
Gitelmanův syndrom jako častá příčina hypokalemie a hypomagnezemie
Gitelman´s syndrome as common cause of hypokalemia and hypomagnesemia
The Gitelman syndrome (GS) is an autosomal recessive disorder characterized by hypokalemic metabolic alkalosis and presence of hypocalciuria and hypomagnesemia. It is one of the most common congenital “salt-wasting” tubulopathies, where the impairment of function of the Na+-Cl- cotransporter (NCCT) in the distal convoluted tubule is primary and hypokalemia secondary. Hypomagnesemia is caused by the impairment of magnesium reabsorption through TRPM6 channel which is located just by NCCT. Clinically, patients suffer from fatigue and hypotension due to loss of salt and water and also have cramps and tetany. In some patients chondrocalcinosis can be identified which leads to protracted pain and repeated aseptic inflammations in the joints. The course of the disease, though, is typically benign, and it rarely leads to structural changes in the kidneys or renal impairment. In the period of 2004–2006 we commenced examination of patients with suspected GS based on clinical and laboratory findings within a grant project in the Czech Republic, and in the following years this methodology was introduced to the common laboratory practice. By the year 2011 we had identified 7 different causal mutations in the gene SLC12A3 (4 of them new) among the Czech population, which is responsible for the origin of this disease. The majority of patients, whose clinical findings indicated the presence of GS, had the mutation actually detected, specifically in heterozygous form; 4 individuals were then homozygous. Most of the identified mutations were missense mutations and the most common type found among the Czech population was the change 1315 G>A within the geneSLC12A3, which causes impairment of glycosylation of the NCCT transporter. Further a great number of single-nucleotide polymorphisms were found that may be involved in clinical manifestation of the disease.
Key words:
gene mutation – gene sequence – Gitelman´s syndrome – NCC channel – PCR
Autoři:
Romana Ryšavá 1; Jana Reiterová 1; Markéta Urbanová 2; Jitka Štekrová 2; Petr Lněnička 2; Vladimír Tesař 1
Působiště autorů:
Nefrologická klinika 1. LF UK a VFN v Praze
1; Ústav biologie a lékařské genetiky 1. LF UK Praha
2
Vyšlo v časopise:
Vnitř Lék 2016; 62(Suppl 6): 78-83
Kategorie:
Přehledné referáty
Souhrn
Gitelmanův syndrom (GS) je autosomálně recesivní nemocnění, které se vyznačuje hypokalemickou metabolickou alkalózou s přítomností hypokalciurie a hypomagnezemie. Patří mezi nejčastější vrozené „salt-wasting“ tubulopatie, u nichž je primární porucha funkce Na+-Cl- kotransportéru (NCCT) v distálním stočeném kanálku a hypokalemie je sekundární. Hypomagnezemie je způsobena poruchou zpětné reabsorpce hořčíku TRPM6 kanálem, který je lokalizován v těsné blízkosti NCCT. Klinicky trpí pacienti zejména únavou a hypotenzí způsobenou ztrátami soli a vody, dále křečemi a tetanií. U některých nemocných lze prokázat chondrokalcinózu, která vede k vleklým bolestem a opakovaným aseptickým zánětům v kloubech. Průběh onemocnění nicméně bývá benigní a zřídka vede ke strukturálním změnám ledvin či k poruše renální funkce. V letech 2004–2006 jsme v ČR v rámci grantového projektu zahájili vyšetřování nemocných se suspekcí na GS dle klinických a laboratorních nálezů a v následujících letech se tato metodika zavedla do běžné laboratorní praxe. Do roku 2011 jsme identifikovali v české populaci celkem 7 různých kauzálních mutací v genu SLC12A3 (z toho 4 nové), který je zodpovědný za vznik tohoto onemocnění. U většiny nemocných, u nichž svědčily klinické nálezy pro možnost přítomnosti GS, mutace nalezena byla, a to v heterozygotní formě; 4 jedinci pak byli homozygoti. Převaha nalezených mutací byly missense mutace a nejčastějším typem v české populaci byla záměna 1315 G>A v genu SLC12A3, která způsobuje poruchu glykozylace NCCT transportéru. Dále byla nalezena celá řada jednonukleotidových polymorfizmů, které se mohou podílet na klinické manifestaci onemocnění.
Klíčová slova:
genová mutace – Gitelmanův syndrom – NCCT kanál – PCR – sekvenace
Úvod
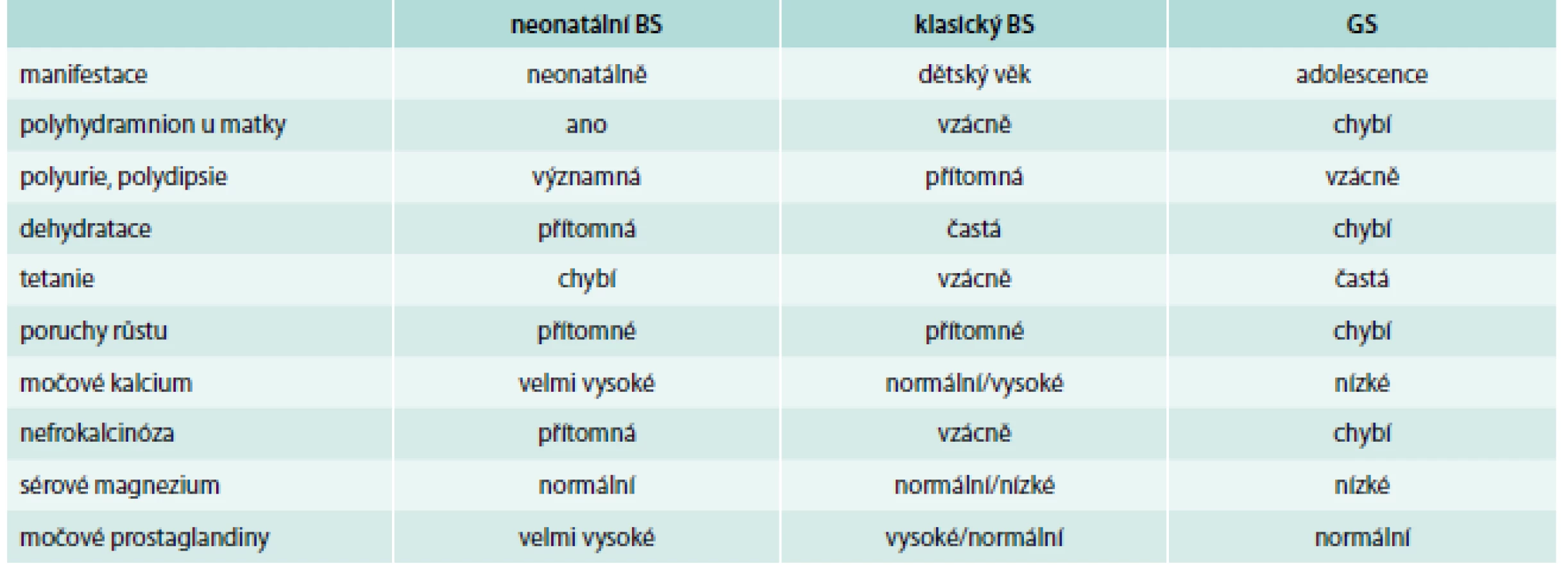
Bartterův (BS) a Gitelmanův syndrom (GS) byly původně popsány jako jedna choroba, proto se GS vstupně řadil mezi onemocnění zahrnovaná pod název Bartter´s-like syndrom [1,2]. Gitelman popsal členy rodiny, kteří kromě hypokalemie a metabolické alkalózy trpěli karpopedálními spazmy a měli sklony k tetanii, která byla vyvolána hypomagnezemií. Na základě biochemických rozdílů a později především na podkladě molekulární genetiky došlo k rozdělení na 2 rozdílné choroby s typickými znaky. Obě choroby jsou přenášeny autosomálně recesivně. U postižených osob je přítomna těžká metabolická alkalóza s výraznou hypokalemií, výrazné ztráty sodíku močí, normální hodnoty krevního tlaku se sklonem k hypotenzi a zvýšená aktivace osy renin-angiotenzin-aldosteron (RAAS). BS byl původně rozdělen na 4 typy (neonatální BS typ 1 a 2, klasický BS, neonatální BS s hluchotou), v současné době se dělí na 5 typů (BS I, II, III, IVA a IVB a V) způsobených mutacemi v 6 různých genech [3]. Většina nemocných s GS má mutace v jednom genu, které vedou k projevům GS.
Gitelmanův syndrom
GS je nemocnění, které se vyznačuje hypokalemickou metabolickou alkalózou, přítomností hypokalciurie, hypomagnezemie a hypermagneziurie. Odhadovaná prevalence onemocnění v populaci je 25 případů/1 milion (v Asii asi vyšší), přičemž se předpokládá, že heterozygotů pro GS může být v kavkazoidní/evropoidní populaci až 1 %. Tím se toto onemocnění řadí mezi nejčastější vrozené tubulopatie u dospělé populace. Gen SLC12A3Z, odpovědný za onemocnění, je lokalizován na dlouhém raménku 16. chromosomu a obsahuje 26 exonů [4]. Přímou analýzou (PCR sekvenace) bylo nalezeno již více než 250 mutací v tomto genu, přičemž 75 % z nich jsou mutace missense typu (záměna jedné báze následně vedoucí k záměně aminokyseliny); místo nejčastějších mutací nebylo zjištěno [5]. Častěji se mutace nachází na C-terminálním konci proteinu. Za použití novějších metod testování jako je MLPA (multiplex legation-dependent probe amplification) lze zachytit i rozsáhlé delece genu.
Nebyla zjištěna korelace mezi typem mutace a klinickým průběhem onemocnění. V jedné studii byl popsán těžší klinický průběh GS u mužů ve srovnání s ženami [6]. Přestože je onemocnění přenášeno autosomálně recesivně, bývá z nejasných důvodů postiženo častěji více sourozenců, než by odpovídalo předpokládaným 25 %.
Gen kóduje protein tvořící tiazid senzitivní Na+-Cl- kotransportér, označovaný jako NCCT kanál, lokalizovaný v distálním stočeném kanálku ledvin. Tento kanál je blokovatelný tiazidovými diuretiky (proto je také často nazýván jako TSC kanál – thiazide sensitive cotransporter). Membránový protein, obsahující 1 021 aminokyselin, je tvořen 12 transmembránovými doménami, N a C konce proteinu jsou lokalizovány intracelulárně. Trojrozměrná struktura transportéru není přesně známá, ale studie z poslední doby ukazují, že jeho funkci může regulovat kináza s označením WNK (with-no-lysine), přičemž zejména WNK-4 prostřednictvím fosforylace ovlivňuje funkci proteinu (WNK-1 má pravděpodobně vliv nepřímý, zprostředkovaný přes WNK-4 kinázu) [7]. Protein bez mutace je po správné glykosylaci umístěn do plazmatické membrány buňky, zatímco mutovaný protein s poruchou glykozylace zůstává většinou lokalizován v endoplazmatickém retikulu. Byly popsány i mutace, které nevedou k poruchám glykozylace a částečně funkční glykoprotein je přítomen v plazmatické membráně [8]. V současné době je popsáno 5 různých tříd mutací, které různým způsobem redukují či inhibují správnou funkci proteinu. Patří mezi ně:
- i porucha syntézy proteinu (endoplazmatické retikulum)
- ii porucha zpracování proteinu (Golgiho aparát)
- iii uložení jinak funkčního proteinu v cytoplazmě
- iv porucha funkce proteinu, který je jinak správně ukotven na membráně
- v porucha endocytózy či internalizace proteinu [7]
U malé části nemocných s GS byla prokázána přítomnost mutace v CLCNKB genu, který kóduje chloridový kanál ClC-Kb, lokalizovaný jak v distálním stočeném kanálku (těsně vedle NCCT kanálu), tak také ve vzestupné části Henleovy kličky [9]. To může způsobit, že tito jedinci mají jakýsi „překryvný“ syndrom mezi BS a GS s velmi variabilní fenotypovou manifestací (od neonatálního BS na jedné straně ke GS na straně druhé) [10]. Mutace v tomto genu by měly být vyšetřovány u všech nemocných s GS, zejména pak v případech, v nichž nebyla prokázána mutace v SLC12A3 genu.
GS je považován za benigní formu „salt-wasting“ či také „salt-losing“ nefropatií, které se většinou začnou manifestovat v adolescentním až dospělém věku. Zvýšené ztráty Na+ a Cl- způsobují hypovolemii a sekundární aktivaci systému RAAS. V důsledku toho se kompenzatorně zvyšuje reabsorpce Na+ ve sběrných kanálcích vybalancovaná zvýšenou sekrecí K+ a H+ iontů, což je příčinou hypokalemické alkalózy.
Zvýšená exkrece Mg2+ do moči je způsobena snížením jeho reabsorpce v distálním stočeném kanálku TRPM6 kanálem, jehož exprese je u GS defektní (etiologie není zcela jasná, ale je zde možný spolupodíl mutací v genu pro claudin 16) [11]. Vzhledem k tomu, že tento kanál se nachází i v duodenu, ovlivňuje hypomagnezemii pravděpodobně i snížená resorpce Mg2+ ve střevě. Byli popsáni i pacienti s GS bez hypomagnezemie. U nich byla snížena až hladina ionizovaného magnezia, které se běžně nevyšetřuje [12].
U jedné skupiny pacientů s GS byla zjištěna zvýšená kostní denzita, v jiné studii však rozdíl v kostní denzitě potvrzen nebyl. Naopak pravděpodobně hypomagnezemie vede k snížené tvorbě parathormonu a následně snížené tvorbě aktivního vitaminu D [13]. To spolu s volumovou deplecí při ztrátách Na+ a vody (která sekundárně podmiňuje zvýšenou reabsorpci Ca2+ v proximálním tubulu) může být příčinou hypokalciurie [14].
Klinické projevy a manifestace onemocnění
Klinické projevy onemocnění se objevují nejčastěji mezi 20. a 30. rokem věku. Nezřídka je průběh onemocnění asymptomatický a choroba se odhalí až po zjištění mutace u jiného člena rodiny. Nejčastěji u pacientů vznikají epizody svalové slabosti a tetanie (projevy hypomagnezemie a hypokalemie). Fyzikálně jsou pozitivní znaky Chvostkův a Trousseaův. Polyurie a opoždění růstu buď chybí či je mírného stupně. Následkem chondrokalcinózy mohou vzniknout u malého procenta pacientů bolesti kloubů. Při hodnocení kvality života u pacientů s GS však byla zjištěna zhoršená kvalita života, podobně jako u pacientů s hypertenzí nebo diabetes mellitus [15]. Nejčastěji pacienti udávali výskyt křečí, parestezií, nykturie a únavy. U 50 % pacientů s GS byl zjištěn prodloužený interval QT na EKG bez ohledu na hloubku hypokalemie a hypomagnezemie. Poruchy rytmu však nebyly při zátěžovém EKG u pacientů s GS pozorovány [16]. U pacientů byly častěji popsány i poruchy soustředění, a dokonce i některé psychiatrické diagnózy (hlavně deprese), ke kterým jistě přispívá hypokalemie. Histologicky v ledvinách nelze prokázat závažnější patologii s výjimkou mírné hypertrofie juxtaglomerulárního aparátu a při imunohistochemickém vyšetření pak mírnou redukci exprese NCCT kanálů v tubulech. U některých pacientů byla také popsána přítomnost proteinurie.
Diferenciální diagnóza
Kombinace hypokalemické metabolické alkalózy je v organizmu poměrně vzácná, a proto by tyto laboratorní nálezy, spolu s přítomností normotenze, resp. hypotenze, měly vždy vést k úvaze o přítomnosti této tubulopatie. Hlavní odlišení od BS spočívá v nálezu hypokalciurie (poměr močového kalcia k močovému kreatininu je menší než 0,1 v koncentrované moči), hypomagnezemie (pod 0,65 mmol/l) a hypermagneziurie s frakční exkrecí magnézia nad 5 %.
Projevy GS mohou být iatrogenně navozeny chronickým abúzem diuretik tiazidového typu anebo laxativy. Při podezření na abúzus diuretik (zejména u mladých žen) je vhodné opakované vyšetření moči na stanovení jejich metabolitů. Laboratorně může pomoci zejména koncentrace vylučovaných chloridů; při GS jsou chloridy v moči normální nebo jen mírně zvýšeny, zatímco diuretika několikanásobně zvyšují frakční exkreci chloridů nad normu. Pomoci může i test s intravenózně podaným furosemidem, který v případě GS nezvýší natriurézu ani kalciurii, zatímco u abúzu tiazidů se oboje signifikantně zvýší. Abúzus laxativ většinou bývá doprovázen metabolickou acidózou (ztráty bikarbonátů stolicí) a poměr K+/kreatinin v moči bývá pod 1,5.
Hypokalemická metabolická alkalóza s hypertenzí a zvýšenou kaliurií může být projevem primárního hyperaldosteronizmu, Cushingova či Liddleova syndromu, kongenitální adrenální hyperplazie či stenózy renální tepny.
Snížení koncentrace ionizovaného kalcia spolu s hypomagnezemií doprovází časné pooperační období po paratyreoidektomii, kdy ale většinou pozorujeme i hyperfosfatemii.
Familiární hypomagnezemie s hyperkalciurií a nefrokalcinózou bývá způsobená mutací v genu CLDN16 pro claudin 16 (na chromozómu 9q21.13) a ovlivňuje již zmíněný kanál TRPM6. Prognóza tohoto onemocnění je ale výrazně horší než u GS a řada nemocných progreduje (zejména díky nefrokalcinóze) do terminálního renálního selhání. Odlišení těchto dvou chorob za situace, při níž není přítomna nefrokalcinóza, je možné prakticky jen geneticky.
V diferenciální diagnóze je třeba vzít v úvahu i celou řadu dalších chorob, které vedou k tubulointersticiálnímu postižení (TI) a mohou způsobit iontové změny. Vzhledem k tomu, že většinou způsobují strukturální změny v ledvinách, mají nemocní často hypertenzi a metabolickou acidózu. Přítomnost tzv. funkčního GS lze třeba pozorovat u Sjögrenova syndromu, u nějž dominuje TI postižení ledvin. Produkty zánětu zde pravděpodobně blokují funkčnost a správné ukotvení NCCT kanálu [17].
Léčba
V léčbě GS je nutné suplementovat především ionty hořčíku, což rovněž vede ke kompenzaci močových ztrát chloridů (chlorid hořečnatý či glycerofosfát hořečnatý). Trvalá suplementace hořčíku upravuje hypomagnezemii, a je prevencí tetanie. Tím se rovněž snižuje deficit draslíku v organizmu, i když běžně bývá nutné suplementovat i draslík. Podáváme ho zejména ve formě kalium chloridu, což částečně zmírňuje již tak významnou metabolickou alkalózu. V některých studiích je popisován pozitivní efekt léků blokujících osu RAAS, které vedou k zvýšení hladiny draslíku (např. podávání antagonistů aldosteronu). Snížení aktivace této osy může mít určitý kardioprotektivní účinek, ale bývá často spojeno s výskytem symptomatické hypotenze, zhoršením únavy a zvýšením ztrát chloridů do moči. Při použití eplerenonu místo spironolaktonu může pozitivní efekt léčby převažovat; eplerenom byl kazuisticky podáván i u těhotných žen s GS [18]. U velmi těžkých forem onemocnění lze zkusit podávat i inhibitory cyklooxygenázy 1 (indometacin), které mohou částečně snížit vylučování sodíku, a tím následně i sekreci draslíku. Jejich efekt je však omezený, lépe fungují u neonatálních forem BS. Při této léčbě je ale nutné pečlivě monitorovat případné nežádoucí účinku léku na zažívací trakt a také glomerulární filtraci [19]. Dlouhodobá prognóza je příznivá, obvykle s celoživotní potřebou suplementace hořčíku a draslíku. K selhání ledvin dochází výjimečně, dosud bylo popsáno jen několik případů.
Naše zkušenosti s vyšetřováním Gitelmanova syndromu
V letech 2004–2006 jsme v ČR zahájili v rámci grantového projektu IGA skríningové vyšetřování nemocných s hypokalemií, metabolickou alkalózou a suspekcí na vrozenou tubulopatii. Hlavním cílem naší práce bylo provedení mutační analýzy u pacientů se suspektním BS a GS z České a také Slovenské republiky (celkem vyšetřeno kolem 100 pacientů). Díky grantu byla metodika zavedena do praxe a ve vyšetřování nemocných se pokračovalo i v následujících letech již v rámci běžné rutinní diagnostiky hrazené z prostředků veřejného zdravotního pojištění.
Suspekce na GS byla vyslovena na základě definovaných laboratorních parametrů (v séru: metabolická alkalóza: pH > 7,44, bikarbonát > 26 mmol/l, kalium < 3,5 mmol/l, magnezium < 0,65 mmol/l, zvýšená hladina reninu a aldosteronu, v moči: zvýšená exkreční frakce iontů sodíku, kalia, magnezia, chloridů, snížená exkreční frakce iontů kalcia). U všech pacientů byla přítomna hypokalemická metabolická alkalóza, hypomagnezemie a hypokalciurie. Klinické údaje od 16 vybraných pacientů s GS jsou uvedeny v tab. 1.

Po provedení izolace DNA z periferních lymfocytů bylo všech 26 exonů genu SLC12A3 a přilehlých částí intronů naamplifikováno PCR 27 páry popsaných primerů [4]. Následně byly všechny exony od jednotlivých pacientů sekvenovány v obou směrech na sekvenátoru ABI Prism TM 310. Většina vzorků byla také vyšetřena MLPA technikou, aby mohly být zachyceny větší genové delece či inzerce, které se klasickou sekvenací nemusí odhalit. Pokud nebyla odhalena kauzální mutace v tomto genu, pokračovalo se vyšetřováním mutace v CLCNKB genu (gen spojený s výskytem klasické formy BS, vzácněji může způsobovat i GS). Dále bylo zjišťováno, zda přítomnost některých jednonukleotidových polymorfizmů (SNP) může přispívat k manifestaci onemocnění. Pro kontrolu byla použita DNA od 100 zdravých dobrovolníků.
4 ze 7 různých kauzálních mutací nalezených v naší populaci nemocných nebyly dříve zjištěny jinými autory, kteří se zabývali analýzou genu SLC12A3, a tedy byly identifikovány nově [20,21]. Dvě z nich jsou mutace posunové, u nichž je výsledný protein zkrácen, a to v jednom případě na 11 % délky a ve druhém na 25 % délky. Takto zkrácené formy proteinu jsou plně nefunkční. U 4 pacientů byla detekovaná mutace v homozygotním stavu, u většiny nemocných šlo o mutace heterozygotní. Nejčastějším typem mutace nalezené v našem souboru byla mutace 1315 G > A způsobující záměnu aminokyselin Gly439Ser. Ta se nachází v exonu číslo 10 a mění smysl výsledného proteinu. Při analýze genu SLC12A3 v české populaci je proto logické se nejprve zaměřit na vyšetření této nejčastěji se vyskytující mutace a dále pak na další časté mutace, k nimž patří i Arg904Glu a Gly741Arg. Jak lze pozorovat na obr. 1, zjištěné missense mutace se nacházejí v průběhu celého proteinu, avšak s vyšším výskytem v C-terminální oblasti (obr. 1). Z tohoto však nelze usuzovat na jakýsi hot-spot mutací, neboť C-terminální oblast představuje až třetina celého proteinu. Řada námi nalezených variant byla později popsána jakožto SNP polymorfizmy, tedy varianty, které nemají na funkci proteinu zásadní vliv.

U 3 jedinců se nepodařilo nalézt ani jednu mutaci ve zkoumaném genu. Tito pacienti byli dále zkoumáni na přítomnost mutace v jiných kandidátních genech (CLCNKB gen). Naopak jeden nemocný, který klinicky byl zařazen do skupiny klasický BS a neměl prokázánu mutaci v CLCNKB genu, byl dodatečně vyšetřen v genu SLC12A3 a zde byly prokázány 2 patogenetické varianty.
Při sledování korelace mezi konkrétní mutací a fenotypem nebyl zjištěn žádný významný ukazatel. Pokud jsme vyšetřovali příbuzné pacientů, byli vždy nositeli stejných mutací, jako pacient.
Závěr
GS je poměrně častou vrozenou příčinou hypokalemické metabolické alkalózy a při splnění kombinace určitých laboratorních nálezů je záchyt průkazu genetické abnormality poměrně vysoký (tab. 2). To, že se u několika málo pacientů v našem souboru nepodařilo mutaci odhalit, může být způsobeno tím, že mutace mohou ležet v oblasti promotoru genu nebo v oblasti intronů, které potom mohou ovlivňovat sestřih. Tyto části genu se běžně nevyšetřují. Pokud se hypokalemie a hypomagnezemie objeví u adolescentů a jedinců ve věku mezi 20.–30. rokem života, je potřeba na toto onemocnění myslet. Při průkazu mutace u daného pacienta je vždy vhodné vyšetřit jeho blízké příbuzné, zda nejsou nositeli stejné mutace a zda netrpí stejným onemocněním, které v té době může mít asymptomatický průběh.
prof. MUDr. Romana Ryšavá, CSc.
rysavar@vfn.cz
Klinika nefrologie 1. LF UK a VFN v Praze
www.vfn.cz
Doručeno do redakce 26. 8. 2016
Přijato po recenzi 5. 10. 2016
Zdroje
1. Bartter FC, Pronove P, Gill JR et al. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis: a new syndrome. Am J Med 1962; 33 : 811–828.
2. Gitelman HJ, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Transactions Ass Am Phys 1966; 79 : 221–235.
3. Shibli AA, Narchi H. Bartter and Gitelman syndromes: Spectrum of clinical manifestations caused by different mutations. World J Methodol 2015; 5(2): 55–61. Dostupné z DOI: <http://dx.doi.org/10.5662/wjm.v5.i2.55>.
4. Simon DB, Nelson-Willliams C, Bia MJ et al. Gitelman´s variant of Bartter´s syndrome, inherited hypokalemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nature Genetics 1996; 12(1): 24–30.
5. Takeuchi Y, Mishima E, Shima H et al. Exonic mutations in the SLC12A3 gene cause exon skipping and premature termination in Gitelman Syndrome. J Am Soc Nephrol 2014; 26(6): 1–9. Dostupné z DOI: <http://dx.doi.org/10.1681/ASN.2014030260>.
6. Lin SH, Cheng NL, Hsu YJ et al. Intrafamiliar phenotype variability in patients with Gitelman syndrome having the same mutaitons in their thiazide-sensitive sodium/chloride cotransporter. Am J Kidney Dis 2004; 43(2): 304–312.
7. Wang L, Dong Ch, Xi YG et al. Thiaside-sensitive Na+-Cl - contrasporter: genetic polymorphisms and human diseases. Acta Biochim Biophys Sin 2015; 47(5): 325–334. Dostupné z DOI: <http://dx.doi.org/10.1093/abbs/gmv020>.
8. De Jong JC, Van der Vliet WA, Van den Heuvel L et al. Functional expression of mutations in the human NaCl cotransporter: evidence for impaired routing mechanisms in Gitelman´s syndrome. J Am Soc Nephrol 2002; 13(6): 1442–1448.
9. Peters M, Jeck N, Reinalter S et al. Clinical presentation of genetically defined patients with hypokale - mic salt-losing tubulopathies. Am J Med 2002; 112(3): 183–190.
10. Knoers NVAM, Levtchenko EN. Gitelman syndrome. Orphanet J Rare Dis 2008; 3 : 22. Dostupné z DOI: <http://dx.doi.org/10.1186/1750–1172–3-22>.
11. Graziani G, Fedeli C, Moroni L et al. Gitelman syndrome: pathophysiological and clinical aspects. QJM 2010; 103(10): 741–748. Dostupné z DOI: <http://dx.doi.org/10.1093/qjmed/hcq123>.
12. Tosi F, Bianda N, Truttmann AC et al. Normal plasma total magnesium in Gitelman syndrome. Am J Med 2004; 116(8): 573–574.
13. Bianchetti MG, Bettinelli A, Casez JP et al. Evidence for disturbed regulation of calciotropic hormone metabolites in Gitelman syndrome. J Clin Endocrinol Metab 1995; 80(1): 224–228.
14. Nijenhuis T, Vallon V, van der Kemp AW at al. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channels abundance explains thiazide-induced hypocalciuria and hypomagnesemia. Clin Incevt 2005; 115(6): 1651–1658.
15. Cruz DN, Shaer AJ, Bia MJ et al. Yale Gitelman‘s and Bartter‘s Syndrome Collaborative Study Group. Gitelman‘s syndrome revisited: an evaluation of symptoms and health-related quality of life. Kidney Int 2001; 59(2): 710–717.
16. Foglia P, Bettinelli A, Tosetto C et al. Cardiac work up in primary renal hypokalaemia-hypomagnesaemia (Gitelman syndrome). Nephrol Dial Transpl 2004; 19(6): 1398–1402.
17. Chen YC, Yang WC, Yang AH et al. Primary Sjogren´S syndrome associated with Gitelman´s syndrome presenting with muscular paralysis. Am J Kidney Dis 2003; 42(3): 586–590.
18. Moustakakis MN, Bockorny M. Gitelman syndrome and pregnancy. Clin Kidney J 2012; 5(6): 552–555. Dostupné z DOI: <http://dx.doi.org/10.1093/ckj/sfs126>.
19. Blanchard A, Vargas-Poussou R, Vallet M et al. Indomethacin, amiloride, or eplerenone for treating hypokalemia i Gitelman syndrome. J Am Soc Nephrol 2015; 26(2): 468–475. Dostupné z DOI: <http://dx.doi.org/10.1681/ASN.2014030293>.
20. Urbanová M, Reiterová J, Štekrová J et al. Genetic analysis of Gitelman’s syndrome patients from the Czech republic and Slovakia – Three novel mutations found. Kidney and Blood Press Res 2006; 29(6): 360–365.
21. Urbanová M, Reiterova J, Štektova J et al. DNA analysis of renal electrolyte transporter genes among patients suffering from Bartter and Gitelman syndromes – summary of mutation screening. Folia Biologica (Praha) 2011; 57(2): 65–73.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2016 Číslo Suppl 6
- Plicní hypertenze – syndrom mnoha tváří – vyžaduje přesnou diagnostiku a specializovanou léčbu
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Limity glykovaného hemoglobinu a význam dalších glykovaných proteinů
- Korelace dávky levothyroxinu s titrem autoimunitních protilátek u primární hypotyreózy
- Zeolit-jodový komplex pomáhá v péči o infikované rány
Nejčtenější v tomto čísle
- Metabolická acidóza u chronického onemocnění ledvin
- Současné možnosti léčby hyponatremie
- Gitelmanův syndrom jako častá příčina hypokalemie a hypomagnezemie
- Osteoporóza – epidemiologie a patogeneze
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy