
Kolagenofibrotická glomerulopatia - raritná glomerulonefritída
Collagenofibrotic glomerulopathy – rare glomerulonephritis
Glomerulopathies with fibrillary deposits form a heterogeneous group of renal diseases that can be identified only by means of electron microscopy. A case of rare type of such a nephropathy, the collagenofibrotic glomerulopathy with focus on differential diagnostics is presented and a review of current knowledge on this renal disease is given.
Key words:
fibrillary glomerulopathies - collagenofibrotic glomerulopathy - electron microscopy
Autoři:
G. Bernasovská 1; M. Demeš 1; A. Okša 1,2; M. Pavlovič 1; A. Vahančík 1; O. Nyitrayová 3; P. Gomolčák 3; D. Daniš 3
Působiště autorů:
I. interná klinika Slovenskej zdravotníckej univerzity a FNsP Bratislava, pracovisko Kramáre, Slovenská republika, prednosta prim. prof. MUDr. Štefan Hrušovský, CSc., Dr. SVS
1; Oddelenie klinickej a experimentálnej farmakoterapie Slovenskej zdravotníckej univerzity Bratislava, Slovenská republika, vedúci h. doc. MUDr. Martin Gajdoš, CSc.
2; Ústav patológie FNsP Bratislava, pracovisko Kramáre, Slovenská republika, vedúci doc. MUDr. Dušan Daniš, CSc
3
Vyšlo v časopise:
Vnitř Lék 2006; 52(12): 1200-1204
Kategorie:
Kazuistika
Práca bola prednesená 19. 9. 2004 na sympóziu s medzinárodnou účasťou Kvalita starostlivosti o nefrologického, dialyzovaného a transplantovaného pacienta, Zemplínska Šírava, Slovenská republika.
Souhrn
Glomerulopatie s fibrilárnymi depozitmi predstavujú heterogénnu skupinu chorôb obličiek, ktoré sa dajú identifikovať iba pomocou elektrónovej mikroskopie. Autori opisujú kazuistiku vzácnej formy takejto nefropatie - kolagenofibrotickej glomerulopatie s dôrazom na diferenciálnu diagnostiku a podávajú prehľad súčasných poznatkov o tomto zriedkavom postihnutí obličiek.
Kľúčové slová:
fibrilárne glomerulopatie - kolagenofibrotická glomerulopatia - elektrónová mikroskopia
Úvod
Nález glomerulopatií s nepravidelne usporiadanými, fibrilárnymi depozitmi je pri renálnych biopsiách oveľa zriedkavejší ako nález imunokomplexových depozitov - zvyčajne v menej ako 1 % biopsií [7,8]. Okrem primárnych glomerulopatií sa fibrilárne depozity v glomeruloch nachádzajú pri rôznych systémových chorobách ako kryoglobulinémia, monoklonové gamapatie, systémový lupus erytematosus, diabetes mellitus a i., preto je dôležitá ich diferenciálna diagnostika. Klasifikácia fibrilárnych glomerulopatií nie je jednotná, zvyknú sa deliť na amyloidové a neamyloidové, s obsahom imunoglobulínov alebo bez nich [8]. Komplexným histopatologickým vyšetrením (svetelná mikroskopia, imunofluorescencia, najmä však elektrónová, prípadne imunoelektrónová mikroskopia) možno rozlíšiť amyloidózu, kryoglobulinémie, fibrilárnu glomerulonefritídu (v užšom zmysle), imunotaktoidnú/mikrotubulárnu glomerulopatiu, kolagenofibrotickú glomerulopatiu, fibronektínovú glomerulopatiu a depozity kolagénu pri rôznych glomerulopatiách [7]. Klinicky sa jednotlivé formy odlíšiť nedajú. Najčastejším príznakom je proteinúria, ktorá môže byť asi v polovici prípadov súčasťou nefrotického syndrómu. Variabilne je prítomná hematúria, hypertenzia a renálna insuficiencia, ktorá má väčšinou progresívny charakter. U niektorých pacientov sa dá priebeh choroby ovplyvniť kortikosteroidmi alebo cytostatikami. Choroba môže rekurovať v transplantovanej obličke [8].
V práci opisujeme kazuistiku pacientky s rýchlou progresiou renálnej insuficiencie na podklade kolagenofibrotickej glomerulopatie.
Kazuistika
29ročnú pacientku sme prijali v januári roku 2004 na odporučenie praktického lekára pre bližšie neurčenú nefropatiu s renálnou insuficienciou (sérový kreatinín 358 µmol/l, clearance kreatinínu meraný 0,30 ml/s) a proteinúriou 1,2 g/24 h. V anamnéze neboli pozoruhodnosti - pôrod zdravého dieťaťa v roku 1999, potom implantácia intrauterinného telieska (do roku 2002) a v roku 2001 zistená stredne ťažká anémia liečená prípravkom železa. Pre podozrenie na celiakiu mala od jesene roku 2003 ordinovanú bezlepkovú diétu.
Pri prijatí uvádzala asi pol roka trvajúce bolesti v lumbálnej oblasti a slabosť. Fyzikálnym vyšetrením sa nenašli žiadne chorobné zmeny, mala optimálne hodnoty krvného tlaku 120/80 mm Hg. Až v priebehu sledovania sa objavila artériová hypertenzia, ktorá dobre reagovala na liečbu blokátorom kalciových kanálov amlodipínom. Laboratórne sa zistila zrýchlená sedimentácia 40/hod (CRP v referenčnom rozmedzí), normocytová normochrómna anémia stredne ťažkého stupňa s hemoglobínom 87 g/l, pokročilá renálna insuficiencia so sérovým kreatinínom 512 µmol/l a ureou 35,1 mmol/l, meraný clearance kreatinínu 0,17 ml/s, metabolická acidóza (pH 7,279, BE -9,9 mmol), v moči proteinúria do 1,6 g/24 hod, bez hematúrie a bakteriúrie. Ultrasonografia obličiek odhalila iba nešpecifické znaky difúznej nefropatie, ale s normálnou veľkosťou obličiek. Vylúčili sme prerenálnu a postrenálnu príčinu zlyhania obličiek a urobili biopsiu pravej obličky pod ultrazvukovou kontrolou.
V štandardne spracovanom bioptickom materiáli bolo pri svetelnej mikroskopii prítomných 16 glomerulov, z nich 5 úplne sklerotizovaných, v ostatných viacmenej dobre zachovaných sa našla výrazná fibrotizácia a zhrubnutie Bowmanových puzdier, náznak lobularity glomerulov a dvojitá kontúra kapilárnych stien. V celom obraze dominovali rozsiahle tubulárne nahromadenia homogénneho, eozinofilného, PAS-pozitívneho materiálu, hustý lymfocyto-plazmocytový infiltrát, atrofia tubulov s výrazne zhrubnutými, eozinofilnými, homogénnymi stenami a hyalínovými valcami v lumene (obr. 1). Podobné zmeny sa našli na artériách, znaky vaskulitídy neboli prítomné. Farbenie na amyloid bolo negatívne. Pri imunofluorescenčnom vyšetrení sa zachytila ostrá lineárna fluorescencia bazálnych membrán pri IgG (+++), IgA (++) - pozitívne valce v lumenoch tubulov a imunokomplexy C3 (+++) v mezangiu a stenách ciev. IgM, IgD, IgE a C4 boli negatívne. Histopatológ vyslovil podozrenie na tubulointersticiálne poškodenie obličiek na podklade bližšie nešpecifikovanej metabolickej choroby.

Pokračovali sme v diferenciálnej diagnostike so zameraním na glomerulopatie metabolických porúch, kam podľa klasifikácie SZO patrí diabetická glomeruloskleróza, amyloidóza, mnohopočetný myelóm a iné monoklonové gamapatie, kryoglobulinémie, choroby pečene, kosáčikovitá anémia, cyanotizujúce kongenitálne srdcové chyby a pľúcna hypertenzia. Do diferenciálno-diagnostických úvah sme zaradili i chronickú lymfocytovú leukémiu, systémový lupus erytematosus (SLE) a iné systémové choroby spojiva s postihnutím obličiek.
Elektroforetické a imunoelektroforetické vyšetrenie séra i moču vylúčilo prítomnosť paraproteínu. Morfologické vyšetrenie kostnej drene nesvedčilo pre lymfoproliferatívne ochorenie ani plazmocytóm. Zistili sme mierne zvýšenú sérovú koncentráciu kryoglobulínov (128 mg/l, norma do 80,0 mg/l), ale klinické ani laboratórne vyšetrenia nepodporovali diagnózu žiadneho typu kryoglobulinémie. Markery hepatitídy B a C boli negatívne. Elektroforéza hemoglobínu nepotvrdila kosáčikovitú anémiu s patologickým hemoglobínom. Koncentrácia imunoglobulínov a zložiek komplementu v sére bola v referenčnom rozmedzí, autoprotilátky ANA, anti-DNP, ANCA, ACLA, ASMA, AMA, APCA, ATA, ARA, AEmA a ABMT boli negatívne, slabá pozitivita sa našla len pri ABMG. Nedokázal sa reumatoidný faktor, ani pozitivita ASO. Pacientka nespĺňala klasifikačné kritériá pre SLE alebo iné systémové choroby spojiva, resp. vaskulitídu. Ďalšími vyšetreniami sme vylúčili paraneoplastickú etiológiu glomerulopatie: USG brucha, kolonoskopia, gastroskopia, pľúcne, gynekologické vyšetrenie a onkomarkery boli negatívne. Vzhľadom na anamnestický údaj o celiakii bez podrobnejších výsledkov v dokumentácii sme realizovali enterobiopsiu, pri ktorej sa našli len minimálne zmeny v histochemickom vyšetrení disacharidáz, proteináz a fosfatáz, histologicky mierny chronický zápal v sliznici, bez atrofie klkov (klasifikácia Marsh 0).
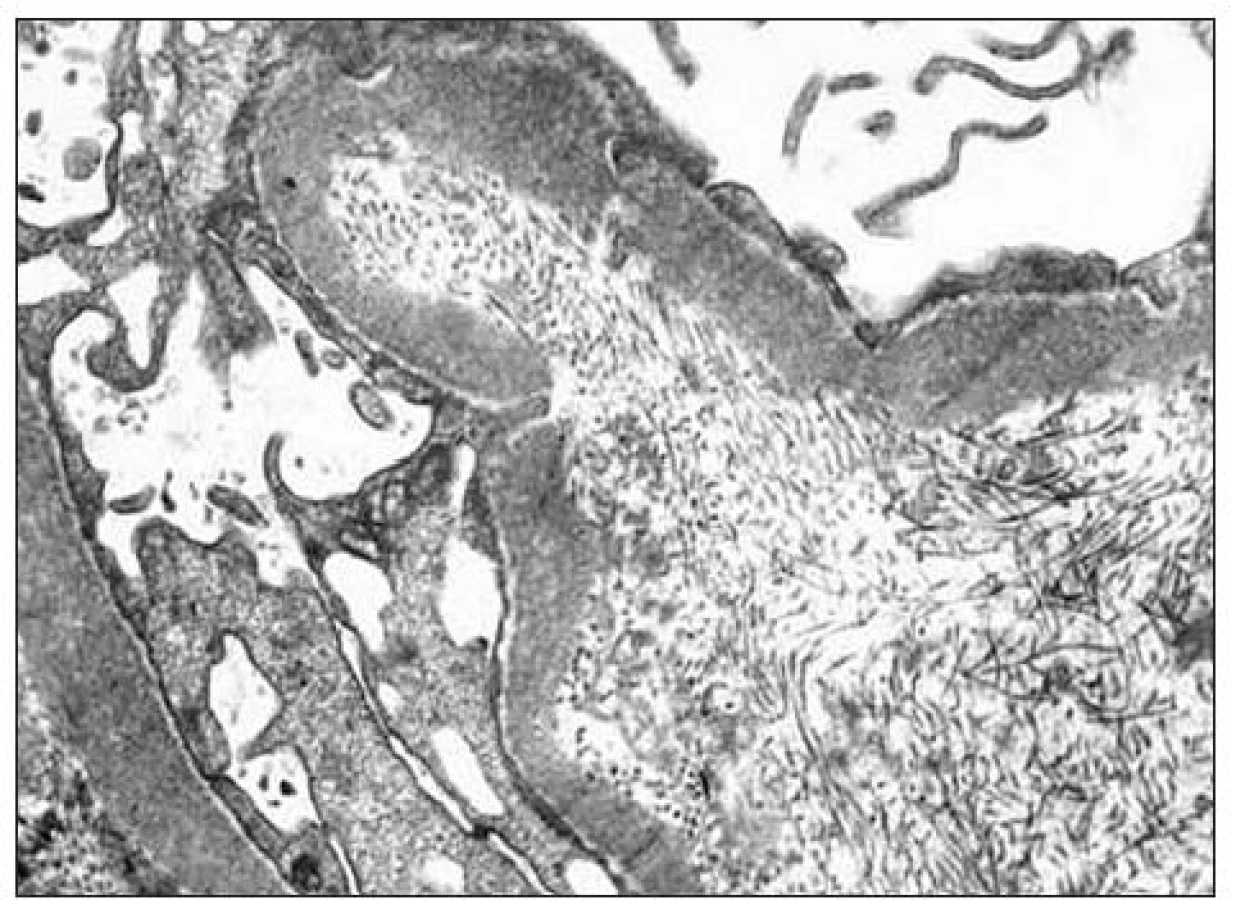
Kľúčový význam pre definitívnu diagnózu malo ultraštruktúrne vyšetrenie vzorky z renálnej biopsie. Pre elektrónovú mikroskopiu sa zachytili 3 glomeruly: v 1. glomerule sa zistili iba nepravidelné glomerulové bazálne membrány bez depozitov, v 2. glomerule sa pod zachovanou lamina densa v subendotelovom priestore a v mezangiu našli masívne ložiská fibrilárneho materiálu (obr. 2) a v 3. glomerule sa zachytili depozity homogénnej hmoty subendotelovo (obr. 3). Zriedkavý morfologický nález zapadal do skupiny chorôb označovaných v patológii ako „choroby s rozlíšiteľnými ultraštruktúrnymi depozitmi“. Fibrilárny materiál tvorili nepravidelne usporiadané, na pozdĺžnych rezoch nerovnomerne široké a miestami špirálovito stočené vlákna s priečnym pruhovaním s periodicitou okolo 50 nm, ktoré na priečnych rezoch nemali obvyklý okrúhly tvar (obr. 4). Tento obraz najviac zodpovedal kolagénu III. typu, ktorý sa normálne v glomeruloch nenachádza, ale je tu prítomný najmä pri kolagenofibrotickej glomerulopatii a v podstatne menšom rozsahu pri iných sklerotizujúcich glomerulopatiách. Doplnili sme preto imunohistochemické vyšetrenie so špecifickou protilátkou proti kolagénu III. typu (Bio Genex, klon HWD 1.1), ktorá však s glomerulami nereagovala.


Pacientku sme liečili nízkobielkovinovou diétou doplnenou ketoanalógmi aminokyselín, anémiu sme korigovali železom a erytropoetínom, hypertenziu amlodipínom, metabolickú acidózu bikarbonátom a sekundárnu hyperparatyreózu kalciom a vitamínom D. Po obdržaní výsledku z elektrónovej mikroskopie sme podali pulznú terapiu metylprednisolonom 500 mg/deň i.v. počas 3 dní a pokračovali v kombinovanej liečbe prednisonom 1 mg/kg/deň p.o. a cyklofosfamidom 100 mg/deň p.o. Po 3-týždňovej hospitalizácii sme zaznamenali nárast koncentrácie hemoglobínu o 20 g/l, pretrvávala renálna insuficiencia s kreatinínémiou 456 µmol/l, clearance kreatinínu 0,22 ml/s, proteinúria 1,2 g/24 hod. Pacientku sme odovzdali do starostlivosti rajónneho nefrológa. Kombinovanú imunosupresívnu liečbu prednisonom v detrahovanej dávke 20 mg/deň (zníženie dávky ku koncu hospitalizácie pre flebitídu na predlaktí po i.v. kanyle) a cyklofosfamidom 100 mg/deň pacientka užívala ďalších 6 mesiacov bez efektu na proteinúriu (0,9-1,2 g/24 hod) a filtračnú funkciu (0,17 ml/s). Počas ambulantného sledovania sporadicky uvádzala nauzeu a zvýšenú tvorbu hematómov a prekonala hnisavú angínu bez vplyvu na renálne funkcie. Pre leukopéniu sa cyklofosfamid po 7 mesiacoch vynechal. Po 8 mesiacoch od stanovenia diagnózy bola pre progresiu renálnej insuficiencie (kreatinín 616 µmol/l, urea 37,8 mmol/l, clearance kreatinínu 0,17 ml/s, K 5,4 mmol/l) zaradená do pravidelného hemodialyzačného programu.
Diskusia
Kolagenofibrotická glomerulopatia (tiež glomerulopatia s kolagénom III. typu, primárna glomerulová fibróza) patrí medzi fibrilárne glomerulopatie s ukladaním depozitov kolagénu III. typu. Väčšina správ o tejto diagnóze pochádza z Japonska, takže sa dá predpokladať zemepisná alebo i rasová podmienenosť [4,10,14-17]. Ide o veľmi zriedkavú chorobu, ktorá sa vyskytuje vo veku od 6 do 72 rokov a nie je závislá od pohlavia [2,4,5,10,13,16]. Z klinického hľadiska ju niektorí delia na dva podtypy: jeden so začiatkom v dospelosti a druhý detský typ [3]. Najčastejšie sa prejavuje edémami s nefrotickou alebo len miernou proteinúriou. Filtračná funkcia je pri manifestácii choroby väčšinou normálna alebo aj zvýšená a asi u jednej tretiny pacientov môže byť hypertenzia a hematúria. V diagnostike môže podľa niektorých autorov pomôcť zvýšená sérová koncentrácia prokolagénu III. typu [16]. Choroba má progresívny charakter, ale rýchlosť progresie je veľmi variabilná [4,7]. Nález pri svetelnej mikroskopii ani imunofluorescencii nie je diagnostický, rozhodujúci význam má ultraštruktúrne vyšetrenie. Pri svetelnej mikroskopii býva akcentovaná lobulárna štruktúra glomerulov s expanziou mezangia a zhrubnutými kapilárnymi stenami, často s dvojitou kontúrou, čo môže pripomínať membránovú alebo membránovo-proliferatívnu glomerulonefritídu (chýba však hypercelularita). Obraz pri štandardnej imunofluorescencii je nešpecifický, nepodporuje imunokomplexovú glomerulopatiu. Pri elektrónoptickom vyšetrení je pre kolagenofibrotickú glomerulopatiu charakteristické masívne nahromadenie kolagénových vlákien v mezangiu a v subendotelovom priestore, ale nie subepitelovo. Vlákna sú priečne pruhované s periodicitou 43-65 nm, tvoria nepravidelné, zakrivené a strapkaté zväzočky s pozdĺžnymi zárezmi a v priečnom reze majú špirálovitý alebo čiarkovitý tvar. Tieto znaky odlišujú atypický kolagén III. typu od normálneho, ktorý má v pozdĺžnych rezoch priame a v priečnych rezoch cirkulárne usporiadanie vlákien a vyskytuje sa bežne v interstíciu a stenách ciev, ale nie v glomeruloch [7,9].
U našej pacientky sme biopsiu obličky urobili až v pokročilom štádiu choroby, kedy bola 1/3 zachytených glomerulov už úplne sklerotizovaná. Nefropatia s renálnou insuficienciou a proteinúriou sa diagnostikovala len krátko pred prijatím a rýchlo progredovala. Na základe nálezu pri svetelnej mikroskopii sa diferenciálna diagnostika zamerala na metabolické a systémové choroby s postihnutím obličiek (pozri vyššie), ktoré sa klinickým a laboratórnym vyšetrením vylúčili. Až elektrónová mikroskopia odhalila v glomeruloch masívne ložiská fibrilárneho materiálu s charakteristickými znakmi kolagénu III. typu. Okrem kolagenofibrotickej glomerulopatie možno podobný obraz nájsť pri syndróme osteoonychodysplázie („nail-patella syndrome“) s autozómovo dominantnou dedičnosťou, kde sú však zväzky kolagénu akumulované prevažne vnútri bazálnej membrány glomerulov, kým pri kolagenofibrotickej glomerulopatii sa nachádzajú subendotelovo a v mezangiu [1]. Klinický obraz oboch jednotiek je navyše celkom odlišný, pri kolagenofibrotickej glomerulopatii (a ani u našej pacientky) neboli dokumentované žiadne zmeny skeletu. Menej nápadné depozity fibrilárneho kolagénu III. (ale aj I.) typu sa niekedy nachádzajú v glomeruloch pri diabetickej nodulárnej skleróze, fokálnej segmentovej glomeruloskleróze, membránovoproliferatívnej glomerulonefritíde a glomerulonefritídach s extrakapilárnou proliferáciou vo forme kosáčikovitých výrastkov [7]. Histologický a imunofluorescenčný nález, ani klinické a laboratórne vyšetrenia u našej pacientky však týmto typom glomerulopatií nezodpovedali. Negatívny imunohistochemický dôkaz kolagénu III. typu môže mať viac príčin:
- Prítomnosť štruktúrne modifikovaného kolagénu III. typu, resp. morfologicky podobného, ale antigénne odlišného kolagénu, ktorý s vysokošpecifickou protilátkou proti štandardnému kolagénu III. typu nereagoval. V tejto súvislosti je zaujímavý nedávno opísaný prípad japonskej pacientky s kolagenofibrotickou glomerulopatiou a nálezom dvoch foriem fibrilárnych zväzkov v mezangiu a subendotelovom priestore glomerulov. Pri imunohistochemickom vyšetrení glomerulové lézie reagovali s protilátkami proti kolagénu III. a V. typu. Imunoelektrónová mikroskopia potvrdila V. typ kolagénu u obidvoch foriem fibríl [11]. Zatiaľ nie je jasné, či ide o dve rozdielne chorobné jednotky, alebo variant jednej choroby. Protilátku proti kolagénu V. typu sme nemali k dispozícii.
- Nedostatočné množstvo kolagénu III. typu vo vzorke, ktorá sa dodatočne vyšetrila imunohistochemicky.
- Metodická chyba pri imunohistochemickej analýze.
Etiológiu ani patogenézu kolagenofibrotickej glomerulopatie sa doteraz nepodarilo objasniť [3]. Autozómovo recesívny typ dedičnosti podporuje opis štyroch súrodeneckých dvojíc z Japonska, z ktorých dve sestry mali morfologicky dokázanú kolagenofibrotickú glomerulopatiu so zvýšenou sérovou koncentráciou prokolagénu III. typu, zatiaľ čo ich rodičia ani ostatní súrodenci nemali proteinúriu ani zvýšenú koncentráciu prokolagénu III [16]. Vyšetrenie prokolagénu III. typu je u nás nedostupné. V patogenéze sa predpokladá syntéza fibrilárneho kolagénu I. alebo III. typu, ktoré sa v normálnom glomerule nenachádzajú, ale sú bežné v interstíciu a stenách ciev, aktivovanými bunkami mezangia alebo migrujúcimi fibroblastami z interstícia, resp. modifikovanými bunkami hladkého svalu cievnej steny [7,12]. Podľa doterajších záznamov je choroba takmer výhradne lokalizovaná v glomeruloch, systémovú manifestáciu dokumentujú len 2 prípady: v jednom šlo o pacienta s kolagenofibrotickou glomerulopatiou a fibrózou pečene s nálezom kolagénu III. typu aj v perisínusoidových priestoroch pečene [10], v druhom bol opísaný výsledok autopsie pacienta s kolagenofibrotickou glomerulopatiou, liečeného 7 rokov ambulantnou peritoneálnou dialýzou, s nálezom masívnej akumulácie typických kolagénových vlákien aj v slezine, pečeni, myokarde a štítnej žľaze [18].
Pre raritný výskyt kolagenofibrotickej glomerulopatie pochopiteľne neexistujú kontrolované štúdie terapie. V literatúre mala kombinácia kortikosteroidov s cytostatikami variabilnú účinnosť, progresiu choroby väčšinou neovplyvnila. Naša pacientka bola napriek kombinovanej imunosupresii a komplexnej liečbe renálnej insuficiencie po 8 mesiacoch zaradená do dialyzačného programu.
Záver
Prostredníctvom kazuistiky sme chceli upozorniť na vzácnu chorobu - kolagenofibrotickú glomerulopatiu, ktorá patrí do skupiny nefropatií s neamyloidovými, neimunoglobulínovými fibrilárnymi depozitmi. Na rozdiel od iných fibrilárnych glomerulopatií sa nespája s diabetom, hematologickou malignitou, kryoglobulinémiou ani systémovým lupusom. Rozhodujúci význam v jej diagnostike má ultraštruktúrne vyšetrenie renálnej biopsie. Podľa našich vedomostí ide o prvý opis tejto diagnózy v domácej odbornej literatúre.
MUDr. Gabriela Bernasovská
www.fnspba.sk
e-mail: gaber@stonline.sk
Doručeno do redakce: 1. 5. 2006
Přijato po recenzi: 9. 9. 2996
Zdroje
1. http://www.gamewood.net/pathcase/895/895-case.htm.
2. Gubbler MC, Dommergues JP, Foulard M et al. Collagen type III glomerulopathy: A new type of hereditary nephropathy. Pediatr Nephrol 1993; 7 : 354-360.
3. Hisakawa N, Yasuoka N, Nishiya K et al. Collagenofibrotic glomerulopathy associated with immune complex deposits. Am J Nephrol 1998; 18 : 134-141.
4. Ikeda K, Yokoyama H, Tomosugi K et al. Primary glomerular fibrosis: A new nephropathy caused by diffuse intra-glomerular increase in atypical type III collagen fibers. Clin Nephrol 1990; 33 : 155-159.
5. Imbasciati E, Gherardi G, Morozumi K et al. Collagen type III glomerulopathy: A new idiopathic glomerular disease. Am J Nephrol 1991; 11 : 422-429.
6. Iskandar SS. Glomerulopathies with organized deposits. http://www.uninet.edu/cin2003/conf /iskandar/iskandar.html.
7. Iskandar SS. Glomerulopathies with Organized Deposits. Seminars in Diagnostic Pathology 2002; 19 : 116-132.
8. Kováč A, Slugeň I, Daniš D et al. Fibrilárne glomerulopatie. Vnitř Lék 1997; 43 : 691-695.
9. Mauiyyedi S, Selig MK, Marion AP et al. Renal Glomerular Disease. In: Dickersin GR. Diagnostic Electron Microscopy: A Text/Atlas. 2nd ed. New York; Springer Verlag 2000 : 859-863.
10. Mizuiri S, Hasegawa A, Kikuchi A et al. A case of collagenofibrotic glomerulopathy associated with hepatic perisinusoidal fibrosis. Nephrology 1993; 63 : 183-187.
11. Morita H, Hasegawa T, Minamoto T et al. Collagenofibrotic glomerulopathy with a widespread expression of type V collagen. Virchows Archiv 2003; 442 : 163-168.
12. Naruse K, Ito H, Moriki E et al. Mesangial cell activation in the collagenofibrotic glomerulonephropathy. Case report and review of the literature. Virchows Archiv 1998; 433 : 183-188.
13. Salcedo JR. An autosomal recessive disorder with glomerular basement membrane abnormalities similar to those seen in the nail patella syndrome: report of kindred. Am J Med Genet 1984; 19 : 579-584.
14. Sanaka T, Nakao H, Matsumura O et al. A case of nephrotic syndrome of collagenofibrotic glomerulopathy. In: Arakawa M, Yamanaka N (eds). Collagenofibrotic Glomerulonephropathy. Niigata; Nishimura 1991 : 51-59.
15. Sugiyama N, Shimizu J, Nakamura M et al. A case of „collagenofibrotic glomerulonephropathy“. In: Arakawa M, Yamanaka N (eds). Collagenofibrotic Glomerulonephropathy. Niigata; Nishimura 1991 : 41-49.
16. Tamura H, Matsuda A, Kidoguchi N et al. A family with two sisters of collagenofobrotic glomerulonephropathy. Am J Kidney Dis 1996; 27 : 588-595.
17. Yamanaka N, Sugisaki Y, Wakamatsu R et al. Collagenofibrotic Glomerulonephropathy. In: Arakawa M, Yamanaka N (eds): Collagenofibrotic Glomerulonephropathy. Niigata; Nishimura 1991 : 59-68.
18. Yasuda T, Imai H, Nakamoto Y et al. Collagenofibrotic glomerulopathy: a systemic disease. Am J Kidney Dis 1999; 33 : 123-127.
Štítky
Diabetologie Endokrinologie Interní lékařstvíČlánek vyšel v časopise
Vnitřní lékařství

2006 Číslo 12
- Prognostický význam hladiny natriuretických peptidů při léčbě empagliflozinem
- Srovnání vlivu omeprazolu a pantoprazolu na antiagregační účinek klopidogrelu
- Omeprazol a mechanismus hojení krvácení z peptického vředu
- Současné postavení a přínos sartanů v klinické praxi
Nejčtenější v tomto čísle
- Aktuální přístupy k diagnostice a terapii hemoptýzy: editorial
- Postavení embolizace bronchiální tepny v terapii klinicky významné hemoptýzy
- Statiny a osteoporóza
- Ultrazvukové mapování žilního systému dolních končetin s ohledem na výskyt a anatomii přední přídatné velké safény
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy