerlin TE, Murphy E, Zhang W, et al. Genomewide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet 2013; 45 : 613–620.
10. Roy MG, Livraghi ‑Butrico A, Fletcher AA, et al. Muc5b is required for airway defence. Nature 2014; 505 : 412–416.
11. Kropski JA, Young LR, Cogan JD, et al. Genetic Evaluation and Testing of Patients and Families with Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Me. 2017; 195 : 1423–1428.
12. Společnost lékařské genetiky a genomiky. http://www.slg.cz. Navštíveno dne 27. 2. 2020.
13. Doubková M, Staňo Kozubík K, Radová L, et al. A novel germline mutation of the SFT ‑ PA1 gene in familial interstitial pneumonia. Hum Genome Var 2019; 6 : 12.
14. Borie R, Kannengiesser C, Gouya L, et al. Pilot experience of multidisciplinary team discussion dedicated to inherited pulmonary fibrosis. Orphanet Journal of Rare Diseases 2019; 14 : 280.
15. Hartmannová H, Piherová L, Tauchmannová K, et al. Acadian variant of Fanconi syndrome is caused by mitochondrial respiratory chain complex I deficiency due to a noncoding mutation in complex I assembly factor NDUFAF6. Hum Mol Genet 2016; 25 : 4062–4079.
16. Donadieu J, Lamant M, Fieschi C, et al. Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica 2018; 103 : 1278–1287.
17. Kazzi B, Lederer D, Arteaga ‑Solis E, et al. Recurrent diffuse lung disease due to surfactant protein C deficiency. Respir Med Case Rep 2018; 25 : 91–95.
18. Silhan LL, Shah PD, Chambers DC, et al. Lung transplantation in telomerase mutation carriers with pulmonary fibrosis. Eur Respir J 2014; 44 : 178–187.
19. Bennett D, Refini RM, Valentini ML,et al. Pirfenidone Therapy for Familial Pulmonary Fibrosis: A Real‑Life Study. Lung 2019; 197 : 147–153.
20. Mangaonkar AA, Ferrer A, Pinto E Vairo F, et al. Clinical Correlates and Treatment Outcomes for Patients With Short Telomere Syndromes. Mayo Clin Proc 2018; 93 : 834–839.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2020 Číslo 6
- Inovace v hojení ran: krytí Zetuvit Plus Silicone Border pro optimální management exsudátu z ran
- Ivabradin zlepšuje kvalitu života starších pacientů se srdečním selháním
- Fixní kombinace kandesartan/amlodipin v terapii arteriální hypertenze
- Clopidogrel je v prevenci kardiovaskulárních příhod přínosnější než kyselina acetylsalicylová
Nejčtenější v tomto čísle
- Infarkt myokardu nebo syndrom zlomeného srdce?
- Postihnutie tráviaceho traktu pri zmiešanej chorobe spojivového tkaniva (Sharpovom syndróme)
- Potravinová alergie a intolerance
- Poruchy v komplementovém systému
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy