
Perzistentní průjmy, hypotenze a polyneuropatie
Persistent diarrhoea, hypotension, polyneuropathy
We present a case report of a 59-year-old man with a history of arterial hypertension and excision of malignant melanoma. He was admitted to the hospital because of two months history of diarrhoea, weight loss and circulatory collapse. In addition, the patient suffered from marked vegetative instability with symptomatic hypotension, polyneuropathy and progression of renal insufficiency, without proteinuria. Complex examination did not reveal neoplasms, endocrine, autoimmune, infectious or neurodegenerative disorders. A serial biopsy of colon failed to provide a clue to the diagnosis. However, AA amyloidosis was found on the kidney biopsy. Neither chronic inflammation nor malignancy was revealed and, hence, no causal treatment could have been established. The patient died from multiple organ failure. The autopsy confirmed systemic AA amyloidosis. The triad consisting of diarrhoea, polyneuropathy and hypotension should rise the suspicion on amyloidosis.
Key words:
amyloidosis – biopsy – hypotension – polyneuropathy – diarrhoea – renal insufficiency
Autoři:
R. Sýkora 1; J. Raděj 1; I. Novák 1; A. Kroužecký 1; J. Mareš 1; I. Irová 2; Š. Hadravská 2; J. Chvojka 1; T. Karvunidis 1; T. Maňáková 2; M. Matějovič 1
Působiště autorů:
I. interní klinika Lékařské fakulty UK a FN Plzeň, přednosta doc. MUDr. Martin Matějovič, Ph. D.
1; Šiklův patologicko‑anatomický ústav Lékařské fakulty UK a FN Plzeň, přednosta prof. MUDr. Michal Michal
2
Vyšlo v časopise:
Vnitř Lék 2008; 54(11): 1106-1110
Kategorie:
Kazuistika
Souhrn
Prezentujeme případ 59letého muže s anamnézou arteriální hypertenze a excize maligního melanomu v minulosti. Byl hospitalizován pro 2 měsíce trvající průjmy, úbytek na váze a kolapsové stavy. Současně trpěl polyneuropatií, nestabilitou autonomního nervového systému se symptomatickými poklesy krevního tlaku a progresí renální insuficience bez proteinurie. Komplexní diagnostikou zaměřenou k vyloučení nádorového, autoimunitního, endokrinního, infekčního i neurodegenerativního onemocnění se nedařilo zjistit původ obtíží nemocného. Po negativním výsledku etážové biopsie střevní sliznice byla prokázána AA-amyloidóza z biopsie ledviny. Chronický zánětlivý proces ani nádorové onemocnění vysvětlující systémové postižení amyloidem nebyly diagnostikovány, proto nebylo možné zahájit kauzální léčbu. Nemocný zemřel pod obrazem multiorgánového selhání. Post mortem byla potvrzena systémová AA‑amyloidóza. Triáda zahrnující průjmy, hypotenzi a projevy polyneuropatie by měla kliniky vést k podezření na amyloidózu.
Klíčová slova:
amyloidóza – biopsie – hypotenze – polyneuropatie – průjmy – renální insuficience
Úvod
Amyloidózu tvoří skupina získaných i dědičných chorob, které vznikají na podkladě poruchy metabolizmu proteinů, kdy dochází k mimobuněčnému generalizovanému nebo lokálnímu ukládání amyloidu do tkání téměř všech orgánových systémů [1]. Amyloid je amorfní hmota, tvořená nerozpustnými depozity fibrilárních proteinů, ve struktuře β-skládaného listu, odolná vůči proteolýze. Doposud bylo popsáno 25 prekurzorových proteinů amyloidu [2]. Onemocnění lze klasifikovat jako:
- a) primární (AL) amyloidózu, způsobenou ukládáním amyloidových fibril odvozených z lehkých řetězců imunoglobulinů, vznikající při lymfoproliferativních onemocněních s plazmocytární diferenciací
- b) sekundární (AA) amyloidózu, jejímž podkladem je ukládání proteinu akutní fáze, tzv. sérového amyloidového A-proteinu (SAA), vznikajícího v podmínkách chronických onemocnění. Příkladem jsou především dlouhodobě aktivní formy revmatoidní artritidy, anyklózující spondylitidy, Crohnovy choroby či některých vaskulitid, zatímco chronické záněty (např. osteomyelitis, tuberkulóza) jako příčina AA-amyloidózy v současnosti ustupují v rozvinutých zemích do pozadí
- c) hereditární a familiární amyloidózy, které jsou převážně autozomálně dědičné; nejčastější příčinou těchto amyloidóz je mutace transthyretinu (ATTR, např. familiární amyloidová polyneuropatie a/nebo kardiomyopatie způsobená ukádáním transthyretinu)
- d) amyloidózu dialyzovaných nemocných, kde prekurzorem amyloidu je β2-mikroglobulin
- e) senilní systémovou amyloidózu
- f ) též jsou popsány vzácné formy orgánově specifických lokálních forem amyloidózy AL i AA typu
Klinický obraz amyloidózy je tvořen různorodými příznaky, které odpovídají dysfunkcím orgánových systémů, ve kterých je amyloid ukládán, např. polyneuropatie, nefrotický syndrom, hypotenze, arytmie a chronické městnavé srdeční selhání (při amyloidové kardiomyopatii), poruchy trávicího traktu, syndrom karpálního tunelu a jiné [2–4]. Postižení různých orgánů vede k souboru příznaků, které jsou často zavádějící nebo se překrývají s jinými, oddalují histologickou diagnostiku, a tím zahájení vhodné léčby nemocnění.
Diagnostiku amyloidózy ztěžuje:
- a) její relativně vzácný výskyt
- b) široká diferenciální diagnostika příznaků
- c) nutné histologické potvrzení diagnózy
Při pátrání po přítomnosti systémové amyloidózy lze zvolit necílený odběr tkáně, např. biopsii rekta, dásně, jazyka. Podobně necílená aspirace podkožního tuku z oblasti břicha vykazuje vysokou senzitivitu [5]. Přetrvává‑li podezření, je vhodné při negativním výsledku biopsie rekta a břišního tuku doplnit cílenou biopsii z orgánu, který nejvíce vykazuje známky postižení. Nejčastěji se jedná o biopsii ledvin, jater, nervus suralis (při neuropatii) či o endomyokardiální biopsii [2,6]. Ve sliznici rekta je důležitý záchyt hlubších vrstev sliznice, případně submukózy.
Preparát je hodnocen světelnou mikroskopií při barvení kongo-červení. V polarizovaném světle je patrná fluorescence barveného amyloidu. Imunohistochemické vyšetření specifickými protilátkami je výtěžné při identifikaci AA a ATTR amyloidu, méně však AL-amyloidu, kdy je vyšetření limitováno ztrátou antigenních epitopů, ke kterým dochází v průběhu amyloidogeneze. Imunoelektronová mikroskopie je rovněž přínosná pro určení typu amyloidu. V případě familiární amyloidózy lze přesnou charakteristiku amyloidu provést vyšetřením sekvence aminokyselin nebo hmotnostní spektroskopií a DNA analýzou amyloidogenní mutace. Tyto metody jsou však dostupné jen ve specializovaných centrech. Scintigrafie s jodem značenou komponentou sérového amyloidu (SAP) s velmi vysokou senzitivitou (90 %) i specifitou (93 %) pro AA - a AL-amyloid není vhodná pro dědičné amyloidózy s TTR a není rutinně dostupná [7].
Pacienti s dokumentovanou amyloidózou, kteří splňují diagnostická kritéria pro mnohočetný myelom či Waldenströmovu makroglobulinemii, nemusí být dále podrobně diagnostikováni. V ostatních případech AL-amyloidózy je nutné určit, zda je přítomna monoklonální plazmocelulární proliferace (vyšetřením kostní dřeně, doplněné o imunohistochemické vyšetření s protilátkou proti κ a λ lehkým řetězcům) společně s detekcí monoklonálního proteinu v séru a moči elektroforézou, imunofixací a ev. kvantifikací volných lehkých řetězců v séru. Posledně jmenované vyšetření lze využít zejména u nemocných s AL-amyloidózou, kteří nemají monoklonální protein v imunofixaci, případně při sledování odpovědi na léčbu.
Podle typu amyloidózy a v případě známé příčiny ukládání fibrilárních depozit amyloidu lze zahájit kauzální léčbu nebo symptomatickou léčbou zlepšit kvalitu života nemocného [8]. V terapii amyloidózy kromě léčby základního onemocnění (např. mnohočetného myelomu) se snažíme symptomaticky ovlivnit také nefrotický syndrom či srdeční selhání. V případech ATTR u familiárních forem lze zvážit transplantaci jater [8]. V současnosti jsou předmětem klinického výzkumu terapeutické látky ovlivňující tvorbu fibrilárních depozit na různých úrovních metabolizmu [9–11]. Bližší popis možností léčby amyloidózy není cílem této práce a zde odkazujeme především k recentním souhrnným článkům [12].
Prognózu pacientů postižených amyloidózou ovlivňuje typ amyloidózy a charakter orgánového postižení [2]. Jen 51 % pacientů s neléčenou AL-amyloidózou přežívá 1 rok, 16 % pak déle než 5 let [4]. Nemocní s AL-amyloidózou, kteří jsou léčeni ve specializovaných centrech, mají v posledních letech významně lepší prognózu (medián přežití 40 měsíců) oproti 13 měsícům popisovaným v roce 1990 [13]. V případě symptomatického postižení srdce dosahuje průměrné přežití 6–8 měsíců. Citlivým prognostickým ukazatelem u těchto forem amyloidózy je vedle echokardiografického vyšetření sledování dynamiky hladin NT-proBNP a troponinu T a I [14].
U AA-amyloidózy jsou přesné údaje o prognóze hůře dostupné vzhledem k rozptýlenosti nemocných mezi různé klinické obory. Prognózu určuje i průběh a léčba základního onemocnění. Dle studií ve skupině neléčených pacientů 4 roky přežívá kolem 50 % nemocných, 10 let pak jen 25 % pacientů [15]. Prognóza ATTR-amyloidózy závisí na typu mutace. Mutace TTR, manifestovaná ve 20–30 letech, vede obvykle ke kratšímu přežití v důsledku rychle progredující kardiomyopatie a neuropatie [2].
Popis případu
Na I. interní kliniku FN Plzeň byl přijat 59letý muž pro 2 měsíce trvající vodnaté průjmy, výrazný váhový úbytek a kolapsové stavy. V osobní anamnéze trpěl arteriální hypertenzí, ischemickou chorobou dolních končetin a depresí. Šest měsíců před hospitalizací mu byl excidován maligní melanom kůže z oblasti sterna, bez známek generalizace. Nemocný byl v důchodu, bez významné pracovní anamnézy, nekuřák, bez abúzu alkoholu.
Zobrazovací, endoskopické ani mikrobiologické vyšetřovací metody trávicího traktu neodhalily příčinu profuzních průjmů. Negativní výsledky vyšetření kardiovaskulárního systému (echokardiografie, holterovská monitorace krevního tlaku a EKG) ukazovaly na vazoregulační příčinu těžké symptomatické hypotenze, vyžadující i nepřímou srdeční masáž, oběhovou podporu vazopresory a opakovaný pobyt na jednotce intenzivní péče. K diagnóze nepřispěla četná biochemická, hematologická, imunologická či endokrinologická vyšetření. Přítomné dysestezie dolních končetin a následně hyperestezie celého těla byly neurologicky vyhodnoceny jako polyneuropatie.
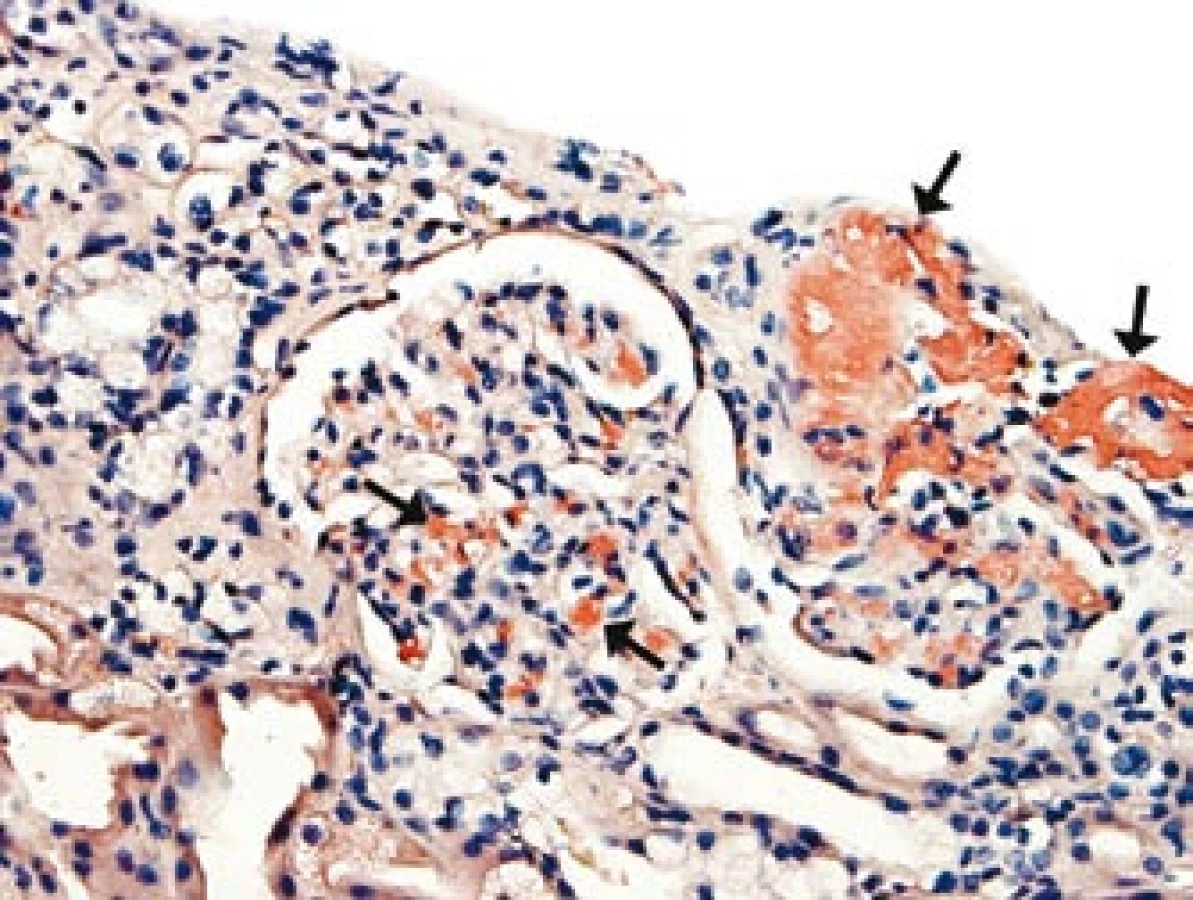
Přetrvávající hypotenze s opakovanými kolapsovými stavy, které nebyly vysvětlitelné srdeční dysfunkcí či nedostatečností nadledvin, vedly k diferenciální diagnostice autonomní dysfunkce (tab. 1). Komplexním neurologickým vyšetřením (EEG, CT, MRI, lumbální punkce) byla vyloučena autonomní dysfunkce s postižením centrální nervové soustavy typu multisystémové atrofie (Shy-Dragerova syndromu). Současně jsme se soustředili na diferenciální diagnostiku autonomního selhání s postižením periferního nervového systému a především na vyloučení amyloidózy. Etážová biopsie sliznice tlustého střeva však neprokázala infiltraci amyloidem. Obtíže nemocného se zhoršovaly, progredovala renální nedostatečnost, avšak bez proteinurie. Následnou biopsií ledviny byla potvrzena depozita charakteru AA-amyloidu v arteriolách (obr. 1).
![Selhání autonomního nervového systému (upraveno podle Freemana) [18].](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image/7dbab7ff2c09807a0d924a6681aa01f4.png)
Nemocný byl opakovaně hospitalizován na jednotce intenzivní péče pro poruchy vědomí a těžké hypotenze při autonomní nestabilitě. Přidružovaly se infekční komplikace. Byla nutná náhrada funkce ledvin, umělá plicní ventilace a vysazování oběhové podpory noradrenalinem bylo možné jen při podávání perorálního α1-sympatomimetika midodrinu (Gutron) a intravenózního terlipresinu (Remestyp). Po 83 dnech hospitalizace pacient zemřel pod obrazem multiorgánového selhání. Histologicky byla potvrzena infiltrace cév AA-amyloidem, především arteriol ledvin, srdce (obr. 2), lymfatických uzlin, gastrointestinálního traktu, jater, plic, nadledvin, prostaty a kostní dřeně.

Diskuze
Komplexní diagnostikou se nedařilo zjistit původ obtíží nemocného. Amyloid nebyl zastižen ve standardních bioptických vzorcích střevní sliznice z následujících důvodů:
- a) AA-amyloid v našem případě postihoval především arterioly střevní sliznice.
- b) Infiltrace cév amyloidem nebyla pravděpodobně zachycena vzhledem k nehomogenní distribuci AA-amyloidu v trávicím traktu, kterou potvrzuje i v literatuře popisovaná senzitivita rektálních biopsií mezi 75 a 85 % [16].
Amyloidóza byla následně diagnostikována z biopsie ledviny a na základě histologického, imunofluorescenčního, elektronmikroskopického a imunohistochemického zhodnocení byl amyloid klasifikován jako typ AA. Převažující postižení arteriol (obr. 1) a prerenální faktory mohou v tomto případě vysvětlit progredující renální insuficienci bez proteinurie. Chronický zánětlivý infekční i neinfekční proces či malignita vysvětlující systémové postižení amyloidem AA byly vyloučeny laboratorními i zobrazovacími metodami, které byly ve vyšetřovacím procesu doplněny také o octreotidovou scintigrafii, celotělovou pozitronovou emisní tomografii – CT, vyšetření periferní krve průtokovou cytometrií, ale též pitvou. SAP scintigrafie nebyla při diagnostice amyloidózy a distribuce amyloidu pro nedostupnost metody provedena. Dominující postižení arteriol všech orgánových systémů, odhalené histologickým vyšetřením, bylo odpovědné za těžké symptomatické hypotenze, průjmy i polyneuropatii.
U našeho nemocného lze diskutovat diagnózu familiární, resp. dědičné formy amyloidózy z následujících důvodů:
- a) Familiární amyloidózy jsou nejvíce heterogenní skupinou onemocnění, často autozomálně dominantně dědičné. Nejčastějším zástupcem je familiární amyloidová polyneuropatie, která však není typická pro naši geografickou oblast. Nicméně lze připustit, že se jednalo o sporadický výskyt familiární amyloidózy i vzhledem k faktu, že otec nemocného zemřel také ve věku kolem 60 let na blíže nevysvětlené neurologické onemocnění projevující se dle popisu rodiny obdobnými příznaky, bližší informace však rodina nemocného neposkytla. Perineurální šíření amyloidu, charakteristické pro familiární amyloidovou polyneuropatii, však nebylo pitvou prokázáno, jemná depozita amyloidu v periferních nervech jsou oproti infiltraci cév hůře průkazná při barvení kongo-červení [6].
- b) Po extenzivní diagnostice a pitvě nebyl zjištěn chronický zánětlivý proces ani malignita svědčící pro sekundární AA-amyloidózu. U familiární amyloidózy je prekurzorem amyloidu TTR, jehož vlákna však mohou být kombinovaná s AA proteinem, nicméně přesné stanovení typu amyloidu u vzácných forem familiárních amyloidóz je nemožné bez speciálních metod molekulární biologie [17], které v našem případě nebyly použity vzhledem k rychlé progresi onemocnění a špatné prognóze nemocného. Metody zaměřené na diagnostiku familiárních typů amyloidóz jsou dostupné v ČR na Ústavu dědičných a metabolických poruch v Praze. Také je nutné připustit možnost, že proces chronické inflamace nebyl zjištěn, přestože existoval.
- c) V literatuře není popisován vztah maligního melanomu a amyloidózy. Amyloidóza je spojovaná s endokrinními karcinomy štítné žlázy, pankreatu, feochromocytomem a nediferencovaným karcinomem žaludku, které byly vyloučeny.
Závěr
Triáda zahrnující průjmy, hypotenzi a projevy polyneuropatie by měla lékaře vždy vést k podezření na amyloidózu. Negativní nález v bioptickém materiálu diagnózu amyloidózy nevylučuje a vyžaduje opakované odběry či biopsie jiných postižených tkání.
Práce byla podpořena Výzkumným záměrem MSM 0021620819 „Náhrada a podpora funkce některých životně důležitých orgánů“.
doc. MUDr. Martin Matějovič, Ph.D.
www.fnplzen.cz
e‑mail: matejovic@fnplzen.cz
Doručeno do redakce: 12. 5. 2008
Přijato pro recenzi: 18. 8. 2008
Zdroje
1. Westermark P, Benson MD, Buxbaum JN et al. Nomenclature Committee of the International Society of Amyloidosis. Amyloid: toward terminology clarification. Report from the Nomenclature Committee of the International Society of Amyloidosis. Amyloid 2005; 12 : 1–4.
2. Adam Z, Ščudla V. Klinické projevy a diagnostika AL-amyloidózy a některých dalších typů amyloidóz. Vnitř Lék 2001; 47 : 36–45.
3. Brychta T, Pařenica J, Zatočil T et al. Restriktivní kardiomyopatie jako projev primární amyloidózy. Vnitř Lék 2004; 50 : 66–71.
4. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Semin Hematol 1995; 32 : 45–59.
5. Maysouye I. Diagnostic screening of systemic amyloidosis by abdominal fat aspiration: an analysis of 100 cases. Am J Dermatopathol 1997; 19 : 41–45.
6. Li K, Kyle RA, Dyck PJ. Immunohistochemical characterization of amyloid proteins in sural nerves and clinical associations in amyloid neuropathy. Am J Pathol 1992; 141 : 217–226.
7. Hazenberg BP, Bijzet J, Limburg PC et al. Diagnostic performance of amyloid A protein quantification in fat tissue of patients with clinical AA amyloidosis. Amyloid 2007; 14 : 133–140.
8. Ryšavá R. Perspektivy: Současné terapeutické postupy u amyloidózy ledvin. Postgrad Nefrol 2006; 4 : 50–51.
9. Dember LM. Emerging treatment approaches for the systemic amyloidoses. Kidney Int 2005; 68 : 1377–1390.
10. Lachmann H, Obici L, Berber L et al. Results of multi‑center, randomized, placebo-controlled trial for the treatment of amyloid A (AA) amyloidosis associated renal disease with NC-503 (eprodisate disodium). Nephrol Dial Transplant 2006; 21 (Suppl 4): iv294.
11. Gillmore JD, Hawkins PN. Drug insight: emerging therapies for amyloidosis. Nature Clin Pract Nephrol 2006; 2 : 263–270.
12. Wechalekar AD, Hawkins PN, Gillmore JD. Perspectives in treatment of AL amyloidosis. Br J Haematol 2008; 140 : 365–377.
13. Palladini G, Russo P, Nuvolone M et al. Treatment with oral melphalan plus dexamethasone produces long‑term remissions in AL amyloidosis. Blood 2007; 110 : 787–788.
14. Dispenzieri A, Gertz MA, Kyle RA et al. Serum cardiac troponins and N‑terminal pro‑brain natriuretic peptide: a staging system for primary systemic amyloidosis. J Clin Oncol 2004; 22 : 3751–3757.
15. Bohle A, Wehrmann R, Eissele R et al. The long‑term prognosis of AA and AL renal amyloidosis and the pathogenesis of chronic renal failure in renal amyloidosis. Path Res Pract 1993; 189 : 316–331.
16. Hachulla E, Grateau G. Diagnostic tools for amyloidosis. Joint Bone Spine 2002; 69 : 538–545.
17. Kebbel A, Röcken C. Immunohistochemical classification of amyloid in surgical pathology revisited. Am J Surg Pathol 2006; 30 : 673–683.
18. Freeman R. Autonomic dysfunction. In: Samuels M, Feske S (eds). The Office Practice of Neurology. 2nd ed. Philadelphia: Churchill-Livingstone 2003; 14 : 145.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2008 Číslo 11
- Plicní hypertenze – syndrom mnoha tváří – vyžaduje přesnou diagnostiku a specializovanou léčbu
- Proces hojení ran krok za krokem a co ho může zkomplikovat
- Limity glykovaného hemoglobinu a význam dalších glykovaných proteinů
- Korelace dávky levothyroxinu s titrem autoimunitních protilátek u primární hypotyreózy
- Zeolit-jodový komplex pomáhá v péči o infikované rány
Nejčtenější v tomto čísle
- Hypokalemická periodická paralýza u pacientů s hypertyreózou
-
Histiocytóza z Langerhansových buněk u osob dospělého věku – nemoc s mnoha tvářemi.
Zkušenosti jednoho pracoviště a přehled projevů nemoci - Bezpečnost dlouhodobého podávání losartanu v běžné klinické praxi: neintervenční studie NCT-CZ 14/04/LOZ
- Význam stanovování inhibinu B v klinické andrologické praxi
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy