
Příznaky a projevy Erdheimovy-Chesterovy choroby
Symptoms and manifestations of Erdheim-Chester disease
Erdheim-Chester disease (ECD) is a rare form of non-Langerhans-cell histiocytosis, associated in more than 50% of cases to BRAFV600E mutations in early multipotent myelomonocytic precursors or in tissue-resident histiocytes. It encompasses a spectrum of disorders ranging from asymptomatic bone lesions to multisystemic, life-threatening variants.
CNS involvement occurs in 56 % of ECD patients. Diabetes insipidus, visual disturbances, pyramidal and extra-pyramidal syndromes were the most recurrent neurological signs, whereas concomitant pituitary involvement, retro-orbital masses and axial lesions in the presence of symmetric bilateral osteosclerosis of long bones depicted the typical ECD clinical picture. Patients with CNS infiltration show a lower occurrence of heart involvement and a higher incidence of bone, skin, retro-peritoneal, lung, aortic and renal infiltration. ECD pathophysiology, clinical features and diagnostic are presented in this paper.
Keywords:
Erdheim-Chester disease
Autoři:
Z. Král 1; M. Krejčí 1; Marta Ježová 2; S. Ševčíková 3; Z. Řehák 4; R. Koukalová 4; K. Starý 5; J. Neubauer 6; Z. Adam 1
Působiště autorů:
Interní hematologická a onkologická klinika LF MU a FN Brno
1; Ústav patologie LF MU a FN Brno
2; Ústav patologické fyziologie, LF MU, Brno
3; Oddělení nukleární medicíny, Masarykův onkologický ústav, Brno
4; Interní gastroenterologická klinika LF MU a FN Brno, endokrinologické oddělení
5; Klinika radiologie a nukleární medicíny LF MU a FN Brno
6
Vyšlo v časopise:
Transfuze Hematol. dnes,26, 2020, No. 3, p. 167-176.
Kategorie:
Souhrnné/edukační práce
Souhrn
Erdheimova-Chesterova choroba (Erdheim-Chester disease, ECD) je vzácnou nemocí ze skupiny histiocytóz. Dříve byla zařazena do skupiny „non Langerhans cell histiocytosis“, případně do skupiny juvenilního xantogranulomu, ovšem v dnešní době patří do stejné skupiny jako histiocytóza z Langerhansových buněk. Důvodem této změny jsou nové molekulárně genetické poznatky. V 50 % případů je ECD asociována s mutací BRAFV600E v multipotentním myelomonocytovém prekurzoru a v patologickém ložisku histiocytů. V míře závažnosti a agresivitě průběhu jsou mezi pacienty s ECD velké rozdíly. Na jedné straně spektra příznaků jsou náhodné asymptomatické nálezy ve skeletu dlouhých kostí, na druhé straně ECD s agresivním průběhem. ECD je obvykle multisystémové onemocnění, u 56 % pacientů s ECD je postižen centrální nervový systém (CNS). Nejčastější neurologické příznaky a projevy ECD jsou poruchy zraku, dysartrie a ataxie, pyramidální a extrapyramidální symptomy, postižení hypofýzy nebo retroorbitální infiltráty, případně diabetes insipidus. Ve většině případů je přítomna pro ECD typická symetrická bilaterální osteoskleróza dlouhých kostí dolních končetin. Pacienti s ECD a CNS infiltrací mají jen vzácně postižení srdce, zato mají časté kostní, kožní, retroperitoneální postižení, postižení plic, aorty a infiltrace perirenálního tuku. Článek se podrobně zabývá patofyziologií, klinickým obrazem a diagnostikou ECD.
Klíčová slova:
Erdheimova-Chesterova choroba
ÚVOD
Erdheimova-Chesterova nemoc (Erdheim-Chester disease, ECD) byla poprvé popsána v roce 1930 Jakobem Erdheimem a Williamem Chesterem a pojmenována jako lipoidní granulomatóza [1]. Později bylo rozpoznáno, že ECD je tvořena buňkami histiocytární řady, a byla zařazena do skupiny „non-Langerhans cell histiocytosis“.
V roce 2012 byla poprvé popsána přítomnost specifické mutace genu BRAFV600E u ECD [2, 3], od roku 2012 byly popsány četné další mutace. Toto poznání vedlo k léčebným postupům cílícím na BRAF a MEK mutace [4, 5]. Objev těchto mutací byl pak reflektován v poslední klasifikaci histiocytárních chorob [6].
ECD je charakterizovaná infiltráty z histiocytů, které střádají lipidy, a proto má jejich cytoplazma v mikroskopu bledý či pěnitý vzhled (foamy histiocytes). Pěnité buňky jsou imunohistochemicky CD68-pozitivní, ale CD1a-/S100-negativní.
Podle klasifikace Mezinárodní zdravotnické organizace (WHO) je ECD neoplastická choroba odvozená od histiocytů. Dříve do objevu typických mutací probíhala dlouhá diskuse o tom, zda na chorobu nahlížet jako na maligní, nebo na reaktivní polyklonální proliferaci. ECD může působit velmi různorodé klinické obtíže, jako jsou bolesti kostí, diabetes insipidus, neurologické poruchy typu dysartrie a ataxie, ale také kožní, kardiovaskulární a plicní problémy. Příznaky této nemoci mohou přivést pacienty k lékařům všech odborností. Choroba má obvykle pozvolný průběh. Charakteristickým znakem ECD jsou osteosklerotické změny dlouhých kostí dolních končetin.
V roce 2014 bylo popsáno celosvětově asi 500–700 případů ECD [7]. V roce 2019 se počet popsaných ECD případů zvýšil na 1 500 [8]. Na našem pracovišti jsme v průběhu 30 let diagnostikovali ECD u 4 lidí, z toho 3 pacienti byli léčeni a jedna nemocná léčbu odmítla.
Díky vzácnosti této nemoci nejsou známa epidemiologická data. Většina pacientů je dospělého věku, asi v 70 % se jedná o muže. Medián věku při stanovení diagnózy je 48–56 let. Pediatrické případy jsou vzácné a obvykle jsou asociovány s histiocytózou z Langerhansových buněk (LCH) [7, 9, 10].
PATOFYZIOLOGIE A KLINICKÝ OBRAZ ECD
Histopatologické nálezy
Pro stanovení diagnózy ECD je zásadní odebrání vzorku tkáně na histopatologické a imunohistochemické vyšetření. Ložiska ECD mají při mikroskopickém vyšetření charakter fibro-inflamatorních infiltrátů, obsahujících aktivované pěnité histiocyty, často provázené Toutonovými gigantickými buňkami, které vznikají fúzí jednojaderných histiocytů (obr. 1). Toutonovy buňky mají několik jader ve věnečku kolem eozinofilního středu a pěnitý vnější lem cytoplazmy. Při imunohistochemickém barvení je prokazatelná pozitivita histiocytárních markerů CD68, CD163 a faktoru XIIIa. V tkáňovém vzorku ECD jsou naopak negativní znaky Langerhansových buněk, tj. není přítomen znak CD1a a není prokazatelný znak CD207 (langerin). Pozitivita S100 je vzácná, asi ve 20 % případů. ECD histiocyty jsou morfologicky a imunohistochemicky identické s histiocyty juvenilního xantogranulomu, a proto byla ECD považována za variantu juvenilního xantogranulomu.

Pokud je to možné, doporučuje se provést biopsii kožních xantelesmat nebo biopsii perirenálního tuku, protože tyto dvě lokalizace bývají nejvíce výtěžné [7]. U našich 4 případů byla diagnóza ECD stanovena z odběru vzorku tkáně z postižené kosti.
Přítomnost mutace BRAF V600E může patolog odhalit dvěma metodami. První a jednodušší je imunohistochemie s protilátkou anti-BRAF V600E VE1. Mutované buňky se barví cytoplazmaticky s různou intenzitou. Výsledek by měl být ověřen ještě jinou metodou.
Pro vyšetření mutace BRAF V600E molekulárně genetickými metodami je vhodnější nativní tkáň, ale lze použít i fixovaného materiálu. V praxi se provádí nejčastěji z bloků standardně fixovaných 10% formolem a zalitých do parafinu. Také je možné vyšetřovat řezy z bloků ve zkumavce nebo na podložním skle. Toto vyšetření je dostupné v laboratořích molekulární patologie, které jsou součástí patologických pracovišť při velkých fakultních nemocnicích. Nejvhodnější materiál k vyšetření vybírá vždy patolog, který kvalifikovaně odhadne procentuální zastoupení nádorových buněk. Vzorky kosti, které prošly procesem odvápňování za působení kyseliny mravenčí, jsou obecně méně vhodné k dalšímu vyšetření než vzorky měkkých tkání. Pokud lze, vybíráme již během blokování měkčí součásti nádorového ložiska, které by odvápňování nepotřebovaly.
Trepanobiopsie většinou obsahují menší podíl nádorových buněk, proto při negativním nálezu mutace se doporučuje zopakovat vyšetření z jiného dostupného mimokostního ložiska.
Zánětlivé projevy ECD
Erdheimovu-Chesterovu nemoc provázejí fibrózní a zánětlivé změny systémové i lokální, které způsobují poškození orgánů. Zánětlivé změny s typicky zvýšenou hodnotou C-reaktivního proteinu (CRP) jsou přítomné nejméně u 80 % pacientů s ECD [11].
Pouze několik publikací se věnuje zánětlivým procesům provázejícím ECD, popisují komplexní síť cytokinů a chemokinů, regulující recruitment histiocytů a jejich akumulaci v ECD ložiscích [12]. Bylo prokázáno, že interleukin 6 (IL-6) stimuluje v ložisku ECD produkci tumor-nekrotizujícího faktoru (TNF-alfa). Interleukin 8 (IL-8) působí jako chemoatraktant pro polymorfonukleáry a monocyty. Chemokinový ligand 18 (CCL18) má významný spolupodíl na procesu fibrotizace a jeho 3–4násobné navýšení bylo prokazatelné v séru 20 pacientů s ECD. Výše hladin CCL18 korelovala se závažností fibrotizace [13, 14].
Při analýze 23 cytokinů v séru 37 pacientů s ECD byly prokázány vysoké hladiny řady z těchto cytokinů (interferonu alfa, IL-1/IL-1RA, IL-6, IL-12 a dalších), což signalizuje systémovou imunitní odpověď dominantně Th1 typu [15].
U solidních tumorů je mutace BRAFV600E asociována s onkogenem indukovanou senescencí, charakterizovanou zástavou dělení a zvýšenou expresí tumor suprimujícího proteinu, jako je p16INK4a a p21 [16, 17].
Buňky podléhající senescenci indukují komplexní zánětlivou odpověď, která má mnoho společného s ECD. Imunohistochemické analýzy ECD ložisek odhalily, že BRAFV600E mutované histiocyty byly pozitivní pro marker senescence p16INK4a [18].
Podle těchto výsledků mohou buňky s mutací BRAFV600E indukovat zánětlivé změny v histiocytech cestou senescenčního programu.
Patogeneze ECD – aktivace MAPK regulační cesty
Mutace BRAFV600E je přítomna ve více typech tumorů. Tato onkogenní mutace aktivuje signální cestu RAS-RAF-MEK, která je důležitá pro průběh proliferace, apoptózy, angiogeneze a migrace. V roce 2010 testování onkogenů ve tkáních Langerhansovy histiocytózy (LCH) prokázalo mutace BRAFV600E u 35 z 61 testovaných tkáňových vzorků [19]. V roce 2012 byla tato mutace prokázána u 54 % případů ECD, a to v typických pěnitých buňkách a v Toutonových gigantických buňkách, ale nebyla přítomna v lymfocytech, fibroblastech či endoteliálních buňkách [2, 3].
Kombinovaná celoexomová a transkriptomová analýza tkáňových vzorků od pacientů s ECD prokázala přítomnost dalších mutací MAPK signální cesty, a to u genů MAP2K1, ARAF, NRAS a KRAS a dále translokace zahrnující BRAF, ALK a NTRK geny. Mutace v genu MAP2K1 byly nalezeny asi u 30 % pacientů, zatímco KRAS a NRAS mutace byly přítomny jen ve 27 % [20].
Ale MAPK signální cesta není jedinou důležitou signální cestou u ECD. Aktivující mutace v genu PIK3CA byly nalezeny u 11 % pacientů [5]. Tyto mutace aktivují signální dráhu PI3K-AKT, která může být aktivována cestou MAPK signalizace. A postupně jsou nacházeny i další mutace [21–23].
Tkáňové makrofágy mají dvojí možný původ, část z nich vychází z embryonální krvetvorby a má schopnost sebeobnovy. Dále při zánětu vstupují monocyty z krve do tkání a akumulují se v postižených tkáních.
Původ mutovaných histiocytů je zřejmě z klonálních prekurzorových hematopoetických buněk, protože popsaná mutace BRAFV600E byla nalezená v cirkulujících hematopoetických buňkách. Dále xenotransplantace CD34+ buněk od pacienta s ECD do imunokompromitované myši dala vznik pěnitým histiocytům v tomto příjemci. Z toho vyplývá, že mutované histiocyty u ECD jsou odvozené z hematopoetických buněk kostní dřeně [24, 25].
A společný původ pěnitých histiocytů v mutované kmenové hematopoetické buňce vedl k analýze 189 pacientů, která prokázala, že u 10 % byly nalezeny překryvné znaky s myeloidními neoplaziemi, nejčastěji s chronickou myelomonocytární leukemií (CMMoL), dále pak s myeloproliferativními neoplaziemi a myelodysplastickým syndromem (MDS) [26, 27].
Patogeneze myeloproliferativních nemocí, MDS a ECD má některé společné mechanismy, ale mutace MAPK signální dráhy vede směrem k ECD [27].
Toto poznání patofyziologie změnilo pohled na tuto nemoc jako na klonální proces, u něhož může být zásadním přínosem cílená léčba BRAF inhibitory (vemurafenib), nebo anti-TNFα monoklonální protilátkou (infliximab) anebo IL-1R antagonistou (anakinra).
Klinická manifestace ECD
ECD je obvykle multisystémové onemocnění a postižen může být prakticky každý orgán, i když frekvence postižení je odlišná, jak ukazuje tabulka 1.

Kostní postižení
Kostní postižení je přítomné u 74 %, podle některých údajů až u 80–95 % všech ECD pacientů [8]. Nejčastěji jsou postiženy dlouhé kosti dolních končetin. Při rentgenovém (rtg) či CT (computer tomography) zobrazení je zřetelná symetrická diafyzální osteoskleróza. V těchto oblastech je zvýšené vychytávání technecia (Tc) při scintigrafii skeletu, ale také zvýšená akumulace radioaktivně značené glukózy (FDG) při PET/CT vyšetření (positron emission tomography). V roce 2020 se již preferuje PET/CT zobrazení před kostní scintigrafií, protože zobrazí jak rozsah postižení kosti, tak i metabolickou aktivitu tohoto postižení [29]. Uvedené kostní změny mohou být klinicky němé, pouze u 40 % všech pacientů s ECD jsou přítomny mírné kostní bolesti. Kostní postižení dokumentované různými zobrazovacími metodami (CT, scintigrafie skeletu a PET/CT) ilustrují obrázky 2–4.



Mozkové, mozečkové, faciální a orbitální manifestace
Postižení CNS může být velmi různorodé a neexistuje žádný zcela typický obraz. Nervový systém bývá postižen u 25–50 % nemocných s prokázanou ECD.
Symptomem mozkového postižení může být dysartrie a ataxie mozečkového původu a také pyramidální syndromy. Z dalších možných projevů ECD v neurologické oblasti je třeba zmínit bolesti hlavy, neuropsychické poruchy včetně psychóz, deprese, různé poruchy kognice, senzorické poruchy a poruchy funkce hlavových nervů. Výjimečně byly zaznamenány i případy ECD s intrakraniální hypertenzí, nevolností, edémem papily (2,5 %). Některá drobnější ložiska v CNS mohou být asymptomatická.
Infiltráty při ECD je možné s pomocí MR (magnetic resonation) zobrazení s aplikací gadolinia nalézt kdekoliv v CNS, ale i v míše. Obraz je nespecifický a nálezy u ECD mohou být zaměněné za primární či metastatický nádor, demyelinizační onemocnění či leukodystrofii. Ložiska ECD při MR zobrazení se mohou také podobat meningeomům nebo granulomatózním chorobám. Podobné typy intrakraniálního postižení byly rovněž popsány u juvenilního xantogranulomu. Také ložiska histiocytózy z Langerhansových buněk (LCH) mají podobnou distribuci a obraz jako ložiska ECD. Meningeální ložiska jsou v případě ECD více expanzivní a častější než u LCH. U pacientů s ECD, u nichž byla prováděna MR zobrazení CNS, bylo nalezeno postižení mening u 23 % z nich. Často se projevovalo jako difuzní zesílení durální masy.
V případě LCH obvykle nedochází k postižení míchy na rozdíl od ECD, kde vzniknout může. Degenerativní atrofický proces v mozečku je popisován u LCH při dlouhodobém průběhu nemoci. Podobné degenerativní onemocnění mozečku bylo popsáno v 17 % případů ECD [30, 31]. Výjimečně byly popsány poruchy typu hemiparézy či paraparézy (7 %), tento typ poruchy způsobila ECD ložiska v míše, proto se doporučuje provádět nejen MR zobrazení CNS, ale i míchy. Asi 17 % pacientů má nespecifické změny při MR zobrazení nebo jsou přítomna ložiska v bílé hmotě mozkové. Více než 50 % pacientů má více ložisek v CNS.
Přítomnost CNS postižení u ECD představuje negativní prognostický faktor [32]. Postižení CNS u jednoho z našich pacientů ilustrují obrázky 5 a 6.


Dalším projevem může být infiltrace orbit, často bilaterální. Tato infiltrace způsobí exoftalmus, který může být i značně masivní. Exoftalmus bývá až u 37 % případů ECD. Infiltrace orbity může způsobovat bolest pociťovanou v orbitě, dále obrnu očních nervů a v nejhorším případě i slepotu.
Diferenciálně diagnostiky je nutno myslet na Gravesovu chorobu, granulomatózní choroby, lymfomy a obrovskobuněčnou arteritidu [33].
Také vedlejší nosní dutiny jsou často infiltrovány ECD, nejčastěji maxilární a sfenoidální sinusy, méně často etmoidální [31].
Endokrinní manifestace
V podstatě může dojít k postižení kterékoli endokrinní žlázy, ale nejčastější je diabetes insipidus, který bývá přítomen u 33 % pacientů a často je prvním příznakem ECD. V případě postižení hypofýzy u ECD se nemusí jednat jen o postižení sekrece vazopresinu. Při analýze dalších hypofyzárních hormonů byl u nemocných s ECD přítomný deficit somatotropinu (78,6 %), přítomnost hyperprolaktinemie (44,1 %), deficit gonadotropinů (22,2 %), deficit tyreotropinu (9,5 %) a deficit ACTH (3,1 %) [34].
Zřetelná infiltrace stopky hypofýzy při MR zobrazení byla prokazatelná jen u 24,4 % pacientů s přítomným hormonálním deficitem při ECD. Infiltraci stopky hypofýzy u našeho pacienta ilustruje obrázek 7.

U části pacientů s ECD byla snížena hladina gonadotropinu a testosteronu a současně vznikla testikulární insuficience (53 %). Sonograficky byla testikulární infiltrace prokazatelná u 29 % vyšetřených, obvykle oboustranná.
Podle CT vyšetření byla infiltrace nadledvinek přítomná u 39 % vyšetřených s ECD, ale k adrenální insuficienci dochází výjimečně. Až u 72 % pacientů s diabetes insipidus při ECD jsou diagnostikovány i další poruchy, například diplopie s nálezem ložisek retrobulbárně nebo v oblasti chiasmatu [34].
Retroperitonální a renální postižení
Retroperitonální postižení je detekovatelné pomocí CT zobrazení až u 60 % pacientů s ECD. Často může být asymptomatické. Je způsobeno infiltrací tuku v okolí ledvin (perirenální oblast) a způsobuje obraz tzv. vlasatých ledvin (hairy kidney). Infiltrace ledvin s případnou poruchou drenáže se popisuje u 6 % pacientů s ECD. Perirenální fibrózu, obraz tzv. vlasatých ledvin ilustruje obrázek 8.
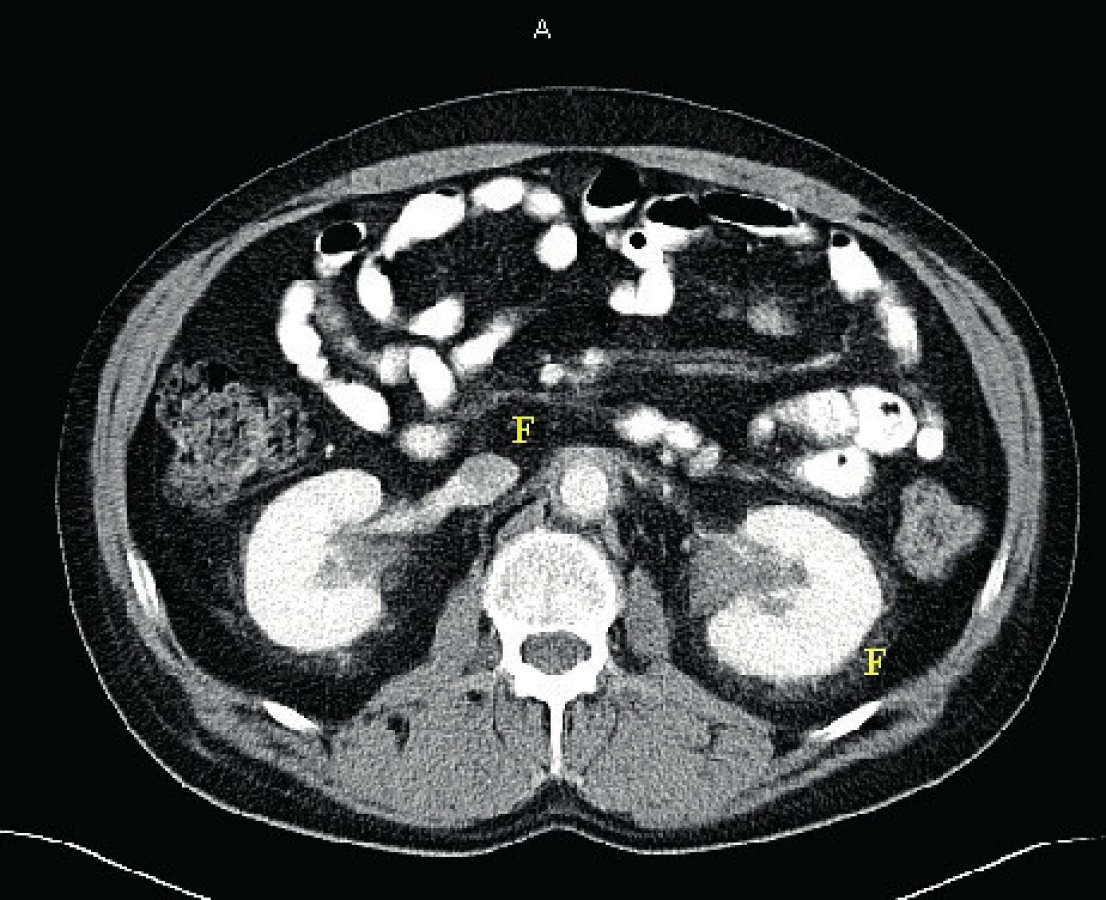
Asi u jedné třetiny pacientů je přítomna i retroperitoneální fibróza, která někdy způsobuje poruchu drenáže ledvin a vývoj hydronefrózy. Uvádí se, že při ECD dochází k zesílení stěny aorty po celém obvodu, zatímco u idiopatické retroperitoneální fibrózy je zadní část aorty postižena jen výjimečně [10 ].
Postižení plic a kardiovaskulárního aparátu
U 46–62 % pacientů s ECD je diagnostikováno kardiovaskulární postižení dominantně hrudní a břišní aorty. Má obraz periadventiciální infiltrace (coated aorta) a může přecházet na hlavní aortální tepny. Infiltrace srdce či koronárních tepen, případně pleurální a perikardiální infiltrace, je prokazatelná v 9–11 % ECD pacientů. Perikardiální infiltrace může způsobit tamponádu.
Nález na aortě typu coated aorta s periarteriální infiltrací obvykle nezpůsobuje žádné příznaky. Pokud však přechází na koronární tepny, může způsobit oblenění průtoku a infarkt myokardu.
Relativně často bývá při MR zobrazení srdce u ECD detekovatelný v pravé síni nádoru podobný infiltrát (atriální pseudotumor) ve stěně pravé síně, je prokazatelný až u 40 % pacientů. Častější je tato komplikace u ECD nemoci s mutací BRAFV600E. Pro detekci kardiálního postižení se za nejpřínosnější považuje MR zobrazení, zatímco pro postižení typu coated aorta je vhodnější CT zobrazení [35].
Infiltrace plicního parenchymu je popisována u 18 % pacientů. Postižení plic při HRCT (high-resolution computer tomography) zobrazení je prokazatelné u 30–50 % nemocných. Běžný rentgenový snímek plic obvykle nic patologického nezachytí, ale HRCT může prokázat zesílení interlobulárních sept nebo různé formy plicních opacit. V tekutině získané při bronchoalveolární laváži mohou být prokazatelné makrofágy a pěnité histiocyty. Plicní postižení je obvykle asymptomatické, vzácně se projeví jako kašel a dušnost. Při spirometrickém vyšetření obvykle bývá zjištěna restriktivní ventilační porucha se sníženou difuzní kapacitou [37].
Kožní postižení
Kožní postižení bylo detekováno u 25–30 % pacientů s ECD. Nejčastější formou jsou kožní ložiska typu xantelesma-like podobná specifickým kožním infiltrátům v oblasti očních víček (xanthelesma palpebrarum). Kožní postižení našeho pacienta ilustruje obrázek 9.
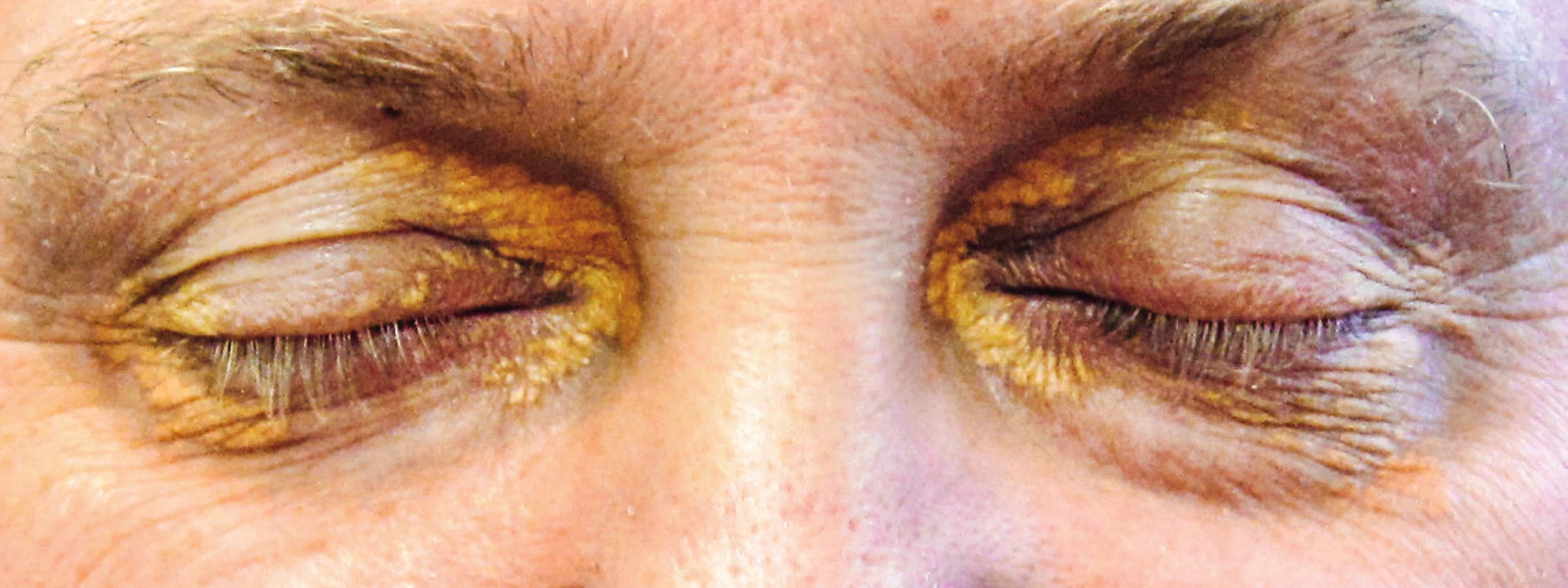
Jinak ECD ložiska se mohou nespecificky objevit kdekoli na kůži, obvykle jako papolunodulární ložiska, často na nohou, zádech a trupu [38]. V některých případech došlo ke vzniku četných drobných hemangiomů. Předpokládá se, že příčinou je zvýšená koncentrace vaskulárního endoteliálního růstového faktoru (VEGF) [39].
Asociace s dalšími chorobami
Nejčastěji je tato nemoc asociována s histiocytózou z Langerhansových buněk (LCH), četnost se odhaduje na 10–20 %. Nejčastějšími projevy LCH jsou osteolytická kostní ložiska či kožní projevy. Ložiska LCH a ECD obvykle nesou tu samou mutaci BRAFV600E [40].
Také Rosaiho-Dorfmanova nemoc může být asociována s ECD [41].
Doporučená vyšetření a navržená klasifikace ECD
Pro stanovení diagnózy ECD je histopatologické vyšetření základem jako u všech maligních chorob. V našich případech byla diagnóza stanovena z kostní biopsie. Problémem je, že biopsie kosti pro hodnocení vyžaduje jako standardní krok odvápnění. A proces odvápnění změní materiál tak, že může být nevhodný ke genetické analýze. Pokud jsou při typickém klinickém obraze dostupná kožní ložiska, je možnost diagnózu stanovit odběrem materiálu z kožních ložisek. Pokud je více patologických ložisek, doporučuje se současný odběr z více ložisek, protože vlivem zánětlivých změn typických pro ECD je histologický obraz měnlivý. Počet pěnitých histiocytů může být malý a v biopsii mohou převažovat nespecifické zánětlivé změny s fibrózou nebo fibrotické změny s ojedinělými histiocyty, jejichž cytoplazma není typicky pěnitá. Podle recentních mezinárodních doporučení pro diagnostiku a léčbu ECD [7, 8] je vhodné provést odběry na histopatologické vyšetření z více suspektních ložisek, protože nálezy mohou být divergentní. V tabulce 2 jsou uvedeny anamnestické a klinické údaje, dále zobrazovací a laboratorní vyšetření důležitá pro stanovení diagnózy ECD, vycházející z mezinárodních doporučení z roku 2014.
![Diagnostika ECD – mezinárodní doporučení z roku 2014 [7]](https://pl-master.mdcdn.cz/media/cache/media_object_image_large/media/image_pdf/4c9e1c09877b49c2ce1abde16a709f35.png)
V mezinárodním doporučení pro diagnostiku a léčbu ECD byla navržena klasifikace uvedená v tabulce 3, které může pomoci při rozhodování o léčebném postupu. Tato klasifikace reflektuje rozdíly v míře agresivity, rychlosti průběhu a v dominujícím postižení [7]. Při dlouhodobějším sledování bylo zjištěno, že 70 % pacientů žije déle než 5 let po stanovení diagnózy [12].

Sledování aktivity ECD
Hodnoty CRP jsou zvýšené u 50–80 % pacientů s ECD, jiné laboratorní změny jsou vzácnější. Anémie byla přítomna u 67 % pacientů, zatímco změny počtu destiček nejsou specifické, u 20 % jsou mírně snížené, ale mohou být také zvýšené, podobně jako leukocyty, protože jsou také reaktantem akutního zánětu. Počet lymfocytů bývá ve 25 % snížen. Tyto změny bývají důsledkem chronické zánětlivé reakce, ale mohou být také projevem současně probíhajícího myeloproliferativního onemocnění nebo MDS [8, 24]. Jaterní enzymy bývají zvýšeny jen u 5–10 % pacientů, ale až u 30 % pacientů bylo prokázáno zvýšení VEGF.
Výše uvedené laboratorní změny lze použít pro hodnocení aktivity nemoci. Zásadnější je ale přínos zobrazovacích vyšetření, především PET-CT vyšetření, které zobrazí jak místa aktivity nemoci, tak změny akumulace FDG [29]. Obrázek 10 ilustruje možnost dokumentovat léčebnou odpověď pomocí ET/CT. Existují také kritéria hodnotící neurologické a psychické symptomy ECD [42].

ZÁVĚR
Erdheimova-Chesterova choroba je velmi vzácná nemoc vycházející z buněk krvetvorby. Její přítomnost se však manifestuje ne odchylkami v počtu krvinek, ale poškozením řady důležitých orgánů a tkání, klinické projevy a symptomy jsou velmi různorodé, nejčastější je postižení dlouhých kostí dolních končetin a postižení CNS. K projevům ECD patří také endokrinní manifestace, postižení ledvin, kardiovaskulární a kožní postižení a řada dalších projevů. Pacienti s ECD přicházejí k ambulantním specialistům různých odborností a je na těchto lékařích, aby zorganizovali vhodné diagnostické vyšetření a v indikovaných případech odběry vzorků na histopatologické vyšetření.
Ke stanovení diagnózy ECD může pomoci provedení PET/CT vyšetření, původně indikovaného pro zvýšené teploty či horečky nejasného původu (FUO – fever of unknown origin). PET/CT může odhalit pro ECD charakteristický obraz zvýšené akumulace FDG v dlouhých kostech a CT zobrazení může odhalit pro ECD typické zesílené kortikalis, perirenální fibrózu či změny typu coated aorta. Pokud se ECD prokáže, PET/CT má potenciál citlivě vyhodnocovat léčebnou odpověď. Odběr materiálu na diagnostiku ECD je vhodné dělat z ložisek s vysokou akumulací FDG a vždy požádat o stanovení mutace BRAFV600E, protože průkaz této mutace umožnuje použít cílenou léčbu [43–45].
Podíl autorů na přípravě rukopisu
KZ, AZ – příprava rukopisu
KM, JM, SK, ŠS – korekce a revize rukopisu
ŘZ, KR, NJ – korekce rukopisu, dodání obrazové dokumentace
Čestné prohlášení autorů
Autoři práce prohlašují, že v souvislosti s tématem, vznikem a publikací tohoto článku nejsou ve střetu zájmů a vznik ani publikace článku nebyly podpořeny žádnou farmaceutickou firmou.
Do redakce doručeno dne 20. 1. 2020.
Přijato po recenzi dne 31. 3. 2020.
prof. MUDr. Zdeněk Adam, CSc.
Interní hematologická a onkologická klinika LF MU a FN Brno
Jihlavská 20
25 00 Brno
e-mail: adam.zdenek@fnbrno.cz
Zdroje
1. Chester W. Über lipoidgranulomatose. Virchows Arch Pathol Anat. 1930;279 : 561–602.
2. Haroche J, Charlotte F, Arnaud L, et al. High prevalence of BRAF V600E mutations in Erdheim-Chester disease but not in other non-Langerhans cell histiocytoses. Blood. 2012;120 : 2700–2703.
3. Blombery P, Wong SQ, Lade S, et al. Erdheim-Chester disease harboring the BRAF V600E mutation. J Clin Oncol. 2012;30:e331–332.
4. Diamond EL, Durham BH, Haroche J, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov. 2016;6 : 154–165.
5. Emile J-F, Diamond EL, Hélias-Rodzewicz Z, et al. Recurrent RAS and PIK3CA mutations in Erdheim-Chester disease. Blood. 2014;124 : 3016 – 3019.
6. Emile J-F, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127 : 2672–2681.
7. Diamond E, Dogna L, Hyman DM, et al. Consensus guidelines for diagnosis and clinical management of Erdheim Chester disease. Blood. 2014;124 : 483–492.
8. Papo M, Emile JE, Maciel TT, et al. Erdheim-Chester disease: a concise review. Current Rheumatol Rep. 2019;21–66.
9. Cohen-Aubart F, Emile J-F, Carrat F, et al. Phenotypes and survival in Erdheim-Chester disease: Results from a 165-patient cohort. Am J Hematol. 2018;93:e114–117.
10. Estrada-Veras JI, O’Brien KJ, Boyd LC, et al. The clinical spectrum of Erdheim-Chester disease: an observational cohort study. Blood Adv. 2017;1 : 357–366.
11. Haroche J, Amoura Z, Wechsler B, et al. Erdheim-Chester disease. Presse Med. 2007;36 : 1663–1668.
12. Stoppacciaro A, Ferrarini M, Salmaggi C, et al. Immunohistochemical evidence of a cytokine and chemokine network in three patients with Erdheim-Chester disease: implications for pathogenesis. Arthritis Rheum. 2006;54 : 4018–4022.
13. Dagna L, Girlanda S, Langheim S, et al. Erdheim-Chester disease: report on a case and new insights in its immunopathogenesis. Rheumatology (Oxford). 2010;49 : 1203–1206.
14. Pacini G, Cavalli G, Tomelleri A, et al. The fibrogenic chemokine CCL18 is associated with disease severity in Erdheim-Chester disease. Oncoimmunology. 2018;7: e1440929.
15. Arnaud L, Gorochov G, Charlotte F, et al. Systemic perturbation of cytokine and chemokine networks in Erdheim-Chester disease: a single-center series of 37 patients. Blood. 2011;117 : 2783–2790.
16. Michaloglou C, Vredeveld LCW, Soengas MS, et al. BRAFE600 - associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436 : 720–724.
17. Kriegl L, Neumann J, Vieth M, et al. Up and downregulation of p16(Ink4a) expression in BRAF-mutated polyps/adenomas indicates a senescence barrier in the serrated route to colon cancer. Mod Pathol. 2011;24 : 1015–1022.
18. Cangi MG, Biavasco R, Cavalli G, et al. BRAFV600E-mutation is invariably present and associated to oncogene-induced senescence in Erdheim-Chester disease. Ann Rheum Dis. 2015;74 : 1596–1602.
19. Badalian-Very G, Vergilio J-A, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116 : 1919–1923.
20. Diamond EL, Durham BH, Haroche J, et al. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov. 2016;6 : 154–165.
21. Durham BH, Lopez-Rodrigo E, Abramson DH, et al. Activating mutations in CSF1R and additional receptor tyrosine kinases in sporadic and familial histiocytic neoplasms. Blood. 2018;132 : 49–55.
22. Janku F, Vibat CR, Kosco K, et al. BRAF V600E mutations in urine and plasma cell-free DNA from patients with Erdheim–Chester disease. Oncotarget. 2014;11 : 3607–3610.
23. Aitken SJ, Presneau N, Tirabosco R, et al. An NRAS mutation in a case of Erdheim Chester disease. Histopathology. 2016;66 : 316 – 319.
24. Durham BH, Roos-Weil D, Baillou C, et al. Functional evidence for derivation of systemic histiocytic neoplasms from hematopoietic stem/progenitor cells. Blood. 2017;130 : 176–180.
25. Milne P, Bigley V, Bacon CM, et al. Hematopoietic origin of Langerhans cell histiocytosis and Erdheim-Chester disease in adults. Blood. 2017;130 : 167–175.
26. Papo M, Diamond EL, Cohen-Aubart F, et al. High prevalence of myeloid neoplasms in adults with non-Langerhans cell histiocytosis. Blood. 2017;130 : 1007–1013.
27. Haroche J, Poulain S, Marceau-Renaut A, et al. Clonal hematopoiesis in Erdheim-Chester Disease. Blood. 2017;130 : 3788–3779.
28. Cohen-Aubart F, Emile J-F, Carrat F, et al. Phenotypes and survival in Erdheim-Chester disease: Results from a 165-patient cohort. Am J Hematol. 2018;93:E114–117.
29. Arnaud L, Malek Z, Archambaud F, et al. 18F-fluorodeoxyglucosepositron emission tomography scanning is more useful in followup than in the initial assessment of patients with Erdheim-Chester disease. Arthritis Rheum. 2009;60 : 3128–3138.
30. Lachenal F, Cotton F, Desmurs-Clavel H, et al. Neurological manifestations and neuroradiological presentation of Erdheim-Chester disease: report of 6 cases and systematic review of the literature. J Neurol. 2006;253 : 1267–1277.
31. Drier A, Haroche J, Savatovsky J, et al. Cerebral, facial, and orbital involvement in Erdheim-Chester disease: CT and MR imaging findings. Radiology. 2010;255 : 586–594.
32. Arnaud L, Hervier B, Néel A, et al. CNS involvement and treatment with interferon-α are independent prognostic factors in Erdheim - -Chester disease: a multicenter survival analysis of 53 patients. Blood. 2011;117 : 2778–2782.
33. Alper MG, Zimmerman LE, Piana FG. Orbital manifestations of Erdheim-Chester disease. Trans Am Ophthalmol Soc. 1983;81 : 64 – 85.
34. Courtillot C, Laugier Robiolle S, et al. Endocrine manifestations in a monocentric cohort of 64 patients with Erdheim-Chester disease. J Clin Endocrinol Metab. 2016;101 : 305–313.
35. Cohen-Aubart F, Guerin M, Poupel L, et al. Hypoalphalipoproteinemia and BRAFV600E mutation are major predictors of aortic infiltration in the Erdheim-Chester disease. Arterioscler Thromb Vasc Biol. 2018;38 : 1913–1925.
36. Haroche J, Amoura Z, Dion E, et al. Cardiovascular involvement, an overlooked feature of Erdheim-Chester disease: report of 6 new cases and a literature review. Medicine (Baltimore). 2004; 83 : 371 – 392.
37. Brun A-L, Touitou-Gottenberg D, Haroche J, et al. Erdheim - -Chester disease: CT findings of thoracic involvement. Eur Radiol. 2010;20 : 2579–2587.
38. Chasset F, Barete S, Charlotte F, et al. Cutaneous manifestations of Erdheim-Chester disease (ECD): clinical, pathological, and molecular features in a monocentric series of 40 patients. J Am Acad Dermatol. 2016;74 : 513–520.
39. Legendre P, Norkowski E, Le Pelletier F, et al. Glomeruloid haemangioma: a possible consequence of elevated VEGF in POEMS and Erdheim-Chester disease. Eur J Dermatol. 2018;28 : 784–789.
40. Hervier B, Haroche J, Arnaud L, et al. Association of both Langerhans cell histiocytosis and Erdheim-Chester disease linked to the BRAFV600E mutation. Blood. 2014;124 : 1119–1126.
41. Razanamahery J, Diamond EL, Cohen-Aubart F, et al. ErdheimChester disease with concomitant Rosai-Dorfman like lesions: a distinct entity mainly driven by MAP2K1. Haematologica. 2020;105: e5–e8.
42. Diamond EL, Reiner AS, Buthorn JJ, et al. A scale for patient-reported symptom assessment for patients with Erdheim-Chester disease. Blood Adv. 2019;3 : 934–938.
43. Mergancová J, Kubes L, Elleder M. A xanthogranulomatous process encircling large blood vessels (Erdheim-Chester disease?). Czechoslovak Med. 1988;11(1):57–64.
44. Kinkor Z. Závažné plicní postižení u Erdheim-Chesterovy nemoci (kazuistika). Česko-slov Patol Soud Lék. 2001;37 : 114–117.
45. Řehák Z, Koukalová R, Vašina J, et al. 18F-FDG PET/CT obraz Erdheimovy-Chesterovy nemoci – přehled českých pacientů. Nukleární medicína. 2018;7 : 50–56.
Štítky
Hematologie a transfuzní lékařství Interní lékařství OnkologieČlánek vyšel v časopise
Transfuze a hematologie dnes

2020 Číslo 3
- Srovnání vlivu omeprazolu a pantoprazolu na antiagregační účinek klopidogrelu
- Ivabradin zlepšuje kvalitu života starších pacientů se srdečním selháním
- Fixní kombinace kandesartan/amlodipin v terapii arteriální hypertenze
- Současné postavení a přínos sartanů v klinické praxi
- Prognostický význam hladiny natriuretických peptidů při léčbě empagliflozinem
Nejčtenější v tomto čísle
- Příznaky a projevy Erdheimovy-Chesterovy choroby
- Doporučený postup při řešení a vyšetřování potransfuzních reakcí
- Charakteristika a výsledky léčby mladších nemocných s akutní myeloidní leukemií pod 60 let: Analýza reálných dat z české databáze DATOOL-AML
- Aktuální pohled na léčbu nově diagnostikovaných nemocných s periferními T-lymfomy a novinky v této oblasti s důrazem na využití brentuximab vedotinu
Zvyšte si kvalifikaci online z pohodlí domova
Mazová zátka a její řešení
nový kurzVšechny kurzy